

Abstract
Human Immunodeficiency Virus 1 (HIV-1) infection frequently results in HIV-1 Associated Neurocognitive Disorders (HAND), and is characterized by a chronic neuroinflammatory state within the central nervous system (CNS), thought to be driven principally by virally-mediated activation of microglia and brain resident macrophages. HIV-1 infection is also accompanied by changes in cerebrovascular blood flow (CBF), raising the possibility that HIV-associated chronic neuroinflammation may lead to changes in CBF and/or in cerebral vascular architecture. To address this question, we have used a mouse model for HIV-induced neuroinflammation, and we have tested whether long-term exposure to this inflammatory environment may damage brain vasculature and result in rarefaction of capillary networks. In this paper we describe a method to quantify changes in cortical capillary density in a mouse model of neuroinflammatory disease (HIV-1 Tat transgenic mice). This generalizable approach employs in vivo two-photon imaging of cortical capillaries through a thin-skull cortical window, as well as ex vivo two-photon imaging of cortical capillaries in mouse brain sections. These procedures produce images and z-stack files of capillary networks, respectively, which can be then subjected to quantitative analysis in order to assess changes in cerebral vascular architecture.
Keywords: Neuroscience, Issue 107, Two-photon Microscopy, Capillary morphology, Cerebral cortex, Cerebral vasculature, In vivo imaging, Ex vivo imaging, Amira, Thin-skull cortical window, Arterial catheterization, Capillary skeletonization, Capillary density, Mouse
Introduction
Human Immunodeficiency Virus 1 (HIV-1) invades the brain during the acute phase of virus infection, and productively infects both microglia and brain resident macrophages, leading to their activation - and the release of both host-derived inflammatory mediators and soluble HIV-1 virotoxins such as Tat and gp120 (reviewed in 1,2). As a consequence, a chronic neuroinflammatory state becomes established in the CNS, which is thought to contribute to the pathogenesis of HIV-1 Associated Neurocognitive Disorders (HAND)3-5.
Chronic overexpression of HIV-1 Tat or interleukin (IL)-17A within the CNS of mice has been shown to result in microvascular rarefaction6,7. This raises the possibility that chronic neuroinflammation may contribute to the pathogenesis of HAND through effects on the cerebral vasculature. In order to further examine this question, we have developed methods to quantify cerebral vascular structures.
This paper describes a method for quantifying the number of capillary nodes, capillary segments, mean segment length, total segment length, mean capillary diameter, and total capillary volume using in vivo imaging of capillary networks through a thin skull cortical window (modified from previously described protocols)8,9, as well as ex vivo imaging of brain sections, using two-photon microscopy. This combined approach provides for a holistic quantitation of cerebral vascular parameters, since the in vivo thin-skull cortical window allows for the preservation of the cerebral environment, while ex vivo imaging of capillary networks in brain slices enables the reconstruction of complete, three-dimensional capillary networks - which can then be quantified using commercially available software.
Protocol
The University of Rochester's University Committee on Animal Resources approved all procedures performed in this paper.
1. Pre-surgical Preparation (and Mice)
Prepare the surgical area with all required equipment. Sterilize all tools used during the procedure beforehand using 70% ethanol. Optionally, use a glass-bead sterilizer or autoclave to sterilize the tools.
Place the mouse in an isoflurane induction chamber connected to an isoflurane vaporizer. Set isoflurane levels to 4% at a rate of 1 L/min. Note: for the experiments reported here, mice with a doxycycline (DOX) inducible HIV-1 Tat transgene driven by the astrocyte-specific glial fibrillary protein (GFAP) promoter (HIV Tat-Tg mice)10 were used to examine the effect of HIV-1 induced neuroinflammation on cerebral vascular structure, as previously described7,10.
Gently tip the induction chamber to observe the mouse's righting reflex. Once the righting reflex has been suppressed, set the isoflurane level to 2% and redirect the airflow from the induction chamber to the nose cone at the surgical bench.
