

Abstract
Intricate gene regulatory networks orchestrate biological processes and developmental transitions in plants. Selective transcriptional activation and silencing of genes mediate the response of plants to environmental signals and developmental cues. Therefore, insights into the mechanisms that control plant gene expression are essential to gain a deep understanding of how biological processes are regulated in plants. The chromatin immunoprecipitation (ChIP) technique described here is a procedure to identify the DNA-binding sites of proteins in genes or genomic regions of the model species Arabidopsis thaliana. The interactions with DNA of proteins of interest such as transcription factors, chromatin proteins or posttranslationally modified versions of histones can be efficiently analyzed with the ChIP protocol. This method is based on the fixation of protein-DNA interactions in vivo, random fragmentation of chromatin, immunoprecipitation of protein-DNA complexes with specific antibodies, and quantification of the DNA associated with the protein of interest by PCR techniques. The use of this methodology in Arabidopsis has contributed significantly to unveil transcriptional regulatory mechanisms that control a variety of plant biological processes. This approach allowed the identification of the binding sites of the Arabidopsis chromatin protein EBS to regulatory regions of the master gene of flowering FT. The impact of this protein in the accumulation of particular histone marks in the genomic region of FT was also revealed through ChIP analysis.
Keywords: Plant Biology, Issue 107, Developmental Biology, Arabidopsis thaliana, Chromatin ImmunoPrecipitation (ChIP), transcriptional regulation, flowering time
Introduction
During recent years a wide range of genetic, molecular and genomic tools have been developed in the model species Arabidopsis thaliana. This technology has facilitated enormously the progress in understanding how plant development is regulated. Among the developmental processes studied using Arabidopsis as a model, the genetic control of flowering time has been extensively analyzed. These studies have shown that plants modulate very precisely the time of flowering in response to endogenous cues such as hormones and the age of the plant, and also to environmental signals such as photoperiod and temperature that synchronize flowering time with the natural cycle of seasons1,2. The isolation and characterization of Arabidopsis mutants with alterations in the time of flowering has been determinant in unveiling a complex network of genes that regulate the flowering time in response to endogenous and environmental factors. These genetic circuits are integrated at the level of a few master genes that act as switches of flowering, and the exact timing of the floral initiation depends on the balance of flowering promoting and repressing activities that work upstream of the floral integrator genes1,3.
The functional characterization of genes identified for their role in the control of flowering initiation, aided by the recent use of genomic approaches, have revealed the central role of transcriptional regulation in the modulation of flowering time. In fact, many of the master genes of flowering encode transcription factors4. In addition, a number of chromatin remodeling protein complexes influence the expression of master genes of flowering. A number of the Arabidopsis mutants isolated for their altered flowering time turned out to carry mutations in genes encoding a variety of chromatin modifiers. Different chromatin remodelers that introduce posttranslational modifications in histone tails, reposition nucleosomes relative to the DNA or exchange canonical histones by histone variants are necessary for the proper regulation of flowering in Arabidopsis5,6. Some of these chromatin remodeling activities catalyze the deposition or removal of covalent modifications such as acetylation or methylation in specific histone residues. These histone marks are specifically recognized by specialized effectors that recruit other chromatin remodeling complexes, transcription factors or components of the transcriptional machinery to regulate the transcriptional activity of flowering genes.
Chromatin ImmunoPrecipitation (ChIP) allows the identification of in vivo DNA-binding sites for proteins of interest (Figure 1). This procedure takes advantage of the ability of certain chemicals to cross-link the proteins to the DNA. The resulting DNA-protein complexes can be then immunoprecipitated by using specific antibodies against transcription factors, chromatin-binding proteins, or particular modifications and heterologous epitopes (commonly referred to as “tags”) attached to the protein of choice. The DNA purified from these immunoprecipitates can be used as a template in quantitative PCR (qPCR) reactions to assess for enrichment of particular sequences of interest. In this way, the binding sites of transcription factors or the distribution of histone marks in particular genes can be established7,8. In addition, combined with Next Generation Sequencing (NGS) that enables massive parallel sequencing, ChIP technology has made possible the genome-wide identification of the binding sites of transcription factors as well as unveiling epigenomic landscapes of histone modifications. Furthermore, the simultaneous analysis of gene expression allows monitoring how the binding of transcriptional regulators or the deposition of particular histone marks correlate with the transcriptional activity of genes9.
The use of ChIP protocols in Arabidopsis has allowed assessing the impact that a variety of transcriptional regulators have on the chromatin organization of master genes of flowering and how these structural changes influence gene expression5,6. Previous results showed that the Arabidopsis locus EARLY BOLTING IN SHORT DAYS (EBS) acts as a repressor of flowering and mutants in this gene show an acceleration of flowering and upregulation of the master gene of flowering FT. In addition, loss-of-function mutations in FT fully suppress the early flowering phenotype of the ebs mutant plants, indicating that FT is required for the premature flowering of ebs mutants and that EBS is necessary for the repression of this master gene of flowering10,11. EBS encodes a PHD-containing protein that can specifically bind histone H3 di- and trimethylated in the lysine 4 residue (H3K4me2/3), suggesting a role for EBS in the chromatin-mediated repression of FT12. The use of the ChIP approach demonstrated that the Arabidopsis PHD-containing protein EBS10,11 binds regulatory regions of the floral integrator gene FT to repress its expression12. Additional data obtained through the use of ChIP technology showed that this protein is required to maintain low levels of histone acetylation, a hallmark of active transcription, in the chromatin of this master gene of flowering during initial stages of Arabidopsis development. These observations, together with genetic and gene expression data, demonstrate that this Arabidopsis PHD-containing protein has a central role in the fine tuning of flowering time by modulating the expression of the floral integrator gene FT12. The work presented here provides an optimized method useful not only for the analysis of histones but also for other chromatin associated proteins, and with increased efficiency and reduced experimental time. Furthermore, this report illustrates how the use of ChIP protocols has provided new insights into the relationship between changes in chromatin modifications and transcriptional states of plant genes, and how these chromatin-mediated mechanisms of gene expression control impact on the onset of flowering in Arabidopsis.
