

Abstract
Chronic alcohol consumption and withdrawal leads to anxiety, escalated alcohol drinking behavior, and alcohol dependence. Alterations in the function of key structures within the cortico-limbic neural circuit have been implicated in underlying the negative behavioral consequences of chronic alcohol exposure in both humans and rodents. Here, we used chronic intermittent ethanol vapor exposure (CIE) in male C57BL/6J mice to evaluate the effects of chronic alcohol exposure and withdrawal on anxiety-like behavior and basal synaptic function and neuronal excitability in prefrontal cortical and extended amygdala brain regions. Forty-eight hours after four cycles of CIE, mice were either assayed in the marble burying test (MBT) or their brains were harvested and whole-cell electrophysiological recordings were performed in the prelimbic and infralimbic medial prefrontal cortex (PLC and ILC), the lateral and medial central nucleus of the amygdala (lCeA and mCeA), and the dorsal and ventral bed nucleus of the stria terminalis (dBNST and vBNST). Ethanol-exposed mice displayed increased anxiety in the MBT compared to air-exposed controls, and alterations in neuronal function were observed in all brain structures examined, including several distinct differences between subregions within each structure. Chronic ethanol exposure induced hyperexcitability of the ILC, as well as a shift toward excitation in synaptic drive and hyperexcitability of vBNST neurons; in contrast, there was a net inhibition of the CeA. This study reveals extensive effects of chronic ethanol exposure on the basal function of cortico-limbic brain regions, suggests that there may be complex interactions between these regions in the regulation of ethanol-dependent alterations in anxiety state, and highlights the need for future examination of projection-specific effects of ethanol in cortico-limbic circuitry.
Keywords: bed nucleus of the stria terminalis, central amygdala, chronic alcohol exposure, limbic system, neuronal excitability, prefrontal cortex, synaptic transmission, patch-clamp electrophysiology
1. Introduction
Alcoholism is a chronic disease that has a high degree of comorbidity with multiple neuropsychiatric diseases, particularly those related to negative affect and anxiety. Chronic alcohol exposure to intoxication produces withdrawal-induced anxiety that is linked with craving for alcohol and increased rates of alcohol drinking (Becker, 2013; Fatseas et al., 2015; Heilig and Koob, 2007; Koob, 2003a; Roberts et al., 2000). This feed-forward cycle of alcohol exposure and withdrawal leads to alcohol dependence and co-morbid anxiety, which may be mediated by aberrant plasticity in brain regions that regulate emotional and reward-seeking behaviors (Burgos-Robles et al., 2013; Everitt et al., 2003; Feder et al., 2009; Herman, 2012; Pleil et al., 2015). In particular, limbic brain regions, including the central nucleus of the amygdala (CeA) and bed nucleus of the stria terminalis (BNST) in the extended amygdala, and cortical regions such as the prefrontal cortex (PFC), have previously been shown to mediate alcohol drinking, negative affect, and anxiety behaviors in humans and rodents (e.g., Bogg et al., 2012; Davis et al., 1997; Drevets, 2001; Gass et al., 2014; Johnstone et al., 2007; Kissler et al., 2014; Lowery-Gionta et al., 2012; Phillips et al., 2003; Pleil et al., 2015; Ressler and Mayberg, 2007; Sailer et al., 2008; Sparrow et al., 2012). Because the CeA and BNST are primary output structures of the extended amygdala, projecting to downstream brain regions that directly control stress and reward behaviors, and the PFC provides top-down modulation of their function during these behaviors, they may be critical sites of neuronal and synaptic adaptations of chronic alcohol exposure that contribute to anxiety and other long-term negative behavioral consequences.
Chronic intermittent ethanol vapor exposure (CIE) is a well-characterized mouse model of alcohol exposure that elicits an abstinence-induced escalation in voluntary ethanol consumption similar to that observed in human alcoholics (Becker, 2013; Becker and Lopez, 2004; Becker and Ron, 2014; Carrara-Nascimento et al., 2013; Crabbe et al., 2014; DePoy et al., 2013; Griffin, 2014; Griffin et al., 2009; Kissler et al., 2014; Lopez and Becker, 2005; Lopez et al., 2014; Repunte-Canonigo et al., 2014), as well as deficiencies in several other learned and affective behaviors, including attention set-shifting (Kroener et al., 2012), fear extinction (Holmes et al., 2012), reversal learning (Badanich et al., 2011), and anxiety and negative affect (Lowery-Gionta et al., 2014). These behaviors have been shown to be dependent on the PFC, CeA, and BNST, and withdrawal from CIE in mice produces alterations in several aspects of neural function in these regions. For example, altered endocannabanoid signaling (Pava & Woodward, 2014), NMDA receptor function and plasticity (Holmes et al., 2012; Kroener et al., 2012), and intracellular GABAergic neuroactive steroid levels (Maldonado-Devincci et al., 2014), have been described in the PFC. In addition, functional alterations reported during CIE withdrawal in the CeA and BNST include NMDA receptor expression and function (Kash et al., 2009), peptide signaling (Kash, 2012; Pleil et al., 2015), and synaptic transmission (Kash et al., 2009; Repunte-Canonigo et al., 2014; Silberman et al., 2013; Wills et al., 2012). In addition, extensive behavioral and neurochemical changes associated with withdrawal from chronic ethanol exposure in rats using similar CIE paradigms have been reported and reviewed elsewhere (e.g., Gilpin and Roberto, 2012; Heilig and Koob, 2007; Meinhardt and Sommer, 2015; Roberto et al., 2012).
While previous studies have characterized the effects of CIE withdrawal on specific neuronal or synaptic functions within individual brain regions of mice, very little research has examined the functional alterations in basal synaptic transmission or neuronal excitability across discrete brain regions that differentially govern ethanol-related outcomes. The goal of the current study was to provide a comprehensive characterization of alterations in basal neuronal function across brain regions shown to be important in regulating anxiety associated with withdrawal from chronic ethanol exposure, in order to provide a basis for future research and for the development of potential therapeutic approaches to treating alcoholism. Specifically, we measured the synaptic function and intrinsic excitability of neurons in the prelimbic and infralimbic PFC (PLC and ILC, respectively), lateral and medial CeA (lCeA and mCeA, respectively), and dorsal and ventral BNST (dBNST and vBNST, respectively) of C57BL/6J mice 48 hr following four cycles of CIE using slice electrophysiology, a time point we show is associated with increased anxietylike behavior in the marble burying test.
2. Materials and methods
2.1 Subjects
Adult male C57BL/6J mice (6–9 weeks old, Jackson Laboratories) were group-housed in a colony room with 12:12 hr light-dark cycle with lights on at 7 a.m. Mice had ad libitum access to rodent chow and water. All procedures were approved by the Institutional Animal Care and Use Committee of the University of North Carolina at Chapel Hill and performed in accordance with the National Institutes of Health guide for the care and use of laboratory animals.