Quickly move the mouse from the induction chamber to the water infused heating pad. Continue to apply isoflurane (1.5-2%) through the nose cone. Adjust anesthesia according to individual mouse tolerance assessed through respiratory rate (RR) monitoring. Note: The depression of RR can perturb physiologic gas parameters altering cerebral blood flow and vessel diameter assessment. Attempt to keep RR between 55-65 breaths per minute. Heart rate (HR) can also be used assess depth of anesthesia; keep HR between 300-450 beats per minute to prevent physiologic changes to vessel diameter. Typically, anesthesia will be adequate between 1.5-2.0% isoflurane at 1 L/min for a given mouse.
Perform a toe pinch to further check the depth of anesthesia. If the mouse does not respond, commence the surgical procedures.
2. Preparation of the Thin-skull Cranial Window
Apply artificial tear gel to the eyes of the mouse. Completely remove the hair from the scalp of the mouse using scissors or an electric razor.
Sterilize the scalp by applying povidone-iodine solution; allow to dry. Then, under a light microscope, remove the skin from the scalp of the mouse, completely exposing the parietal bones, caudal frontal bones, and bregma marking.
Apply a small quantity of a 10% ferric chloride solution to the skull in order to dry the membranes for easy removal. Remove the dried membranes with the #5/45 forceps by gently scraping the skull.
Apply a thin layer of glue around the headplate window. Gently press the headplate against the skull of the mouse, keeping the area of interest in the center of the window. Apply a drop of dental cement to the headplate to polymerize the glue.
Apply a thin layer of glue along the edge of the headplate window to create a reservoir in which to hold saline. Once the glue has dried, screw the headplate into the mouse headplate harness. Note: the mouse headplate harness used in this procedure is a structure with a metal base and two metal columns. On each column there is one spring and one wing nut. The headplate is placed on top of the spring, and the wing nut is tightened until the head is level and does not move.
- Remove any glue from the headplate window using a drill bit attached to a microtorque drill set at 6,000 rpm. Stop every 10-15 sec to prevent overheating of mouse cranium that can result in structural damage to vessels.
- Using a new drill bit, place the microtorque drill 1.5 mm laterally from midline (one-third of the diameter of the headplate window). Begin to thin the skull around this location at 4,000 rpm. Move the drill gently across the skull with no direct downward pressure.
- Cease drilling every 10-15 sec to prevent overheating the mouse cranium. During this time, remove the accumulated skull dust using a compressed air canister.
- Use drops of RT saline to cool cranium for 5-10 sec. Gently remove all fluid with a soft tissue to continue skull thinning.
For the final thinning of the skull, place a small volume of saline in the headplate reservoir and lightly sweep a dental microblade over the area of interest until small vessels are clearly visible.
3. Monitoring of Physiological Parameters
Once the skull is completely thinned, detach the mouse from the holder and place it on its back. Gently tape down both hind legs to clearly expose the medial thighs.
Remove the hair over both medial thighs with scissors or an electric shaver. Disinfect the surgical site by covering the thighs with povidone-iodine.
Pinch the skin on the medial right thigh above the femoral vein and artery using the #5 forceps. Gently pull the skin upwards and use scissors to cut and remove the skin. Note: the left thigh is reserved in the event of surgical complications (vascular anomalies, ruptured vessels).
Apply approximately 3-5 drops of 0.9% saline to surgical site. This allows for greater magnification of vessels, as well as keeping the site moisturized and preventing drying of the vessels. Separate the femoral vein from artery by bluntly dissecting into the femoral neurovascular bundle using two #5/45 forceps.
Place two, 3 cm pieces of surgical suture underneath the femoral artery approximately 1 cm apart. Twist the upper suture clockwise to create a vascular tourniquet that will help to prevent excessive blood loss during catheterization.
Make a small incision in the femoral artery using the spring scissors. Place the catheter in the artery through the incision and advance until the catheter is securely within the artery.
Untwist the upper suture (tourniquet) to assess for leaks and proper catheter placement. Secure the catheter in the femoral artery by tying two knots around the catheter within the vessel using the sutures.
Inject 10 µl of heparin (100 IU/ml) through the catheter to prevent blood clotting. Then, surgically close the right thigh by sewing the skin together with a 4-0 suture in a simple interrupted pattern.