Protocol
1. Crosslinking of the Plant Material (1 hr)
Grow the Arabidopsis lines used in the experiment (wild type — WT — versus mutants, and/or lines expressing the tagged version of your protein of interest versus lines expressing the tag not fused to any protein) for 12-18 days on large Petri dishes (150 mm) with MS-agar medium (1 L: 1x Murashige & Skoog salts, 10 g sucrose, 0.5 g MES, pH 5.7 (KOH), 1% agar). Alternatively, grow plants on pots containing 3:1 mix of soil and vermiculite. CAUTION! Formaldehyde is toxic by inhalation, in contact with skin and if swallowed, and should be handled in a chemical hood wearing suitable personal protective equipment. Steps 1.2 – 1.6 should be performed under the fume hood.
Make little holes in the lid of the 50 ml tubes with a laboratory burner-heated needle. Add 40 ml of 1x PBS buffer and 1.08 ml of formaldehyde to a final concentration of 1% (stock 37% solution). Collect 1.5 g of plant material in those 50 ml tubes. Keep the tubes on ice during crosslinking.
Close the 50 ml tubes with the lid. Place the tubes in a desiccator and apply vacuum. Vacuum infiltrate the tissue for 10 min, then release the vacuum carefully, to prevent churning up the solution. Mix the sample. Repeat twice. After infiltration, observe the plant material and confirm that it becomes slightly translucent and tend to sink to the bottom of the tube (Figure 2).
Add 2.5 ml of 2 M Glycine to achieve a final concentration of 0.125 M. Apply vacuum for 5 min. Glycine acts as a competitive inhibitor of formaldehyde crosslinking.
Discard the PBS formaldehyde-glycine solution by inverting the closed 50 ml tube with holes in the lid.
Rinse the plantlets twice with 1x PBS and once with water discarding the washing solution as described in step 1.5. Dry them on a paper towel and freeze in liquid nitrogen. NOTE: Frozen tissue can be kept at –80 °C for weeks.
2. Preparation of the Antibodies (1 day)
NOTE: For immunoprecipitation, the use of magnetic beads conjugated with antibodies via a protein G or protein A linker with high affinity for the constant domain of the antibody heavy chain is recommended. The DNA-protein complexes can attach unspecifically to the surface of the beads-antibody conjugates. For that reason, it is necessary to perform controls without specific antibodies to quantify the non-specific background binding.
For each immunoprecipitation, prepare 15 µl of magnetic beads in a 1.5 ml microcentrifuge tube. The amount of beads depends on the quantity of the chromatin used for immunoprecipitation and also on the antibodies.
To wash, add 1 ml of Chromatin Immunoprecipitation (ChIP) dilution buffer to the beads, mix by rotation and place the 1.5 ml microcentrifuge tube in a magnetic rack. Wait around 1 min to let the beads attach to the magnet. With the 1.5 ml microcentrifuge tubes still in the rack, remove the supernatant by pipetting. Repeat twice. NOTE: For each plant line two immunoprecipitation sets are needed: one with the beads and antibody (Ab) and a second no antibody (No-Ab) control with magnetic beads only.
Resuspend the beads in 200 µl of ChIP dilution buffer. Add the required amount of the specific Ab to one of the two tubes prepared for each plant line (the exact dilution differs for each Ab and has to be optimized experimentally or, in the case of commercial antibodies, follow the instructions of the manufacturer). Use this sample for immunoprecipitation. Add the same amount of unspecific IgG to the other tube, which will be used as negative control.
Incubate O/N on a rotating wheel at 4 °C to let the antibody attach to the beads.
3. Chromatin Extraction (4 hr)
Grind thoroughly the frozen plant material in liquid nitrogen using mortar and pestle until the powder becomes homogenous and light green. Transfer the powder to a new 50 ml tube.
For steps 3.2 – 3.6, work under the fume hood. Add 30 ml of Extraction buffer 1 (ExB 1), and mix well to soak the tissue powder. From this step on, keep the samples at 4 °C at all times. Frozen tissue and liquid nitrogen leftovers will cause the buffer to freeze. Make sure to thaw the frozen tissue completely by inverting the tubes 4-5 times every 2 min.
Clear the slurry by passing it through a filtration tissue with pore size of 22-25 µm into a new 50 ml tube and centrifuge it at 1,000 x g for 20 min at 4 °C. NOTE: In this step, the nuclei and all the organelles will pellet. ExB 1 contains enough sucrose to provide high density and prevent the disruption of organelle structure.
Gently remove the supernatant by decantation. At this stage, observe a green pellet of around 2 ml.
Resuspend the pellet in 5 ml of ExB 2. Resuspension of the pellet will be difficult at this point. Centrifuge at 1,000 x g for 10 min at 4 °C. NOTE: ExB 2 contains 1% Triton X-100 to help burst the chloroplasts open and remove chlorophyll.
Resuspend the pellet in 300 µl of Extraction Buffer 3 (ExB 3).
In a clean microfuge tube, add 600 µl of ExB 3. Take the resuspended pellet from step 3.6 and carefully layer it on top of the clean ExB 3.