2.2 Chronic intermittent ethanol vapor exposure paradigm
Chronic intermittent ethanol vapor exposure (CIE) was carried out as previously described by our group and others (Becker and Lopez, 2004; Kreifeldt et al., 2013; Lopez and Becker, 2005; Lowery-Gionta et al., 2014; Maldonado-Devincci et al., 2014). Briefly, mice were injected with pyrazole (1mmol/kg, i.p.; Sigma, St. Louis, MO) combined with saline for air-exposed controls (CON, n=17) or ethanol (1.6 g/kg) for ethanol-exposed (EtOH, n=17) mice and placed in cages inserted into the ethanol vapor chamber (La Jolla Alcohol Research, Inc., La Jolla, CA). Half of the cages were exposed to ethanol vapor and the other half to air to serve as control subjects. Mice remained in the vapor chamber for 16 hrs, from 5 p.m. to 9 a.m., daily for four “on” days in a row, constituting one cycle of CIE exposure. Mice underwent four cycles of this CIE exposure, each separated by three “off” days of abstinence in which they remained in their home cages (Figure 1A). This procedure has previously been shown to induce dramatic changes in behavior associated with alcohol abuse (e.g., Becker, 2013; Becker and Lopez, 2004; Holmes et al., 2012). BECs of ethanol-exposed mice were obtained immediately after removal from the vapor exposure periodically across all four cycles of CIE. The submandibular vein was quickly punctured and blood samples were collected in tubes lined with lithium heparin (Becton Dickinson & Company, Franklin Lakes, NJ). For BEC measurement, standards and samples were prepared using a commercially available alcohol dehydrogenase assay (Carolina Liquid Chemistries Corporation, Brea, CA) and loaded into a 96 well pre-read plate. The plate was placed into a reader at the end of the assay and analyzed with SoftMax Pro Software, Version 5 (Molecular Devices Corporation, Sunnyvale, CA). Air ethanol content was monitored in all cages throughout the exposure to ensure that there was no ethanol vapor in the air control cages and that ethanol vapor levels in the ethanol-exposed cages consistently had ethanol concentrations of 19-22 mg/l of air and produced and maintained blood ethanol concentrations (BECs) of 150-250 mg/dl by altering the ethanol dripping rate.
Figure 1.
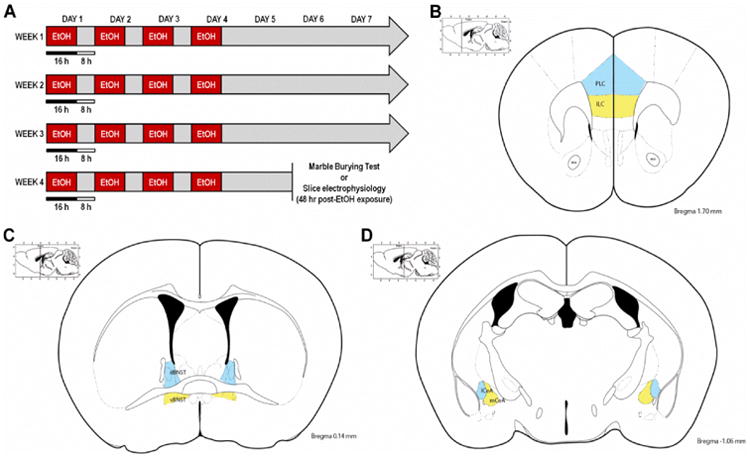
Experimental procedures for chronic intermittent ethanol vapor exposure (CIE), marble burying test (MBT), and slice electrophysiology recordings. A) Timeline for the CIE paradigm, in which mice were exposed to ethanol vapor for 16 hr/day for 4 consecutive days (constituting one cycle of CIE), followed by three days of abstinence, for four consecutive weeks. Mice underwent the MBT or were sacrificed for slice electrophysiology 48 hr after the last ethanol vapor exposure. B-C) Illustrations of brain subregions in which slice electrophysiology recordings were performed, including the: B) prelimbic cortex (PLC; in blue) and infralimbic cortex (ILC; in yellow) of the prefrontal cortex, C) dorsal BNST (dBNST; in blue) and ventral BNST (vBNST; in yellow), and D) lateral CeA (lCeA; in blue) and medial CeA (mCeA; in yellow).
2.3 Marble Burying Test
The marble burying test (MBT) was carried out using procedures modified from previous studies (Amodeo et al., 2012; Perez and De Biasi, 2015; Thomas et al., 2009). Each mouse was removed from its home cage and placed in a polycarbonate testing chamber lined with 3.5 cm of cob bedding (The Andersons, Inc., Maumee, OH) and left for 120 min to habituate. The mouse was briefly removed and placed in a holding chamber while 20 clean, black glass marbles (14mm, Rainbow Turtle, Portland, OR) were placed in the testing chamber in a 5×4 configuration. The mouse was returned to the testing chamber for a 30 min testing session without food or water access and then returned to its home cage. The number of marbles buried ≥75% was assessed and used for statistical analysis. Increased numbers of marbles buried is indicative of an increase in anxiety-like behavior.
2.4 Slice electrophysiology
Whole-cell voltage-clamp and current-clamp electrophysiological recordings were performed in dorsal and ventral BNST, lateral and medial CeA, and prelimbic and infralimbic regions of the PFC from acutely-prepared coronal brain slices according to landmarks based on the Allen Mouse Brain Atlas (Figure 1B-D), as previously described (Holmes et al., 2012; Li et al., 2012; McCall et al., 2013; Pleil et al., 2012; Pleil et al., 2015). Forty-eight hrs after the last ethanol vapor (or air control) exposure, mice were deeply anesthetized with isofluorane and decapitated. Brains were rapidly removed and placed in ice-cold sucrose-artificial cerebrospinal fluid (aCSF) containing (in mM) 194 sucrose, 20 NaCl, 4.4 KCl, 2 CaCl2, 1 MgCl2, 1.2 NaH2PO4, 10.0 glucose, and 26.0 NaHCO3 and saturated with 95% O2/5% CO2. Coronal slices 300 μm in thickness containing the PFC, BNST, and CeA were sectioned on a Leica VT1200 vibratome and stored in a holding chamber with 30°C, oxygenated aCSF containing (in mM) 124 NaCl, 4.4 KCl, 2 CaCl2, 1.2 MgSO4, 1 NaH2PO4, 10.0 glucose, and 26.0 NaHCO3. Slices were transferred to a submerged recording chamber (Warner Instruments, Hamden, CT), where they were perfused with heated, oxygenated aCSF at a rate of approximately 2 ml/min and allowed to equilibrate for 30 min before electrophysiological recordings.