Inject 50 µl of urethane (1.2 mg/g) intraperitoneally into the right lower quadrant. Five minutes later repeat with another 50 µl injection to the left lower quadrant for a total of 100 µl of intraperitoneal urethane. Slowly reduce the isoflurane levels to approximately 0.25% while concomitantly rechecking mouse for the depth of anesthesia. Note: Keep the needle superficial within the peritoneal cavity along the posterior abdominal wall so that it does not perforate the bowel. This can be accomplished by pinching the abdominal rectus muscles away from the viscera and then injecting.
Using a capillary tube, collect a 40 µl sample of arterial blood from the catheter. Attach the catheter to the blood pressure transducer fitted with a saline-filled four-way stopcock and heparin-filled syringe.
Tape the blood pressure transducer to the surgical apparatus to prevent the unintended removal of the catheter. Begin monitoring of mean arterial pressure.
Insert the blood filled capillary tube into a blood gas analyzer entry port. Once all the blood has been extracted, remove the capillary tube from the entry port. The O2 and CO2 blood gas levels will appear on a paper printed from the blood gas analyzer.
4. Injection of the Fluorescent Dye
Pinch the skin on the medial left thigh using the #5 forceps. Gently pull the skin upwards and use scissors to cut and remove the skin. Prepare dextran conjugated red emitting rhodamine dye (70 kDa) (10 mg/kg dissolved in saline).
- Locate the femoral vein.Using a 1 ml syringe with a 30 G needle attached, draw a previously prepared solution of a fluorescent dye appropriate for imaging to the 130 µl line of the syringe. Remove all air bubbles by drawing the dye completely into the syringe and firmly flicking the syringe wall.
- Bend the 30 G needle at an approximately 30 degree angle by pressing it against a hard surface bevel side up. Fill the needle with the dye and discard any excess liquid, leaving a 100 µl sample.
Inject the 100 µl sample of dye slowly into the femoral vein. After removing the needle, apply steady, gently pressure to the site of injection to stop any bleeding. Allow the dye to circulate for 5 min. Note: alternatively, the right femoral vein can be used instead for the fluorescent dye injection to prevent further blood loss through the creation of another surgical site. In addition, if the animal is bleeding uncontrollably from the surgical site, this may be an indication that too much heparin was given to flush the catheter. If there is no clotting within 10-15 min and the mouse is still bleeding, consider restarting the experiment with a new mouse.
Close the left thigh by sewing the skin together with a 4-0 suture, then carefully flip the mouse onto its stomach, and place it into the mouse headplate harness.
5. In Vivo Two-Photon Imaging
Move the surgical apparatus to the two-photon microscope, making sure to maintain continuous anesthesia levels. Place a small quantity of 0.9 % saline into the headplate reservoir and lower the microscope objective so that it comes into contact with the saline. Locate the area of interest using the brightfield-viewing objective
Set the two-photon laser excitation to a wavelength appropriate for the fluorescent dye. Begin two-photon imaging using the 25x objective. Note that in these experiments we used a dextran conjugated to a red emitting rhodamine dye that was excited at a wavelength of 780 nm. A 607/36 bandpass emission filter to detect the fluorescence from the dye, and a 480/20 bandpass emission filter was used to detect arterial autofluorescence
Locate a capillary bed on the view screen, and magnify this area using optical zoom 2. Acquire images of the capillaries using the two-photon imaging software.
After imaging is complete, sacrifice the mouse by cervical dislocation followed by decapitation.
6. Ex Vivo Two-Photon Imaging
Using the extra fine scissors, make an incision in the scalp from the interparietal bones to the frontal bones of the mouse. Secure the skin to the sides of the skull with the pointer finger and thumb.
Place the extra fine scissors underneath the medial interparietal bone and cut the skull along the sagittal suture. Stop cutting the skull approximately 3 mm after the bregma marking. Note: Apply upward pressure when cutting the skull to prevent cutting the brain.