Spin for 1 hr at 16,000 x g at 4 °C. NOTE: This step helps removing the detergent from the sample.
Remove the supernatant by aspirating with a pipette. Resuspend the nuclear pellet in 300 µl of sonication buffer by slowly pipetting up and down to avoid bubble formation, which may affect sonication efficiency. Carefully pipette all the suspension out and transfer it to a clean 1.5 ml microcentrifuge tube. NOTE: This buffer contains SDS, to lyse the nuclei and release the chromatin.
Sonicate the suspension to lyse the nuclei and randomly shear the chromatin into small fragments. Use sonication conditions that render chromatin enriched in fragments between 200-800 bp (20-30 cycles with the settings 30 sec ON / 30 sec OFF at 4 °C for the sonication device available described in the Table of materials/reagents). Check the efficiency of sonication by electrophoretic separation of the obtained DNA fragments on an agarose gel13, loading 2 µl of sonicated chromatin (Figure 3). CAUTION! Make sure to wear ear protection equipment during the sonication step in case the ultrasound device is not contained in a soundproof cabinet. Crucial step! The chromatin fragmentation largely depends on the sonication equipment. The sonication conditions should be adjusted to achieve enrichment in the appropriate DNA sizes. See Figure 3 for an example of how to optimize chromatin fragmentation.
Spin the solution once at 12,000 x g for 10 min at 4 °C. Transfer the supernatant by pipetting to a clean 1.5 ml microcentrifuge tube. Discard the pellet. Take 1/10 of the supernatant to a new 1.5 ml microcentrifuge tube, mark it as Input and freeze at -20 °C.
Dilute the chromatin 10x with ChIP dilution buffer to dilute SDS to final concentration of 0.1%. Note: Samples can be frozen and stored at -20 °C for several weeks.
4. Immunoprecipitation of the Protein of Interest and Rescue of DNA (1 day, 3 hr)
Wash the beads coated with the Ab from step 2.4 three times with 1 ml of ChIP dilution buffer to remove excess of antibody as described in step 2.2. Resuspend in 200 µl of ChIP dilution buffer. Add 50 µl of isolated chromatin. Incubate O/N on a rotating wheel at 4 °C. NOTE: Check the concentration of the chromatin in microvolume UV-Vis spectrophotometer. 50 µl of isolated chromatin should contain more than 10 µg of DNA.
For steps 4.2-4.5 work in 4 °C. Wash the beads twice in 1 ml of Low Salt Washing buffer as described in step 2.2.
Wash the beads once in 1 ml of High Salt Washing buffer.
Wash the beads once in 1 ml of LiCl Washing buffer.
Wash the beads twice in 1 ml of TE buffer. After the last wash, make sure to fully remove the TE buffer left in the tube by aspiration with a pipette.
Take out the INPUT samples (step 3.9). Transfer 5 µl of the INPUT to a new 1.5 ml microcentrifuge tube and continue as with the ChIP samples. Reverse crosslinking by adding 200 µl of 5% chelating ion exchange resin, and incubate for 10 min at 95 °C shaking every 2-3 min. Spin down at 16,000 x g for 30 sec and carefully transfer the supernatant into a new microcentrifuge tube by pipetting. NOTE: While the traditional method for crosslinking reversal involves an O/N incubation with NaCl, the incubation with a chelating ion resin at 95 °C allows for efficient decrosslinking and elution of DNA in 10 min, therefore making the protocol one day shorter.
Add 2 µl of proteinase K (10 mg/ml) and incubate at 37 °C for 30 min to digest proteins and release DNA.
Stop the reaction by incubating at 95 °C for 10 min.
Quickly spin at 16,000 x g for 30 sec and transfer supernatant to a new tube by pipetting.
Clean the DNA isolated using a standard DNA fragments purification kit. NOTE: Proceed according to the manufacturer's instructions.
Transfer the DNA binding column from the kit to a new 1.5 ml microcentrifuge tube. Elute the purified DNA by adding 20 µl of DNase-free water to the center of the column and spinning at 16,000 x g for 60 sec. Stop point: Samples can be stored at -80 °C for several weeks.
5. Measuring Abundance of Binding Sites in the Immunoprecipitated DNA by qPCR (4 hr)
NOTE: DNA isolated from the precipitated chromatin has to be analyzed to determine which DNA fragments have been ChIP-ed from the total chromatin, due to its binding to the protein of interest.
Design primers for the genomic regions of interest with a melting temperature (Tm) of 60 °C and a GC content of 30%-80%. As chromatin is fragmented by sonication, the length of amplified sequence should not be longer than 150-200 nucleotides (Table 1). NOTE: A useful tool for primer design is Primer 3 program (http://bioinfo.ut.ee/primer3-0.4.0/). Additional guidelines for primer design are available in previous reports8,14,15.
Use a BLAST program to make sure that the primers are specific (especially promoters can share similar TF binding sequences) and test the primer efficiency using 1:10 dilutions in water of the input DNA (step 4.9). Certain regions of the genome will be purified better than others. It is therefore important to design PCR primers and check them on the diluted input DNA.
Dilute the purified DNA 1:10 with water and pipette 5 µl of the diluted DNA in a PCR tube for each reaction. NOTE: Dilute only the amount needed, as DNA can attach to the walls of the 1.5 ml microcentrifuge tubes. Multiple thawing-freezing cycles increase the number of attached DNA molecules. For each primer pair, amplification in the input, Ab-ChIP and No-Ab ChIP has to be analyzed.