Recording electrodes (3–5 MΩ) were pulled from thin-walled borosilicate glass capillaries with a Flaming-Brown Micropipette Puller (Sutter Instruments, Novato, CA). Recordings were performed in pyramidal neurons of layer 2/3 of the prefrontal cortical subregions; due to the multitude of neuron types expressed in the extended amygdala regions in addition to the heterogeneous nature of membrane properties within defined neuronal populations in these regions (e.g., Silberman et al., 2013), specific subtypes of neurons we recorded from could not be identified. Excitatory and inhibitory postsynaptic currents (EPSCs and IPSCs, respectively) were measured in voltage-clamp mode using electrodes filled with an intracellular recording solution containing (in mM): 135 Cs-methanesulfonate, 10 KCl, 10 HEPES, 1 MgCl2, 0.2 EGTA, 4 Mg-ATP, 0.3 GTP, 20 phosphocreatine. Lidocaine N-ethyl bromide (1 mg/ml) was included in the intracellular solution to block postsynaptic sodium currents. Neurons were held at -55 mV to isolate glutamatergic synaptic transmission and record spontaneous EPSCs (sEPSCs) or + 10 mV to isolate GABAergic synaptic transmission and record spontaneous IPSCs (sIPSCs) within individual neurons. Tetrodotoxin (TTX, 500 nM) was included in the bath aCSF to eliminate action potential-dependent spontaneous neurotransmission when recording miniature EPSCs (mEPSCs) and IPSCs (mIPSCs). Electrophysiological recordings of synaptic transmission were used to determine PSC frequency and amplitude, as well as to calculate synaptic drive ratios for spontaneous and miniature currents using the following equation: (EPSC frequency × amplitude) / (IPSC frequency × amplitude).
Intrinsic neuronal excitability and current-injected firing was measured in current-clamp mode using electrodes filled with an intracellular recording solution containing (in mM) 135 K-Gluc, 5 NaCl, 2 MgCl2, 10 HEPES, 0.6 EGTA, 4 Na2ATP, and 0.4 Na2GTP. Resting membrane potential (RMP) and current-injected firing of action potentials (using current ramps and steps) were evaluated in current-clamp configuration, including 1) the rheobase (defined as the minimum amount of current required to fire an action potential using a current ramp), 2) the action potential threshold (APT), defined as the membrane potential at which the first action potential fired, and 3) the relationship between increasing steps of current and the number of action potentials fired using a voltage-current plot protocol (V-I plot). These current-injection protocols were performed at both RMP and at a common membrane potential of -70 mV. Additionally, the voltage change produced by a 10 pA hyperpolarizing step from −70 mV was used to calculate input resistance (Res) of all individual neurons using the formula resistance = voltage/current (Res =V/I). Signals were digitized at 10 kHz and filtered at 3 kHz using a Multiclamp 700B amplifier and analyzed using Clampfit 10.3 software (Molecular Devices, Sunnyvale, CA). For all measures, recordings were performed in a maximum of two neurons per subregion, per mouse, and n's reported reflect the number of neurons for each measure.
2.5 Statistical Analysis
Appropriate statistical analyses with α = 0.05 were conducted to MBT and slice electrophysiology measures between EtOH and CON mice. Outliers identified by Grubb's outlier test were excluded from statistical analyses. Two-way repeated-measures ANOVAs were used to compare the relationship between 10 pA steps of current injection and the number of action potentials fired from V-I plots between CON and EtOH mice. Differences between groups for all other dependent measures of intrinsic excitability and synaptic transmission, as well as the number of marbles buried in the MBT, were evaluated using unpaired, two-tailed t-tests. All data are reported as mean ± standard error of the mean (SEM), and all electrophysiological results are presented in Summary Tables 1 and 2.
Table 1. Effects of CIE on synaptic transmission in cortico-limbic brain regions.
| sPSCs | mPSCs | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| EPSC freq | IPSC freq | EPSC amp | IPSC amp | SNR | EPSC freq | IPSC freq | EPSC amp | IPSC amp | SNR | |
| ILC | -- | ↓ | ↑ | -- | -- | -- | -- | -- | -- | -- |
| PLC | -- | -- | -- | -- | -- | -- | -- | -- | -- | -- |
| dBNST | -- | -- | -- | -- | -- | ↑ | -- | -- | -- | -- |
| vBNST | -- | -- | -- | -- | ↑ | ↑ | -- | -- | -- | ↑ |
| lCeA | ↓ | -- | -- | -- | ↓ | -- | -- | -- | -- | -- |
| mCeA | -- | -- | ↓ | -- | -- | -- | -- | ↑ | -- | -- |
Table 2. Effects of CIE on intrinsic neuronal excitability in cortico-limbic brain regions.
| @ RMP | @ -70 mV | |||||||
|---|---|---|---|---|---|---|---|---|
| RMP | Res | APT | Rheobase | VI | APT | Rheobase | VI | |
| ILC | ↑ | ↓ | -- | -- | -- | -- | -- | -- |
| PLC | -- | -- | ↓ | -- | ↑ | -- | -- | -- |
| dBNST | -- | -- | -- | -- | -- | -- | -- | -- |
| vBNST | -- | -- | -- | -- | -- | -- | ↓ | ↑ |
| lCeA | -- | -- | -- | -- | -- | -- | -- | -- |
| mCeA | -- | -- | -- | -- | ↓ | -- | -- | ↓ |
3. Results
3.1 Marble Burying Test
We conducted the marble burying test 48 hr after the final ethanol vapor exposure to assess the effects of chronic ethanol exposure and withdrawal on anxiety-like behavior (Amodeo et al., 2012; Perez and De Biasi, 2015; Thomas et al., 2009). EtOH-exposed mice buried significantly more marbles than air-exposed CON mice (Figure 2; t(10) = 3.673, p = 0.004), consistent with prior reports that chronic ethanol exposure procedures induce withdrawal-associated anxiety (Lowery-Gionta et al., 2015; Perez and De Biasi, 2015), a behavioral state previously shown to be mediated by the function of the cortical and amygdalar brain regions in which we performed slice electrophysiological recordings.
Figure 2.
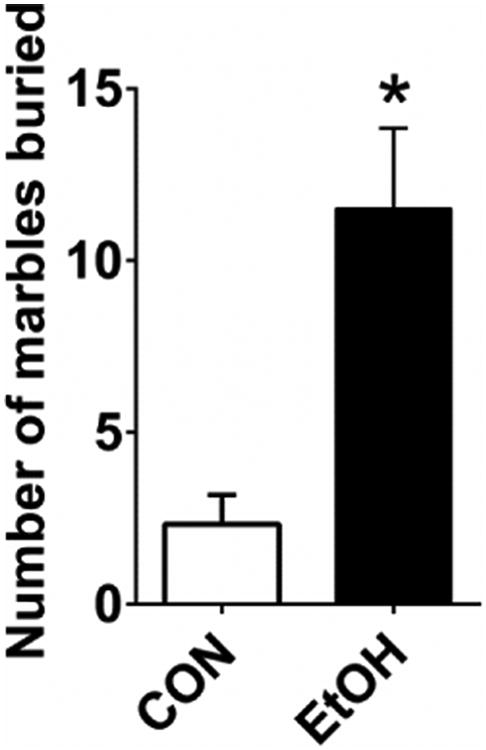
EtOH mice (n=6) buried significantly more marbles than CON mice (n=6) in the MBT 48 hr after the final vapor exposure of four-cycle CIE (t(10) = 3.673, p = 0.004).