Using the #5 forceps, separate the skull from the brain and carefully remove any meninges from the surface of the brain. Remaining meninges can accidently lacerate the brain; extreme care should be taken to avoid this. Gently slide the #5 forceps under the brain advancing slowly forward until brain is free from skull. Note: that downward pressure should be used in order to avoid puncturing the brain.
Place the brain in a brain sectioning tool specific for mice. Wash the brain by applying drops of artificial cerebrospinal fluid over the brain.
Remove a 2 mm coronal section of brain from bregma 0 to bregma -2. Place the coronal section onto a concave glass slide containing artificial cerebrospinal fluid with the 0 bregma (most cranial part of the section) facing upwards. Gently cover the brain slice with a glass cover slip. Note: Avoid pressing the glass once placed on the brain section as this may deform the vascular architecture.
Transfer the slide to the microscope stage and place a small quantity of 0.9% saline on the cover slip. Lower the microscope objective until it comes in contact with the saline. Locate the midline of the brain using the brightfield objective.
Begin two-photon imaging and locate the midline again using the 25x objective. Accomplish this by looking for the longitudinal fissure at the cortical surface of the coronal section. Place the right edge of the imaging screen on the midline and move the viewer screen over laterally three complete frames (approximately 1.5 mm from midline). Note that in these experiments, the imaging parameters were identical to those mentioned in the note in step 5.2.
Locate the depth at which capillaries are barely visible on the view screen. Lower the plane of focus an additional 20 µm to determine the top of the z-stack.
Set the image thickness to 1 µm. Lower the view screen for 100 µm and adjust laser power throughout such that less than 1% of the pixels are oversaturated. Note: the z-stack images are compiled from a series of 100 consecutive 500x500x1 µm images. The larger the z-stack the more accurate the post processing calculations will be. Generally, attempt to collect a z-stack of 100 µm or larger.
Press the XY Repeat icon followed by the adjacent XY icon. Once the z-stack is complete, press the Series Done icon and save the file.
7. Data Processing
- Open an in vivo capillary image in the ImageJ software. Manually set the pixel to distance ratio based on values obtained from the two-photon software.
- Select the *straight* icon from the tools menu. Draw a line extending from one capillary wall to the opposite capillary wall, and record the length provided by ImageJ. Note that it may be necessary to zoom in the image in order to clearly visualize the edges of the capillary walls.
- Repeat step 7.1.1 at different locations along the capillary, as well as for multiple capillaries in the image.
- Open the z-stack file into the analytical software such as Amira. Select the Filament Editor icon from the sub-application toolbar
- Set the viewer thickness to approximately 20. Move the viewer to either the top or bottom of the z-stack.
- Select the Trace icon and move the cursor over the loaded image. Place nodes on the capillaries at the locations described in Figure 2C; segments will automatically appear between adjacent nodes. Trace capillaries throughout the entire z-stack. Note: it will be necessary to place nodes in between end points and branch points in order to trace the capillaries throughout the z-stack.
- To remove these nodes, click the Remove Intermediate icon. Note: This can create erroneous looped segments that must be removed manually by selecting the loop using the Select Single icon, and then selecting the Remove Selected icon. If loops continue to appear in the same area during tracing, it is likely that the nodes were placed on different capillaries.
- Once the tracing is complete, select the Graph Info icon to open a spreadsheet containing the automatically extracted vascular parameters. The number of nodes and segments are available on the main view screen.
Representative Results
The thin-skull cortical window allows for in vivo two-photon imaging of cortical capillaries (Figure 1). A suitable area to image shows numerous, distinct capillaries (Figure 1A). In the same field of view, there is no arterial cell wall autofluorescence, and there may be other fluorescent signals, such as collagen fluorescence, induced by second harmonic generation11 (Figure 1B).
Once the preparation of the brain slice is complete, an imaging area approximately 1.5mm from midline is located (Figure 2A). A 100 µm z-stack produces a three-dimensional image used for analysis in the Amira analytical software (Figure 2B). The capillary network is then manually traced by placing nodes at the beginning and end of a capillary, or any location where one capillary branches into another (Figure 2C). This produces a fully skeletonized image from which morphological parameters are automatically extracted (Figure 2D). The number of capillary nodes, segments, mean segment length, total segment length, and total capillary volume can be extracted using two-photon ex vivo imaging of mouse brain slices (Figure 2).