To complete the PCR mix, add Sybr Green PCR master mix to a final concentration of 1x, and 0.5 µM of each primer. Fill up to 20 µl with DNase-free water.
Perform the qPCR reaction following the instructions of the Sybr Green PCR master mix manufacturer.
6. Data Analysis
NOTE: Among the existing ways of analyzing ChIP-data, two are most commonly used. The first of them is the fold enrichment method, also named as ‘relative enrichment’, 'signal over background' or 'relative to the no-antibody control'. The second method is named as “% of input”.
- Data analysis by fold enrichment method.
- Create a spreadsheet with samples names and raw Ct values that have been obtained after qPCR reaction.
- For each sample, subtract from its raw Ct value the raw Ct value obtained for the corresponding No-Ab control. NOTE: After this mathematical operation, Ct value for No-Ab sample will equal 0.
- Calculate fold enrichment by exponentiation operation with base 2 and exponent (power) equal to the negative remainder obtained in step 6.1.2 (Table 2). fold enrichment = 2- (Ct(sample) - Ct(No-Ab)) NOTE: In this method the abundance of a specific DNA fragment in the ChIP-ed sample is compared with the abundance of this fragment in No-Ab control. The assumption of this method is that the level of background signal is reproducible between different primer sets, samples, and replicate experiments. However, this ´noise´ signal levels do vary between primer sets, samples, and experiments.
- Data analysis by % of input method.
- Create a spreadsheet with samples names and raw Ct values that have been obtained for them after qPCR reaction.
- Adjust raw Ct values for input samples by subtracting a value of logarithm base 2 from the fraction of total extracted chromatin that was taken as an input. NOTE: In this protocol the subtracted value equals 3.32, as a 10% of total chromatin was taken as an input in this protocol. Log2(10) equals 3.32.
- Calculate % of input by multiplying by 100 the value of exponentiation operation with base 2 and exponent (power) equal to remainder of adjusted input values subtracted from raw Ct values of the sample (Table 3). % of input = 100*2(adjusted input – Ct(sample)) NOTE: With this procedure, the relation between specific DNA abundance in the ChIP-ed sample to the total isolated chromatin (input sample) is calculated. Further details on ChIP data analysis can be found in Haring et al.8.
Representative Results
Eight main steps can be singled out in this ChIP protocol for the identification of in vivo protein-DNA interactions, including growing and harvesting of plant material, cross-linking of chromatin, chromatin isolation, chromatin fragmentation, selective isolation of the complexes between DNA and the protein of interest by immunoprecipitation, protein digestion, DNA purification, and qPCR analysis (Figure 1). A crucial step in the ChIP protocol is the fixation of DNA-protein interactions in a cross-linking reaction. Typically, formaldehyde cross-linking is used in plant ChIP. Formaldehyde penetrates the tissue and creates methylene bridges between NH2 groups of proteins or proteins and nucleotides in the DNA16. An efficient fixation of nucleoprotein complexes in vivo requires that formaldehyde reaches the chromatin within plant nuclei, and it is essential that plant tissue is fully submerged in the formaldehyde buffer to ensure that during immunoprecipitation the protein-DNA complexes are effectively pulled down. Arabidopsis leaves are covered in wax layers that difficult the penetrance of the crosslinking agent into the cells. In addition, trichomes favor the formation of air bubbles on the surface of the leaves, preventing the formaldehyde solution from getting inside of the plant tissue. To overcome this limitation, in this step vacuum is applied to the samples, preventing the formation of bubbles in the fixation solution and improving the penetration of formaldehyde into the nuclei of all cells. Seedlings after efficient cross-linking treatment are shown on Figure 2.
Another key step in the ChIP protocol is chromatin shearing. Large chromatin fragments can result in low resolution mapping of the binding sites of the protein of interest in the DNA sequence. In contrast, very small fragments can prevent efficient amplification of the bound DNA by qPCR. According to empirical experience, the optimal chromatin size range for ChIP analysis should be between 200 and 600 bp, and the conditions to achieve the appropriate fragmentation of the chromatin have to be set depending on the sonication device available in the laboratory. Figure 3 illustrates the procedure to optimize chromatin shearing to achieve the maximum enrichment in the desired range of DNA sizes. Before sonication, chromatin consists mainly of very large fragments. An increased number of sonication cycles progressively reduces the average size of the DNA in ChIP samples, reaching the optimum enrichment in the range of 200-600 bp after 20-30 cycles (section 3.8 of the protocol and Figure 3).
Here, the identification of the EBS protein binding sites to regulatory regions of the floral integrator FT is shown as an example of the usefulness of the ChIP technique. The binding of EBS to the FT locus was unraveled through ChIP assays with Arabidopsis lines expressing a cMyc-EBS fusion protein under the control of the native EBS promoter (pEBS::cMyc-EBS). This tagged version of EBS is fully functional as shown by the wild type phenotype of ebs mutant plants expressing this construct. Four regions within the genomic FT locus were tested for EBS binding; one in the promoter region of the gene (FTII), two around the transcriptional start site (FTIV, FTVI), and another one in the first intron of FT (FTVII) (Figure 4A). As shown in Figure 4B, ChIP results demonstrate that the EBS protein can bind a regulatory sequence close to the transcriptional start site of the master gene of flowering FT12. Furthermore, the EBS protein can interact both in vitro and in vivo with histone deacetylases such as HDA6, another chromatin-related protein involved in the regulation of flowering time in Arabidopsis17. In order to assess the possible influence of EBS on the levels of histone acetylation in the genomic region of FT, ChIP assays were performed using an antibody directed against histone H3 acetylated in lysines 9 and 14, a histone mark correlated with transcriptionally active chromatin12. Moreover, these data illustrate how ChIP approaches can contribute to shed light on the molecular mechanisms that modulate the transcriptional regulation of plant developmental processes.