3.2 Infralimbic PFC
We performed whole-cell voltage-clamp recordings from neurons in subregions of the PFC, BNST, and CeA that we hypothesized were involved in mediating the increased anxiety observed in the MBT and previously reported behavioral alterations following CIE. Results from electrophysiological recordings of synaptic transmission in layer 2/3 pyramidal neurons showed that CIE had effects on several aspects of synaptic transmission in the ILC. Neurons from EtOH mice had lower sIPSC, but not sEPSC, frequency than CON mice (Figure 3A,B; sEPSC: p > 0.20, sIPSC: t(16) = 2.45, p = 0.026), and larger sEPSC, but not sIPSC, amplitude than CON mice (Figure 3C; sEPSC: t(15) = 3.77, p = 0.002; sIPSC: p > 0.20). However, ILC neurons from CON and EtOH groups did not differ in sPSC synaptic drive ratio (Figure 3D; p > 0.20). In addition, there were no differences between groups in any mPSC measures (Figure 3E-G; p's > 0.05). These results suggest that there may be both presynaptic disinhibition and a postsynaptic increase in excitatory transmission of ILC neurons after CIE that is network activity-dependent.
Figure 3.
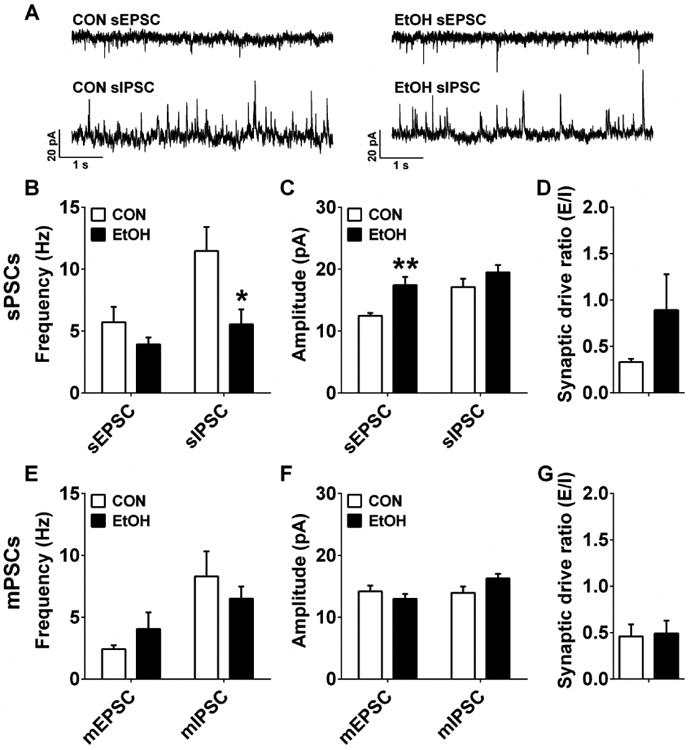
Synaptic transmission measures in the infralimbic cortex (ILC). A) Representative traces of sPSCs in the ILC of CON and EtOH mice. B) ILC neurons in EtOH mice (n=8) had significantly lower sIPSC, but not sEPSC, frequency than those in CON mice (n=10) (sEPSC: p > 0.20; sIPSC: t(16) = 2.45, *p = 0.026). C) ILC neurons in EtOH mice had significantly higher sEPSC, but not sIPSC, amplitude than those in CON mice (sEPSC: t(15) = 3.77, **p = 0.002; sIPSC: p > 0.20). D) There was no difference in sPSC synaptic drive ratio between CON and EtOH groups (p > 0.20). E-G) There were no differences in mEPSC or mIPSC frequency (E) or amplitude (F), nor in mPSC synaptic drive ratio (G) between CON (n=8) and EtOH (n=10) groups (p's > 0.05).
ILC neurons from EtOH mice were depolarized in comparison with those from CON mice (Figure 4A; t(18) = 2.77, p = 0.013), and they had lower Res (Figure 4B; t(13) = 2.32, p = 0.037), suggesting that CIE increased conductance of ILC neurons, leading to a depolarized resting state. However, there was no difference between groups in the APT or rheobase of ILC neurons at RMP (Figure 4C-E; p's > 0.05) or when held at a common potential of -70 mV (data not shown; p's > 0.15). Further, two-way repeated measures ANOVAs on the V-I plots at RMP (Figure 4F,G) and at -70 mV (data not shown) both result in a main effect of current injection magnitude (RMP: F(20,360) = 60.6, p< 0.0001; -70 mV: F(20,200) = 18.1, p < 0.0001), but no effect of CIE or interaction between CIE and current injection steps (p's > 0.70).
Figure 4.
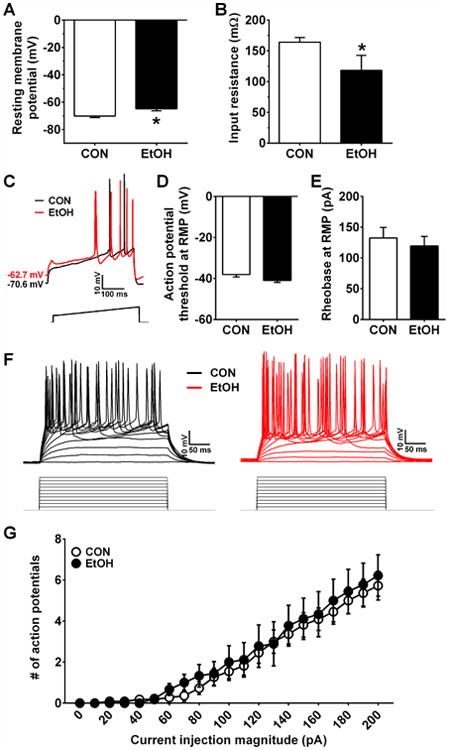
Excitability measures in the ILC. A,B) ILC neurons in EtOH mice (n=10) had a higher RMP (A; t(18) = 2.77, *p = 0.013) and lower Res (B; t(13) = 2.32, *p = 0.037) than those in CON mice (n=11). C) Representative traces of current-injected firing from ILC neurons in CON and EtOH mice during a ramp protocol of 120 pA/1 s at RMP, with stimulus waveform depicted below traces. D-E) There were no differences between CON and EtOH groups in the APT (D) or rheobase (E) during the ramp protocol (p's > 0.05). F) Representative traces of current-injected firing from ILC neurons in CON and EtOH mice during a step protocol of increased current steps of 10 pA/250 ms step, with stimulus waveforms depicted below traces. G) There was no difference between CON and EtOH groups in the relationship between current injection magnitude and the number of action potentials fired during the V-I plot at RMP (p's > 0.70).