Figure 3 shows representative results obtained using the method presented in this paper. In this experiment, a transgenic mouse model of HIV neuroinflammation was used10. Production of the HIV virotoxin Tat was induced in Tat+ mice by doxycycline-infused food for 12 weeks. The control group consisted of Tat- littermates fed the same food. There was no statistically significant difference between Tat+ and Tat- mice in any of the capillary parameters measured. Thus, 12 weeks' exposure of brain tissue to HIV-1 Tat is insufficient to cause rarefaction of the brain microvasculature. In contrast, our previously published data show that more prolonged (20 weeks) chronic CNS expression of Tat results in rarefaction of cerebral vasculature7. Thus, the data shown here (Figure 3) provides valuable new information in regards to the kinetics of Tat-induced vascular remodeling within the CNS of mice.
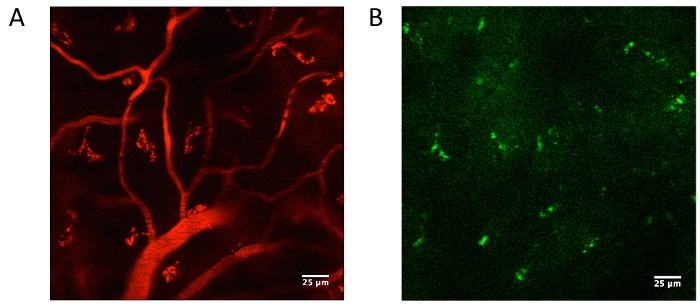 Figure 1. Acquisition of cortical capillary diameter.(A) Following a thin-skull cortical window preparation and intravenous fluorescent dye injection, two-photon microscopy was used to image cerebral vasculature. In our experiments, we used a dextran conjugated to a red emitting rhodamine dye that was excited at 780 nm, and fluorescence was visualized through a 607/36 bandpass emission filter. (B) The same field of view should show no arterial autofluorescence as detected through a 480/20 bandpass emission filter. Second harmonic generation may induce other fluorescent signals to appear through the filters11. Please click here to view a larger version of this figure.
Figure 1. Acquisition of cortical capillary diameter.(A) Following a thin-skull cortical window preparation and intravenous fluorescent dye injection, two-photon microscopy was used to image cerebral vasculature. In our experiments, we used a dextran conjugated to a red emitting rhodamine dye that was excited at 780 nm, and fluorescence was visualized through a 607/36 bandpass emission filter. (B) The same field of view should show no arterial autofluorescence as detected through a 480/20 bandpass emission filter. Second harmonic generation may induce other fluorescent signals to appear through the filters11. Please click here to view a larger version of this figure.
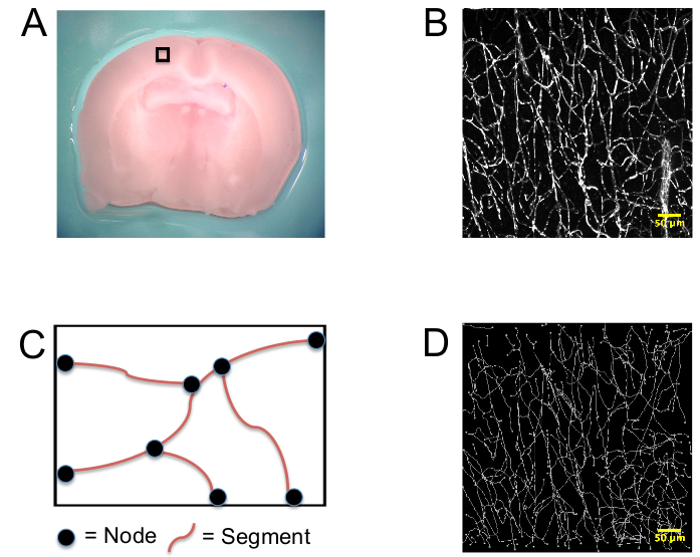 Figure 2. Generation of skeletonized capillary networks. (A) Mice were intravenously injected with a fluorescent dye and sacrificed. A 2 mm coronal section of brain was taken between bregma 0 and bregma -2, and imaging was performed at the 0 bregma approximately 1.5 mm from midline. Arepresentative image of the cortical location chosen for imaging (black box) from a C57BL/6 mouse is shown. This region, the somatosensory cortex, was chosen because we have previously shown that dysregulation of cerebral blood can occur at this site in Tat+ mice12. (B) Representational three dimensional z-stack images of capillary networks. (C) Diagram of the method used to manually trace capillaries during vessel quantification. (D) Representative image of skeletonized z-stack image. Please click here to view a larger version of this figure.