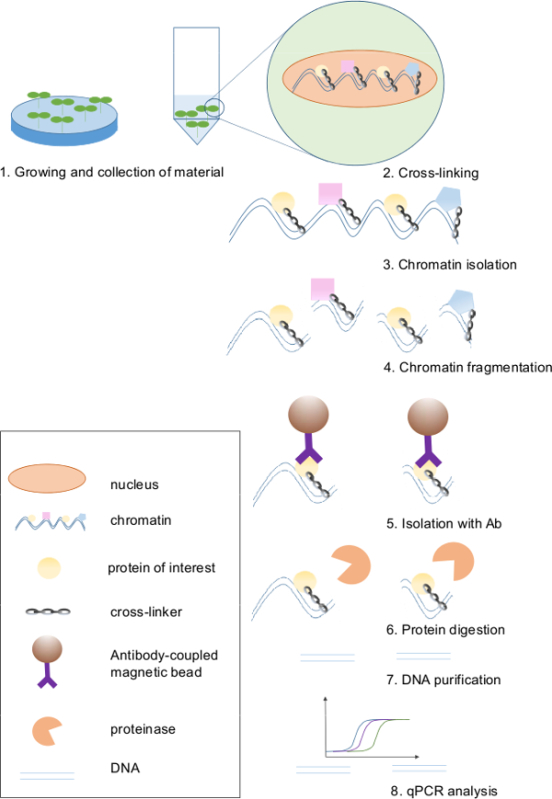 Figure 1. Scheme of the Chromatin Immunoprecipitation protocol. ChIP is a technique commonly used to investigate interactions between proteins and DNA, fixed by a chemical compound. Chromatin with preserved interactions is isolated from the nuclei and fragmented. By using antibodies against the protein of interest, we can select fragments of DNA attached to it. The antibody is conjugated to magnetic beads that enable fast and simple isolation using a magnetic rack. In the next step, proteins are removed by proteinase treatment and DNA is released. Purified DNA fragments are then used as templates in qPCR reactions to measure their relative abundance. Please click here to view a larger version of this figure.
Figure 1. Scheme of the Chromatin Immunoprecipitation protocol. ChIP is a technique commonly used to investigate interactions between proteins and DNA, fixed by a chemical compound. Chromatin with preserved interactions is isolated from the nuclei and fragmented. By using antibodies against the protein of interest, we can select fragments of DNA attached to it. The antibody is conjugated to magnetic beads that enable fast and simple isolation using a magnetic rack. In the next step, proteins are removed by proteinase treatment and DNA is released. Purified DNA fragments are then used as templates in qPCR reactions to measure their relative abundance. Please click here to view a larger version of this figure.
 Figure 2. Example of a tissue after cross-linking with formaldehyde. In order to investigate interactions between DNA and protein, in vivo complexes have to be fixed to make them stable and durable through the multiple steps of the protocol. In this version of the protocol, fixation occurs by formaldehyde treatment. Efficient penetration of formaldehyde through the tissue is crucial for the success of the ChIP method. (A) Before the cross-linking step the plant material floats on the surface of the solution and is covered with small air bubbles; the abaxial and adaxial surfaces of the leaves differ in color. (B) After fixation, plantlets should be visibly soaked in the solution, slightly transparent, and evenly immersed in the crosslinking solution. Please click here to view a larger version of this figure.
Figure 2. Example of a tissue after cross-linking with formaldehyde. In order to investigate interactions between DNA and protein, in vivo complexes have to be fixed to make them stable and durable through the multiple steps of the protocol. In this version of the protocol, fixation occurs by formaldehyde treatment. Efficient penetration of formaldehyde through the tissue is crucial for the success of the ChIP method. (A) Before the cross-linking step the plant material floats on the surface of the solution and is covered with small air bubbles; the abaxial and adaxial surfaces of the leaves differ in color. (B) After fixation, plantlets should be visibly soaked in the solution, slightly transparent, and evenly immersed in the crosslinking solution. Please click here to view a larger version of this figure.
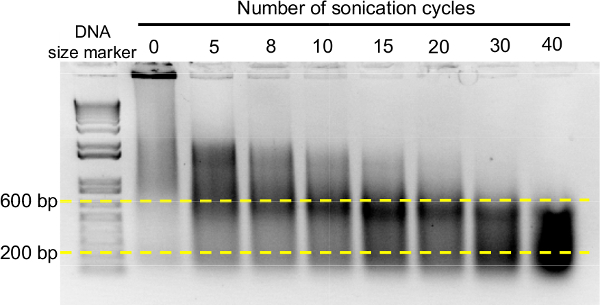 Figure 3. Optimization of the sonication conditions for ChIP analysis. Chromatin isolated from fixed material consists of broad range of fragments lengths with a strong band at the top of the gel representing extremely long chromatin sizes (second line on the left). With increasing number of sonication cycles, the peak of chromatin fragments sizes is shifted towards smaller fragments. In the experiment shown, optimal cycle numbers vary between 20 and 30, after which the optimal chromatin fragments ranging from 200 to 600 bp are more abundant in the ChIP sample. Please click here to view a larger version of this figure.
Figure 3. Optimization of the sonication conditions for ChIP analysis. Chromatin isolated from fixed material consists of broad range of fragments lengths with a strong band at the top of the gel representing extremely long chromatin sizes (second line on the left). With increasing number of sonication cycles, the peak of chromatin fragments sizes is shifted towards smaller fragments. In the experiment shown, optimal cycle numbers vary between 20 and 30, after which the optimal chromatin fragments ranging from 200 to 600 bp are more abundant in the ChIP sample. Please click here to view a larger version of this figure.