3.3 Prelimbic PFC
Analyses of synaptic transmission measures in the PLC revealed no effects of CIE on the frequency, amplitude, or synaptic drive ratios for sPSCs or mPSCs (Figure S1; p's > 0.10). In addition, while the RMP and Res of PLC neurons were not affected by CIE (Figure S2A,B; p's > 0.35), several measures of current-injected firing were (Figure S2C,E). At RMP, the APT, but not rheobase, of PLC neurons from EtOH mice was significantly lower than that of CON mice (APT: Figure S2C, t(16) = 2.56, p = 0.021; rheobase: Figure S2D, p > 0.05). Further, a two-way ANOVA on the V-I plot at RMP revealed a main effect of current injection magnitude (F(20,340) = 39.0, p < 0.0001) and an interaction between CIE and current injection magnitude (F(20,340) = 2.27, p = 0.002), without a main effect of CIE (p > 0.40; Figure S2E). However, when all neurons were held at -70 mV, there were no differences between CON and EtOH groups in APT or rheobase (data not shown; p's > 0.30), and there was only a main effect of current injection magnitude in the ANOVA of the V-I plot (data not shown; current magnitude: F(20,220) = 8.75, p < 0.0001, CIE and interaction: p's > 0.85). Thus, while CIE did not appear to affect synaptic transmission and intrinsic neuronal excitability in the PLC, it did increase the threshold for firing of action potentials.
3.4 Dorsal BNST
Results from electrophysiological recordings of synaptic transmission showed that the dBNST was relatively unaffected by four cycles of CIE. There were no differences between dBNST neurons in CON and EtOH mice in sEPSC or sIPSC frequency or amplitude, nor in sPSC synaptic drive ratio (Figure S3A-C; p's > 0.10). Results from mPSCs showed that neurons from EtOH mice had significantly greater mEPSC amplitude than CON mice (Figure S3E; t(27) = 3.265, p = 0.003), but there were no other differences in mPSC measures (Figure S3D,F; p's > 0.10).
Results from measures of excitability and current-injected firing in the dBNST revealed no differences between CON and EtOH mice for RMP or Res (Figure S4A,B; p's > 0.35), nor for rheobase or APT when neurons were at RMP (p's > 0.20; data not shown) or held at −70 mV (Figure S4C,D; p's > 0.15). A two-way repeated-measures ANOVA on the V-I plot at RMP resulted in a main effect of current injection magnitude (F(20,540) = 72.5, p < 0.0001) but no effect of CIE nor an interaction between the two factors (p's > 0.90; data not shown). A similar result was found when neurons were held at -70 mV—there was a main effect of current injection magnitude (F(20,440) = 22.1, p < 0.0001) but no effect of CIE nor an interaction between the two factors (p's > 0.50; Figure S4E). Altogether, the net effect of chronic ethanol exposure and withdrawal on synaptic transmission and neuronal excitability in the dBNST was small; however this result may reflect somewhat opposing changes in distinct neuronal subtypes within this heterogeneous brain region.
3.5 Ventral BNST
In the vBNST, there were no effects of CIE on sEPSC or sIPSC frequency or amplitude (Figure 5A-C; p's > 0.05), but EtOH mice had significantly greater sPSC synaptic drive ratios than CON mice (Figure 5D; t(18) = 3.28, p = 0.029). There was also an effect of CIE on mEPSC frequency (Figure 5E; t(22) = 3.28, p = 0.026) but not mIPSC frequency (p > 0.60), nor mEPSC or mIPSC amplitude (Figure 5F; p's > 0.45). Similar to sPSCs, vBNST neurons from EtOH mice had significantly greater mPSC synaptic drive ratios than those from CON mice (Figure 5G; t(19) = 2.41, p = 0.026).
Figure 5.
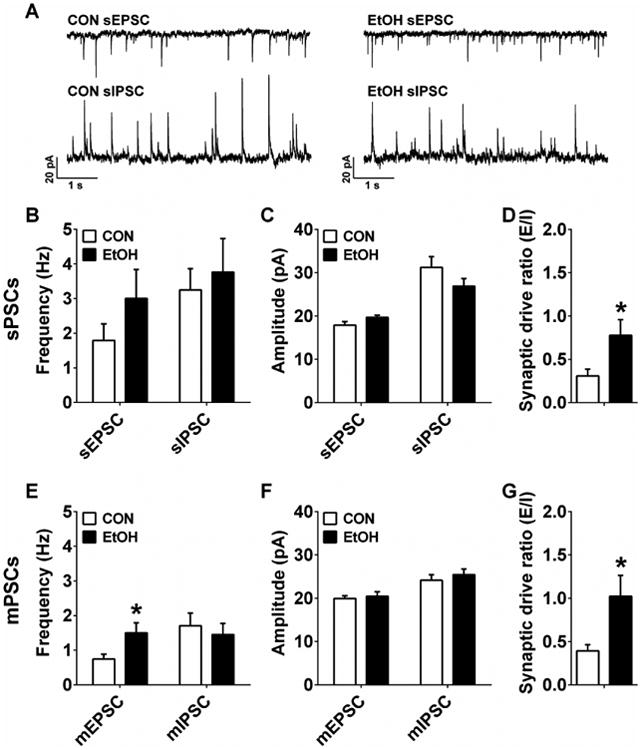
Synaptic transmission measures in the ventral BNST (vBNST). A) Representative traces of sPSC from CON and EtOH mice. B-D) There were no differences between vBNST neurons from CON (n=12) and EtOH (n=10) mice for sPSC frequency (B) or amplitude (C; p's > 0.05), but vBNST neurons in EtOH mice had greater synaptic drive ratios than those in CON mice (D; (t(18) = 3.28, *p = 0.029). E) mEPSC frequency was higher in vBNST neurons from EtOH (n=13) than CON (n=12) mice (t(22) = 3.28, *p = 0.026), but mIPSC frequency was not different (p > 0.60). F) There were no differences between groups in mEPSC or mIPSC amplitude (p's > 0.45). G) The mPSC synaptic drive ratio of vBNST neurons in EtOH mice was significantly higher than those in CON mice (t(19) = 2.41, *p = 0.026).
Excitability results showed that there were no differences in RMP or Res between vBNST neurons in CON and EtOH mice (Figure 6A,B; p's > 0.10), nor in rheobase or APT when neurons were at RMP (p's > 0.10; data not shown). A two-way repeated-measures ANOVA on the V-I plot at RMP resulted in a main effect of current injection magnitude (F(20,380) = 72.5, p < 0.0001) but no effect of CIE nor an interaction between the two factors (p's > 0.80; data not shown). When ventral BNST neurons were held at a common membrane potential of -70 mV, CIE significantly lowered the rheobase (t(22) = 2.45, p = 0.023; Figure 6C,E) but not the APT (Figure 6D; p > 0.10). In addition, an ANOVA on the V-I plot revealed main effects of CIE (F(1,20) = 5.95, p = 0.024) and current injection magnitude (F(20,400) = 80.6, p < 0.0001), as well as an interaction between these two factors (F(20,400) = 4.60, p < 0.0001; Figure 6F,G), indicating greater current-injected firing in vBNST neurons from EtOH mice. Together, results suggest that there is a shift toward excitation in the ventral BNST driven both by increased putative glutamate release onto ventral BNST neurons and increased intrinsic excitability.
Figure 6.