Please click here to view a larger version of this figure.
Figure 2. Generation of skeletonized capillary networks. (A) Mice were intravenously injected with a fluorescent dye and sacrificed. A 2 mm coronal section of brain was taken between bregma 0 and bregma -2, and imaging was performed at the 0 bregma approximately 1.5 mm from midline. Arepresentative image of the cortical location chosen for imaging (black box) from a C57BL/6 mouse is shown. This region, the somatosensory cortex, was chosen because we have previously shown that dysregulation of cerebral blood can occur at this site in Tat+ mice12. (B) Representational three dimensional z-stack images of capillary networks. (C) Diagram of the method used to manually trace capillaries during vessel quantification. (D) Representative image of skeletonized z-stack image. Please click here to view a larger version of this figure.
Please click here to view a larger version of this figure.
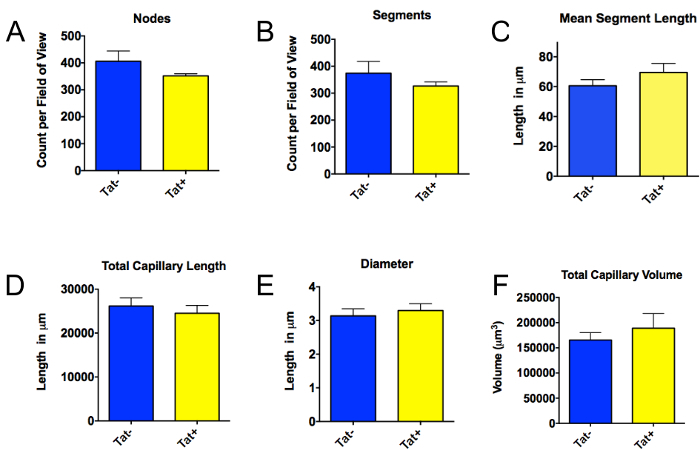 Figure 3. Quantification of cerebral vascular architecture in Tat- mice versus mice exposed to the candidate HIV virotoxin Tat for 12 weeks. Mice with a doxycycline (DOX) inducible HIV-1 Tat transgene driven by the astrocyte-specific glial fibrillary protein (GFAP) promoter (HIV Tat-Tg mice) were used to examine the effect of HIV-1 induced neuroinflammation on cerebral vascular structure, as described7. These mice (Tat+, n=3) were exposed to a chronic (12 week) regimen of Tat induction (i.e., 12 weeks of DOX). Tat- mice (n=4) were used as age-matched controls (these mice correspond to non-transgenic littermates of the Tat+ mice, and therefore do not express HIV-1 Tat). Like the Tat+ mice, the Tat- mice also received 12 weeks of DOX exposure. In the mice exposed to Tat for 12 weeks (Tat+), versus those not exposed to Tat (Tat-), there was no statistically significant difference in the number of nodes (A), the number of segments (B), mean segment length (C), or total capillary length (D), capillary diameter (for the Tat+ mice, we analyzed 14 capillaries in 6 imaging areas; for Tat- mice, we analyzed 7 capillaries in 4 imaging areas) (E), or total capillary volume (F). Since the data were not normally distributed (as determined by the Shapiro-Wilk test), the exact Wilcoxon rank sum test was used to calculate statistical significance (determined in this case as P<0.05). Please click here to view a larger version of this figure.
Please click here to view a larger version of this figure.