 Figure 4. ChIP analyses show that EBS binds FT and ebs mutations alter the level of histone H3 acetylation in this floral integrator gene. (A) Genomic region of FT with black boxes representing exons. Four fragments spanning regulatory regions of this floral integrator gene were tested (grey boxes) by designing specific qPCR primers. (B) EBS binds the FT IV region, located close to the transcriptional start site. Myc-EBS corresponds to ebs mutant plants expressing the cMyc-tagged version of EBS; Myc denotes transgenic plants expressing the cMyc epitope not fused to any Arabidopsis gene. (C) ebs mutant plants display higher abundance of H3K9,14Ac than wild type Landsberg erecta (Ler) in all four FT regions assessed. Error bars show standard deviation (B and C). Modified from previously published data in The Plant Cell12.
Please click here to view a larger version of this figure.
Figure 4. ChIP analyses show that EBS binds FT and ebs mutations alter the level of histone H3 acetylation in this floral integrator gene. (A) Genomic region of FT with black boxes representing exons. Four fragments spanning regulatory regions of this floral integrator gene were tested (grey boxes) by designing specific qPCR primers. (B) EBS binds the FT IV region, located close to the transcriptional start site. Myc-EBS corresponds to ebs mutant plants expressing the cMyc-tagged version of EBS; Myc denotes transgenic plants expressing the cMyc epitope not fused to any Arabidopsis gene. (C) ebs mutant plants display higher abundance of H3K9,14Ac than wild type Landsberg erecta (Ler) in all four FT regions assessed. Error bars show standard deviation (B and C). Modified from previously published data in The Plant Cell12.
Please click here to view a larger version of this figure.
| Forward | Reverse | Region tested |
| TCGTGCAAATGGATGGTTAG | TTTTTTATAAACAAGCGGCC | FT II |
| TGATTTCACCGACCCGAGTTAATGCAAATC | AACTCTGCTTACTATAAGAGGGTCTC | FT IV |
| AAACCACCTGTTTGTTCAAGATC | TCCTGAGGTCTTCTCCACCA | FT VI |
| GGGATTTTTCTTTGTTCCTCC | ATTCCACAACAGAGATTCATCA | FT VII |
Table 1. Primers used in this study.
| Sample | Raw Ct Values | Ct-Ct(No-Ab) | Fold enrichment 2 –(Ct(sample) – Ct(No-Ab)) |
| No-Ab | 34.5 | 0 | 1 |
| Ab | 29.2 | -5.3 | 39.4 |
Table 2. Example of ChIP data analysis by “fold enrichment” method. Ct Values for No-Ab samples represent the background signal caused by unspecific binding of DNA to the Ab/magnetic beads. These Ct values should exceed 30. Difference between Ct values of No-Ab and Ab sample should be higher than 1.
| Step 1 – Adjust the input | |||
| Adjusted input = Raw Ct(input) - Log2(fraction of total chromatin) | |||
| Raw Ct | input from total chromatin | Log210 | |
| Input | 29.5 | 10% | 3.32 |
| Adjusted input = 29.5 – 3.32 = 26.18 | |||
| Step 2 - % of input calculations | |||
| Sample | Raw Ct | Ct(adjusted input) –Ct(sample) | 2 Ct(adjusted input) –Ct(sample) *100% |
| Input | 26.18 | ||
| ChIP-Ab | 31.45 | -5.27 | 2.59 |
| No-Ab | 34.5 | -8.32 | 0.31 |
Table 3. Example of ChIP data analysis by “% of input” method. Inputs represent the total cross-linked chromatin isolated from nuclei, so the Ct values should be equal for all the primer pairs. Conventionally, 5% or 10% of the total chromatin is taken as an input. No-Ab represents a negative control sample — the beads not coated by specific antibodies against the protein or tag of interest. ChIP-Ab stands for the sample after the immunoprecipitation with the antibodies against the protein or tag of interest.
| PBS 10x |
| 1.3 M NaCl |
| 30 mM Na2HPO4 |
| 30 mM NaH2PO4 |
| pH 7 |
| Extraction buffer 1 (ExB 1) |
| 0.4 M sucrose |
| 10 mM Tris-HCl pH 8 |
| 10 mM MgCl |
| 5 mM β-Mercaptoethanol |
| Protease inhibitors |
| Extraction buffer 2 (ExB 2) |
| 0.25 M sucrose |
| 10 mM Tris-HCl pH 8 |
| 10 mM MgCl2 |
| 1% Triton X-100 |
| 5 mM β‑Mercaptoethanol |
| Protease inhibitors |
| Extraction buffer 3 (ExB 3) |
| 1.7 M sucrose |
| 10 mM Tris-HCl pH 8 |
| 0.15% Triton X-100 |
| 2 mM MgCl2 |
| 5 mM β‑Mercaptoethanol |
| Protease inhibitors |
| Sonication buffer |
| 50 mM Tris-HCl pH 8 |
| 10 mM EDTA |
| 1% SDS |
| Protease inhibitors |
| ChIP dilution buffer |
| 1.1% Triton X-100 |
| 1.2 mM EDTA |
| 16.7 mM Tris-HCl pH 8 |
| 167 mM NaCl |
| Protease inhibitors |
| Low Salt buffer |
| 150 mM NaCl |
| 0.1% SDS |
| 1% Triton X-100 |
| 2 mM EDTA |
| 20 mM Tris-HCl pH 8 |
| High Salt Washing buffer |
| 500 mM NaCl |
| 0.1% SDS |
| 1% Triton X-100 |
| 2 mM EDTA |
| 20 mM Tris-HCl pH 8 |
| LiCl Wash buffer |
| 0.25 M LiCl |
| 1% NP-40 |
| 1% sodium deoxycholate |
| 1 mM EDTA |
| 10 mM Tris-HCl pH 8 |
| TE buffer |
| 10 mM Tris-HCl pH 8 |
| 1 mM EDTA |
Table 4. Buffer composition.