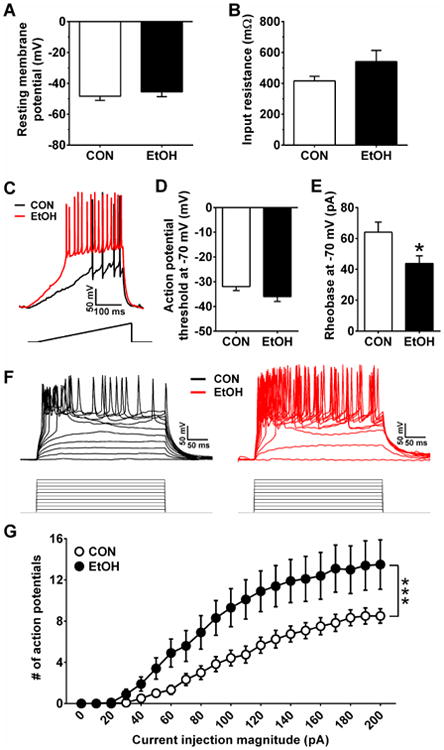
Excitability measures of neurons in the vBNST. A&B) There were no differences in the RMP (A) or Res (B) of vBNST neurons between CON (n=14) and EtOH (n=8) mice (p's > 0.10). C) Representative traces of current-injected firing from vBNST neurons in CON and EtOH mice when held at a common potential of -70 mV during a ramp protocol of 120 pA/1 s at RMP, with stimulus waveform depicted below traces. D&E) While there were no differences between groups in the action potential threshold of vBNST neurons when neurons were held at -70 mV (D; p > 0.10), neurons from EtOH mice had significantly lower rheobase than those from CON mice (E; t(22) = 2.45, *p = 0.023). F) Representative traces of current-injected firing from vBNST neurons in CON and EtOH mice when held at a common potential of -70 mV during a step protocol of increased current steps of 10 pA/250 ms step, with stimulus waveforms depicted below traces. G) Neurons from EtOH mice fired more action potentials per current injection step than neurons from CON mice in the V-I plot, indicated by a significant interaction between CIE and current injection magnitude (F(20,400) = 4.60, ***p < 0.0001).
3.6 Lateral CeA
Synaptic transmission results showed that sEPSCs, but not sIPSCs, in the lCeA of EtOH mice had a significantly lower frequency than those of CON mice (Figure S5A; sEPSC: t(17) = 2.20, p = 0.042; sIPSC: p > 0.85), but there were no differences between CON and EtOH mice in sEPSC or sIPSC amplitude (Figure S5B; p's > 0.20). The sPSC synaptic drive ratio in neurons from EtOH mice was also significantly lower than from CON mice (Figure S5C; t(15) = 3.19, p = 0.006). Results from mPSCs revealed no differences between lCeA neurons from CON and EtOH mice for any measures (Figure S5D-F; p's > 0.05), suggesting that effects of CIE on spontaneous synaptic transmission in the lCeA were network activity-dependent.
While the sPSC synaptic drive ratios in the lateral CeA were affected by CIE, measures of intrinsic excitability and current-injected firing were not (Figure S6). Neither the RMP nor Res of lCeA neurons in EtOH mice differed from those in CON mice (Figure S6A,B; p's > 0.35). There was no difference in the APT or rheobase when neurons were at their RMP (data not shown; p's > 0.15), nor when they were held at a common potential of -70 mV (Figure S6C,D; p's > 0.25). In addition, a two-way repeated-measures ANOVA on the V-I plot at RMP resulted in a main effect of current injection magnitude (F(20,320) = 11.8, p < 0.0001) but no main effect of CIE or interaction (data not shown; p's > 0.95). A similar result was found when neurons were held at -70 mV (Figure S6E; current injection magnitude: F(20,160) = 4.90, p < 0.0001, CIE and interaction: p's > 50).
3.7 Medial CeA
Synaptic transmission in the mCeA was generally unaffected by CIE. The frequency of sEPSCs and sIPSCs were not different between groups (Figure 7A,B; p's > 0.20), but CIE did result in a decrease in sEPSC amplitude without altering sIPSC amplitude (Figure 7A,C; sEPSC: t(18) = 2.51, p = 0.022; sIPSC: p > 0.55). sPSC synaptic drive ratio was not different between CON and EtOH groups (Figure 7D; p > 0.45). mPSC frequency measures were also unaltered in EtOH mice compared to CONs (Figure 7E; p's > 0.25). In contrast to sEPSC amplitude, mEPSC amplitude in mCeA neurons of EtOH mice was significantly larger than in CON mice (Figure 7F; t(16) = 2.19, p = 0.043); there was no difference between groups in mIPSC amplitude (p's > 0.05). Further, there was no effect of CIE on mPSC synaptic drive ratio (Figure 7G; p > 0.30).
Figure 7.
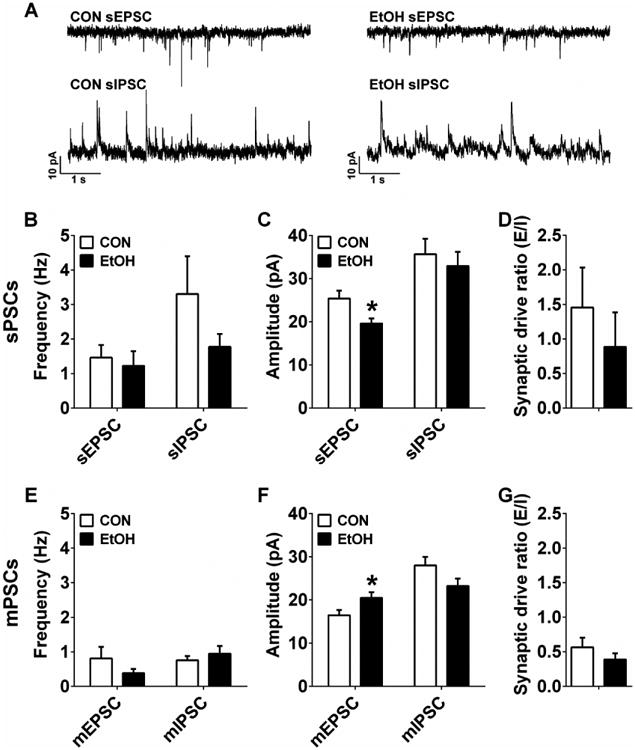
Synaptic transmission measures in the medial CeA (mCeA). A) Representative traces of sPSC from CON and EtOH mice. B There were no differences in sEPSC or sIPSC frequency of mCeA neurons between CON (n=11) and EtOH (n=9) groups (p's > 0.20). C) sEPSC amplitude was significantly smaller in EtOH mice than CON mice (t(18) = 2.51, *p = 0.022), but there was no difference in sIPSC amplitude (p > 0.55). D) There was no effect of CIE on sPSC synaptic drive ratio (p > 0.45). E) There were no differences in mEPSC or mIPSC frequency of mCeA neurons between CON (n=10) and EtOH (n=8_ groups (p's > 0.25); F) mEPSC amplitude was significantly larger in mCeA neurons in EtOH mice than in CON mice (t(16) = 2.19, *p = 0.043), but there was no difference in mIPSC amplitude (p > 0.05). G) There was no effect of CIE on mPSC synaptic drive ratio (p > 0.30).