Figure 3. Quantification of cerebral vascular architecture in Tat- mice versus mice exposed to the candidate HIV virotoxin Tat for 12 weeks. Mice with a doxycycline (DOX) inducible HIV-1 Tat transgene driven by the astrocyte-specific glial fibrillary protein (GFAP) promoter (HIV Tat-Tg mice) were used to examine the effect of HIV-1 induced neuroinflammation on cerebral vascular structure, as described7. These mice (Tat+, n=3) were exposed to a chronic (12 week) regimen of Tat induction (i.e., 12 weeks of DOX). Tat- mice (n=4) were used as age-matched controls (these mice correspond to non-transgenic littermates of the Tat+ mice, and therefore do not express HIV-1 Tat). Like the Tat+ mice, the Tat- mice also received 12 weeks of DOX exposure. In the mice exposed to Tat for 12 weeks (Tat+), versus those not exposed to Tat (Tat-), there was no statistically significant difference in the number of nodes (A), the number of segments (B), mean segment length (C), or total capillary length (D), capillary diameter (for the Tat+ mice, we analyzed 14 capillaries in 6 imaging areas; for Tat- mice, we analyzed 7 capillaries in 4 imaging areas) (E), or total capillary volume (F). Since the data were not normally distributed (as determined by the Shapiro-Wilk test), the exact Wilcoxon rank sum test was used to calculate statistical significance (determined in this case as P<0.05). Please click here to view a larger version of this figure.
Please click here to view a larger version of this figure.
Discussion
The method described here can be applied to analyze brain microvascular structures in a wide range of experimental models/settings. For the success of this method, three critical steps must be mastered. First, the thin-skull window must not damage the skull or underlying brain. It is easy to puncture the skull during thinning, or cause heat induced vascular leakage. This can interfere with imaging as the fluorescent dye will leak into the plane of focus and obscure the capillaries. If the skull frequently breaks during the thin-skull preparation, it is most likely caused by too great a downward pressure. Holding the microtorque drill as close as possible to the burr allows for greater tactile control that will decrease the downward pressure on the skull.
Second, arterial catheterization should occur as quickly as possible once the artery has been cut. Failure to insert the catheter quickly can cause excessive blood loss that will compromise the physiological integrity of the mouse. Difficulty inserting the catheter is usually due to the relatively large size of the catheter compared to the femoral artery. It may be necessary to stretch the catheter in order to reduce its diameter. Additionally, the tips of the #5 forceps can be used to grab and enlarge the opening made in the artery to ease in the placement of the catheter.
Third, the duration of the in vivo surgical procedures should be minimized so that the urethane anesthesia can be administered as soon as possible. The isoflurane anesthesia can cause vasodilation of cerebral blood vessels13, thus skewing the capillary diameter data.
It should be acknowledged that the Amira analytical software can automatically extract the radii of manually traced vessels; however, when using this feature to analyze z-stack images from ex vivo brains, the value is inaccurate. This is because, after removing the brain, the cerebral capillaries are no longer pressurized, and may become morphologically distorted. The in vivo capillary diameter measurement circumvents this problem because the capillaries are pressurized under normal, physiological conditions.
It must also be noted that this method is not without limitations. First, significant training is required to master the in vivo imaging preparation. A researcher must learn to efficiently perform two technically challenging techniques (thin skull cortical window and arterial catheterization); an error in either technique can compromise the experiment and invalidate the data. Second, analysis of the ex vivo z-stacks is time-intensive. The skeletonization of one z-stack image can take up to 2.5 hr. Furthermore, it is advisable that this analysis be completed by an experienced researcher to ensure consistency of the skeletonization process (which involves careful manual tracing of blood vessels).