Discussion
The ChIP protocol described here is a reproducible and powerful technique to analyze interactions between proteins and specific DNA sequences in vivo in Arabidopsis plants. A successful identification of binding sites for the proteins of interest requires an adequate selection of plant organs or developmental stages where the relevant interactions are actually taking place. In addition, it is critical to obtain an appropriate fixation of the plant material and an optimal shearing of the chromatin by sonication. Highly specific antibodies for the efficient immunoprecipitation of the selected protein/tag are also essential.
The use of ChIP approaches in plants faces some difficulties related to the presence of secondary metabolites and cell walls that can potentially interfere with different steps in the protocol. Moreover, the complexity of plant organs, often containing multiple cell types where the DNA-binding protein of interest can be differentially present or active, have hindered the use of this methodology. For that reason, whenever possible it is most advantageous to use in ChIP approaches homogeneous tissues where the presence and/or activity of the protein of interest have been already demonstrated. Complex plant materials containing different tissues tend to cause a dilution of the cell types where the protein actually functions. Tissue sectioning20 have been used in plants for studies in individual tissues or cell types. However, during recent years, different procedures have been developed for the isolation of cell-type specific chromatin in Arabidopsis8 to a range of optimal fragments (see Representative Results section)7. As discussed above, the conditions to achieve this size distribution greatly depend on the specific ultrasound equipment used. In any case, chromatin should be kept at low temperature to prevent reversal of the crosslinking favored by heat generated during sonication. The sonication device of choice should provide a good level of reproducibility, which together with optimal fragmentation conditions, is essential for ChIP analysis (Figure 3).
Another key factor in successful ChIP experiments is the chosen antibody for immunoprecipitation of the protein of interest. Antibodies raised against plant transcription factors can be useful for ChIP analysis, but these sera should be thoroughly tested because antibodies checked in Western blot experiments are not necessarily valid for ChIP. In addition, antibodies raised against different tags are frequently used for this protocol. ChIP-grade antibodies specifically recognizing a variety of tags are commercially available, and have the advantage that they have been tested in several laboratories. The drawback for the use of these commercial antibodies is that the protein of interest has to be fused to the corresponding epitope and expressed in transgenic lines (Figure 4B). In this case, it is advisable to ensure that the chimeric protein is functional by generating Arabidopsis transgenic lines bearing the fusion protein in a genetic background deficient in the gene encoding our protein of interest. The complementation of the phenotypic abnormalities of the mutant line in the transgenic plants will confirm that the fusion protein retains the normal activity of the endogenous protein. When transgenic lines are produced to identify the binding sites of transcriptional regulators, it is preferable to use the endogenous promoter to drive the expression of the tagged protein, preventing the problems associated with the utilization of constitutive strong promoters (see above). Specific antibodies for histone posttranslational modifications such as methylation or acetylation of specific residues are also frequently used in ChIP analyses to determine the epigenomic landscapes of genes involved in the regulation of a number of developmental processes, including the control of flowering time (Figure 4C). As for the antibodies against tags, ChIP-grade commercial antibodies are the preferred option in the case of histone marks.
ChIP experimental approaches coupled to gene expression analyses have been instrumental in increasing our knowledge on how transcription factors, chromatin remodeling proteins, and histone covalent modifications modulate the expression of genes involved in the regulation of plant biological processes and responses. Initially developed as a method to analyze the interaction of proteins present in the chromatin with particular loci or gene regions, the burst of genomic resources and NGS technology has allowed ChIP to become a powerful tool for the genome-wide profiling of DNA-binding proteins such as transcription factors or modified histones and histone variants. ChIP-chip and ChIP-seq have provided a new global dimension to the analyses of interactions between chromatin proteins and DNA in Arabidopsis25. ChIP-seq approaches are considered the alternative of choice to ChIP-chip, which is based on hybridization arrays and introduces some bias, given that these arrays contain a restricted number of probes.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
The authors would like to acknowledge The Plant Cell for allowing the use of some data published in this journal to elaborate the representative results described here in Figure 4. This work was supported by the EU 7FP Marie Curie-Initial Training Network EpiTRAITS (Grant Agreement 316965), and by the Spanish Ministerio de Economìa y Competitividad (grants BIO2010-15589 and BIO2013-43098-R).