Measures of intrinsic excitability in the mCeA were not altered by CIE; neither the RMP (Figure 8A) nor the Res (Figure 8B) of mCeA neurons were different between CON and EtOH mice (p's > 0.35). And, measures of current-injected firing, including APT and rheobase, were not different between groups when measured at RMP (data not shown; p's > 0.10) or at -70 mV (Figure 8C-E; p's > 0.75). However, a two-way repeated-measures ANOVA on the V-I plot at RMP resulted in a main effect of current injection magnitude (data not shown; F(20,340) = 53.4, p < 0.0001) but not CIE (p > 0.05), as well as an interaction between CIE and current injection magnitude (F(20,340) = 4.76, p < 0.0001). When neurons were held at -70 mV, the ANOVA for the V-I plot revealed similar results (Figure 8F,G; current injection magnitude: F(20,280) = 20.9, p < 0.0001, CIE: p > 0.05; interaction: F(20,280) = 3.12, p < 0.0001). Together, these results suggest that only the capacity of for action potential firing was affected by CIE.
Figure 8.
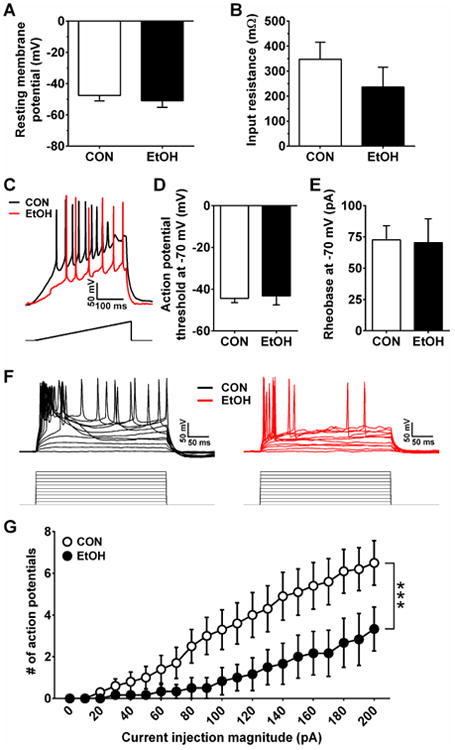
Excitability measures in the mCeA. A,B) There were no differences in the RMP (A) or Res (B) of mCeA neurons between CON (n=14) and EtOH (n=12) mice (p's > 0.35). C) Representative traces of current-injected firing from mCeA neurons in CON and EtOH mice when held at a common potential of -70 mV during a ramp protocol of 120 pA/1 s at RMP, with stimulus waveform depicted below traces. D,E) There were no differences between groups in the APT (D) or rheobase (E) when medial CeA neurons were held at -70 mV (p's > 0.75). F) Representative traces of current-injected firing from mCeA neurons in CON and EtOH mice when held at a common potential of -70 mV during a step protocol of increased current steps of 10 pA/250 ms step, with stimulus waveforms depicted below traces. G) CIE decreased the number of action potentials per current injection step, indicated by an interaction between CIE and current injection step (F(20,280) = 3.12, ***p < 0.0001).
4. Discussion
We found a host of adaptations in prefrontal cortical and extended amygdala nuclei 48 hr following four cycles of CIE, during withdrawal from high level ethanol exposure and at a time point where we also observed increased anxiety-like behavior. Specifically, we observed a net increase in excitability and shift towards excitation in the medial prefrontal cortex and BNST, and the opposite effects in the CeA. Notably, there were alterations in all six subregions of this limbic circuit that we examined, with some marked differences between subregions within each of these structures. While further experiments are required to link neuronal adaptations with the CIE-induced changes in marble burying behavior observed, together, these coincident results demonstrate that withdrawal from chronic ethanol exposure impacts neural function in complex ways relevant to behavioral output. Indeed, our results highlight the broad and severe consequences of chronic ethanol exposure and indicate that adaptations in cortico-limbic circuits may be involved in the negative behavioral effects previously shown to be elicited by this exposure paradigm, including increased alcohol consumption and anxiety and decrements in fear extinction and reversal learning (Badanich et al., 2011; Becker, 2013; Becker and Lopez, 2004; Holmes et al., 2012).
4.1 Methodological Considerations
In this study, we used chronic intermittent ethanol vapor exposure, a well-described model of high ethanol exposure widely used to model escalation of alcohol intake observed during the development of alcoholism in humans. We chose to examine function 48 hours following ethanol exposure, at a time point that was beyond acute withdrawal and allowed us to begin to examine chronic adaptations driven by this exposure and withdrawal. While this provides only a “snapshot” of the changes in neural function that may underlie the negative consequences of chronic ethanol exposure, these may be persist for days or weeks following CIE. Several previous studies have reported results that were consistent across the 48-72 hr and one week post-CIE time points (e.g., Lopez et al., 2014), suggesting the persistence of this effect across the protracted withdrawal time window. Given the many robust changes we observed in these cortico-limbic brain areas, investigation of the persistence of these adaptations will be of critical importance in future studies. In addition, teasing apart the effects of high ethanol exposure and the potentially stressful aspect of this and other non-voluntary administration paradigms is also warranted in subsequent studies.
Additionally, we chose to examine synaptic transmission using a biophysical approach that allowed us to measure both excitatory and inhibitory transmission from each individual neuron. While common methodological approaches examine excitatory and inhibitory synaptic transmission in separate subsets of neurons, our approach allows for both of these measurements within individual neurons, conferring the additional ability to directly assess the balance of excitatory and inhibitory synaptic drive onto individual neurons. Previous research has shown that that this balance is a critical aspect of neuronal circuit function that contributes to the regulation of a number of behaviors (Xue et al., 2014). Indeed, using this biophysical approach in the current study provided essential information about the effects of chronic ethanol exposure on cellular and circuit function in the context of behavioral output. However, because inhibitory synaptic transmission using this approach is measured when neurons are in a depolarized state, it is possible that our ability to measure some alterations in function may have been obscured. Comparisons of our results with those from other studies using classical approaches are addressed below.
4.2 Prefrontal Cortex
We identified both synaptic and excitability alterations in the PFC following CIE that indicated a net increase in basal excitation of the mPFC, consistent with neuroimaging studies in abstinent human alcoholics that report greater metabolic activity in the ventromedial PFC at rest (Seo et al., 2013), however these were manifested differently in the prelimbic and infralimbic subregions. While PLC neurons in EtOH mice had a general decrease in the threshold for firing action potentials (Figure S2), more robust changes were observed in the ILC (Figure 4). There was a decrease in putative presynaptic spontaneous inhibition of ILC neurons, with concurrent increase in putative postsynaptic excitatory transmission (Figure 3). Due to the lack of effect on mPSCs, we hypothesize that these effects are network activity-dependent. Additionally, neurons from EtOH mice were depolarized at rest, with lower input resistance, signifying that CIE increased basal neuronal conductance that led to a depolarized resting state in ILC neurons.