Once all aspects of this protocol are mastered, the data obtained provide a more in-depth and physiologically accurate quantitation of vascular parameters in comparison to other traditionally used methods that show capillary density as capillaries per unit volume, or capillaries per field of view. The in vivo imaging technique allows for the study of other parameters including, but not limited to, red blood cell velocity, arteriole dilation, and red blood cell flux7,12, which may also be able to be examined in longitudinal studies. Furthermore, it can easily be adapted and modified to study research questions related to brain microvascular structure. Such studies could include quantitating pathogenic changes in capillary morphology in other neurocognitive diseases, or measuring age-related changes in capillary density. Therefore, the method presented in this paper is a versatile and powerful tool for the quantitative analysis of cerebral vascular architecture.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank Maria Jepson, Dr. Paivi Jordan, and Dr. Linda Callahan at the University of Rochester Multiphoton Core for technical advice throughout the completion of this protocol. We also thank Dr. Changyong Feng for expert statistical advice, and Dr. Maiken Nedergaard at the University of Rochester Medical Center for the headplate design used in this paper. This work was supported in part by grants T32GM007356 and R01DA026325 from the National Institutes of Health (NIH); and by the University of Rochester Center for AIDS Research grant P30AI078498 (NIH).
References
- Kraft-Terry SD, Buch SJ, Fox HS, Gendelman HE. A coat of many colors: neuroimmune crosstalk in human immunodeficiency virus infection. Neuron. 2009;64(1):133–145. doi: 10.1016/j.neuron.2009.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghafouri M, Amini S, Khalili K, Sawaya BE. HIV-1 associated dementia: symptoms and causes. Retrovirology. 2006;3:28. doi: 10.1186/1742-4690-3-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antinori A, et al. Updated research nosology for HIV-associated neurocognitive disorders. Neurology. 2007;69(18):1789–1799. doi: 10.1212/01.WNL.0000287431.88658.8b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifford DB, Ances BM. HIV-associated neurocognitive disorder. The Lancet. Infectious diseases. 2013;13(11):976–986. doi: 10.1016/S1473-3099(13)70269-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindl KA, Marks DR, Kolson DL, Jordan-Sciutto KL. HIV-associated neurocognitive disorder: pathogenesis and therapeutic opportunities. Journal of neuroimmune pharmacology : the official journal of the Society on NeuroImmune Pharmacology. 2010;5(3):294–309. doi: 10.1007/s11481-010-9205-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann J, et al. CNS-targeted production of IL-17A induces glial activation, microvascular pathology and enhances the neuroinflammatory response to systemic endotoxemia. PloS one. 2013;8(2):e57307. doi: 10.1371/journal.pone.0057307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva JN, et al. Chronic central nervous system expression of HIV-1 Tat leads to accelerated rarefaction of neocortical capillaries and loss of red blood cell velocity heterogeneity. Microcirculation. 2014;21(7):664–676. doi: 10.1111/micc.12145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marker DF, Tremblay ME, Lu SM, Majewska AK, Gelbard HA. A thin-skull window technique for chronic two-photon in vivo imaging of murine microglia in models of neuroinflammation. Journal of visualized experiments : JoVE. 2010. [DOI] [PMC free article] [PubMed]
- Kleinfeld D, Mitra PP, Helmchen F, Denk W. Fluctuations and stimulus-induced changes in blood flow observed in individual capillaries in layers 2 through 4 of rat neocortex. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(26):15741–15746. doi: 10.1073/pnas.95.26.15741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce-Keller AJ, et al. Morphine causes rapid increases in glial activation and neuronal injury in the striatum of inducible HIV-1 Tat transgenic mice. Glia. 2008;56(13):1414–1427. doi: 10.1002/glia.20708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoumi A, Yeh A, Tromberg BJ. Imaging cells and extracellular matrix in vivo by using second-harmonic generation and two-photon excited fluorescence. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(17):11014–11019. doi: 10.1073/pnas.172368799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva J, et al. Transient hypercapnia reveals an underlying cerebrovascular pathology in a murine model for HIV-1 associated neuroinflammation: role of NO-cGMP signaling and normalization by inhibition of cyclic nucleotide phosphodiesterase-5. Journal of neuroinflammation. 2012;9:253. doi: 10.1186/1742-2094-9-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farber NE, et al. Region-specific and agent-specific dilation of intracerebral microvessels by volatile anesthetics in rat brain slices. Anesthesiology. 1997;87(5):1191–1198. doi: 10.1097/00000542-199711000-00024. [DOI] [PubMed] [Google Scholar]