References
- Andres F, Coupland G. The genetic basis of flowering responses to seasonal cues. Nat Rev Genet. 2012;13(9):627–639. doi: 10.1038/nrg3291. [DOI] [PubMed] [Google Scholar]
- Capovilla G, Schmid M, Pose D. Control of flowering by ambient temperature. J Exp Bot. 2015;66(1):59–69. doi: 10.1093/jxb/eru416. [DOI] [PubMed] [Google Scholar]
- Jarillo JA, Piñeiro M. Timing is everything in plant development. The central role of floral repressors. Plant Science. 2011;181(4):364–378. doi: 10.1016/j.plantsci.2011.06.011. [DOI] [PubMed] [Google Scholar]
- Pajoro A, et al. The (r)evolution of gene regulatory networks controlling Arabidopsis plant reproduction: a two-decade history. J Exp Bot. 2014;65(17):4731–4745. doi: 10.1093/jxb/eru233. [DOI] [PubMed] [Google Scholar]
- He Y. Chromatin regulation of flowering. Trends Plant Sci. 2012;17(9):556–562. doi: 10.1016/j.tplants.2012.05.001. [DOI] [PubMed] [Google Scholar]
- Jarillo JA, Pineiro M, Cubas P, Martinez-Zapater JM. Chromatin remodeling in plant development. International Journal of Developmental Biology. 2009;53(8-10):1581–1596. doi: 10.1387/ijdb.072460jj. [DOI] [PubMed] [Google Scholar]
- Saleh A, Alvarez-Venegas R, Avramova Z. An efficient chromatin immunoprecipitation (ChIP) protocol for studying histone modifications in Arabidopsis plants. Nat Protoc. 2008;3(6):1018–1025. doi: 10.1038/nprot.2008.66. [DOI] [PubMed] [Google Scholar]
- Haring M, et al. Chromatin immunoprecipitation: optimization, quantitative analysis and data normalization. Plant Methods. 2007;3:11. doi: 10.1186/1746-4811-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann K, et al. Chromatin immunoprecipitation (ChIP) of plant transcription factors followed by sequencing (ChIP-SEQ) or hybridization to whole genome arrays (ChIP-CHIP) Nat Protoc. 2010;5(3):457–472. doi: 10.1038/nprot.2009.244. [DOI] [PubMed] [Google Scholar]
- Gomez-Mena C, et al. early bolting in short days: An Arabidopsis mutation that causes early flowering and partially suppresses the floral phenotype of leafy. Plant Cell. 2001;13(5):1011–1024. [PMC free article] [PubMed] [Google Scholar]
- Piñeiro M, Gomez-Mena C, Schaffer R, Martinez-Zapater JM, Coupland G. EARLY BOLTING IN SHORT DAYS is related to chromatin remodeling factors and regulates flowering in Arabidopsis by repressing FT. Plant Cell. 2003;15(7):1552–1562. doi: 10.1105/tpc.012153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Gonzalez L, et al. Chromatin-dependent repression of the Arabidopsis floral integrator genes involves plant specific PHD-containing proteins. Plant Cell. 2014;26(10):3922–3938. doi: 10.1105/tpc.114.130781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor. Laboratory Press; 1989. [Google Scholar]
- Lorenz TC. Polymerase Chain Reaction: Basic Protocol Plus Troubleshooting and Optimization Strategies. J. Vis. Exp. 2012. p. e3998. [DOI] [PMC free article] [PubMed]
- Wong ML, Medrano JF. Real-time PCR for mRNA quantitation. Biotechniques. 2005;39(1):75–85. doi: 10.2144/05391RV01. [DOI] [PubMed] [Google Scholar]
- Dedon PC, Soults JA, Allis CD, Gorovsky MA. A simplified formaldehyde fixation and immunoprecipitation technique for studying protein-DNA interactions. Anal Biochem. 1991;197(1):83–90. doi: 10.1016/0003-2697(91)90359-2. [DOI] [PubMed] [Google Scholar]
- Yu CW, et al. HISTONE DEACETYLASE6 interacts with FLOWERING LOCUS D and regulates flowering in Arabidopsis. Plant Physiology. 2011;156(1):173–184. doi: 10.1104/pp.111.174417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, et al. Genome-wide profiling of histone H3 lysine 9 acetylation and dimethylation in Arabidopsis reveals correlation between multiple histone marks and gene expression. Plant Mol Biol. 2010;72(6):585–595. doi: 10.1007/s11103-009-9594-7. [DOI] [PubMed] [Google Scholar]
- Lafos M, et al. Dynamic regulation of H3K27 trimethylation during Arabidopsis differentiation. PLoS Genet. 2011;7(4):e1002040. doi: 10.1371/journal.pgen.1002040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcala M, Fenoll C, Escobar C. Laser microdissection of cells and isolation of high-quality RNA after cryosectioning. Methods Mol Biol. 2012;883:87–95. doi: 10.1007/978-1-61779-839-9_6. [DOI] [PubMed] [Google Scholar]
- Carter AD, Bonyadi R, Gifford ML. The use of fluorescence-activated cell sorting in studying plant development and environmental responses. Int J Dev Biol. 2013;57(6-8):545–552. doi: 10.1387/ijdb.130195mg. [DOI] [PubMed] [Google Scholar]
- Deal RB, Henikoff S. The INTACT method for cell type-specific gene expression and chromatin profiling in Arabidopsis thaliana. Nat Protoc. 2011;6(1):56–68. doi: 10.1038/nprot.2010.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng PY, Vakoc CR, Chen ZC, Blobel GA, Berger SL. In vivo dual cross-linking for identification of indirect DNA-associated proteins by chromatin immunoprecipitation. Biotechniques. 2006;41(6):694–698. doi: 10.2144/000112297. [DOI] [PubMed] [Google Scholar]
- Das PM, Ramachandran K, vanWert J, Singal R. Chromatin immunoprecipitation assay. Biotechniques. 2004;37(6):961–969. doi: 10.2144/04376RV01. [DOI] [PubMed] [Google Scholar]
- Kaufmann K, et al. Target genes of the MADS transcription factor SEPALLATA3: integration of developmental and hormonal pathways in the Arabidopsis flower. PLoS Biol. 2009;7(4):e1000090. doi: 10.1371/journal.pbio.1000090. [DOI] [PMC free article] [PubMed] [Google Scholar]