The robust effects of CIE on ILC neurons as compared to PLC neurons in mice may be related to their differing connectivity and roles in several behaviors mediated by the PFC (Fitzgerald et al., 2014; Holmes et al., 2012). For example, while PLC activation promotes fear behavior, ILC neuronal activity promotes extinction (Fitzgerald et al., 2014). We have previously shown that CIE impairs fear extinction mediated by a reduction in NMDA receptor-dependent burst firing of ILC, but not PLC, neurons (Holmes et al., 2012), and we have also observed a decrease in expression of the neuroactive steroid 3α-hydroxy-5α-pregnan-20-one (3α,5α-THP), a positive allosteric modulator of GABAA receptors, in the PFC (Maldonado-Devincci et al., 2014). Together with our finding that basal excitability of ILC neurons is increased after CIE, we speculate that there may be a loss of control of coordinated firing of ILC neurons after chronic alcohol exposure that has detrimental effects on the appropriate coding of information by the PFC, such as those during extinction of learned behaviors. Future examination of whether this contributes to the persistence of maladaptive behaviors such as sustained fearfulness (Holmes et al., 2012) would be informative. Interestingly, impaired fear extinction is a hallmark of post-traumatic stress disorder in humans, which is commonly co-morbidly expressed with alcoholism. Thus, the harmful effects of multiple cycles of intoxication and withdrawal in PFC function in alcohol-dependent rodents is consistent with the human literature that has elegantly shown that human alcoholics with multiple withdrawals/detoxifications have impaired frontal cortical-dependent decision-making and executive function and enhanced impulsivity, which may contribute to relapse behavior (Little et al., 2005; Loeber et al., 2009; Loeber et al., 2010; Stephens and Duka, 2008).
4.3 Bed nucleus of the stria terminalis
Similar to the PFC, we detected a number of changes in synaptic transmission and neuronal excitability in the vBNST following CIE that indicated an overall increase in excitation. In particular, we found increased synaptic drive ratios for both sPSCs and mPSCs. Given that we also observed increased mEPSC frequency in EtOH mice, increased excitatory drive may be due to increased local, network-independent putative glutamate release onto vBNST neurons (Figure 5). This increased excitatory drive may contribute to the greater sensitivity of these neurons to current injection, as illustrated by the lower rheobase and greater current injected firing capacity of vBNST neurons in EtOH mice (Figure 6). However, given the interconnected nature of the dorsal and ventral portions of the BNST (Dong et al., 2001; Dong and Swanson, 2004), it was surprising that we found that the dBNST was resistant to the effects of chronic ethanol exposure (Figures S3 and S4), with only a small increase in mEPSC amplitude observed following CIE. The lack of effect of CIE on the general dBNST neuronal population is likely due to the cell type and projection target heterogeneity of the subregion, as well as the density of inhibitory interneurons that contribute to its complex microcircuit (Crestani et al., 2013; Dong et al., 2001; Dong and Swanson, 2004; Kash, 2012; Kash et al., 2015; Magableh and Lundy, 2014; Pleil et al., 2015). Thus, effects of chronic alcohol exposure in the dBNST may be specific to discrete populations of neurons or modulatory systems there (Kash, 2012; Pleil et al., 2015; Silberman et al., 2013). For example, previous work has shown that BNST neurons that produce the neuropeptide corticotropin-releasing factor (CRF) are vulnerable to the effects of chronic alcohol exposure, as is CRF modulation of BNST neurons that project to the midbrain (Silberman et al., 2013). In contrast, while containing overlapping targets and cell types, the vBNST is more homogenous, with a large proportion of neurons identified as projection neurons to discrete downstream regions, such as the ventral tegmental area and nuclei of the hypothalamus (Herman, 2012; Herman et al., 1994; Jennings et al., 2013). Thus, measurements taken from genetically-identified neuronal populations in the BNST, particularly the dBNST, may be required to more discretely characterize the effects of chronic alcohol exposure and withdrawal on BNST neuronal function in the future.
4.4 Central amygdala
Alterations in CeA neuronal function were the opposite of those observed in the PFC and BNST. In the lCeA, sEPSC frequency and sPSC synaptic drive ratios were decreased in EtOH mice (Figure S5). In the mCeA, sEPSC amplitude was decreased (Figure 7) and there was a decrease in current-injected action potential firing in the VI plot (Figure 8). While we did not find an increase in basal GABAergic transmission after CIE as others have reported in rats and mice (Repunte-Canonigo et al., 2014; Roberto et al., 2012; Roberto et al., 2004), our finding that there was a net inhibitory effect of CIE on CeA function is generally consistent with a suppression of CeA activation, albeit through a different mechanism. There are several potential factors contributing to this difference, including species/background strain used, biophysical electrophysiology approach described above, time point of electrophysiological measurements post-CIE, and incorporation of intermittent voluntary ethanol drinking throughout the vapor exposure paradigm. Interestingly, increased basal inhibitory tone in the CeA has been shown to mediate escalation of alcohol drinking in dependent rats (Roberto et al., 2010). Further, there is convincing evidence that pharmacological manipulations of stress hormone and peptide receptors, as well as their downstream signaling molecules, in the CeA that modulate GABAergic transmission also regulate abstinence-induced escalation of drinking behavior after CIE in rats and mice (Funk and Koob, 2007; Funk et al., 2006; Kissler et al., 2014; Repunte-Canonigo et al., 2015; Vendruscolo et al., 2012). Therefore our results are consistent with the conceptual framework that modulation of inhibition in the CeA is critical component in the relationship between chronic ethanol exposure and ethanol-related behaviors.
4.5 Implications for cortico-limbic circuits
In this study, we report many effects of CIE and withdrawal on the basal function of prefrontal cortical and extended amygdalar nuclei, as well as anxiety-like behavior that is mediated by the function of these brain regions. Many previous studies have shown that pharmacological manipulations in these regions can modulate the negative behavioral consequences of this dependence-inducing ethanol exposure, including anxiety and excessive alcohol consumption. However, lesioning the CeA or BNST does not block this escalation of drinking behavior (Dhaher et al., 2008). Thus, the importance of these structures in negative affect and alcohol and anxiety-related behaviors may be dependent upon their dense anatomical and functional connections with one another (Hoover and Vertes, 2007; Johnstone et al., 2007; Vertes, 2004; Wager et al., 2008). For example, while control subjects have increased PFC activation to engage top-down suppression of the amygdala to regulate negative emotion (Ochsner et al., 2004; Wager et al., 2008), humans with major depressive disorder, anxiety disorders, and alcoholism are unable to recruit this circuitry and commonly display concurrent hypoactivation of the mPFC and hyperactivation of the amygdala (Bremner, 2006; Drevets, 2000a, b, 2001; Johnstone et al., 2007; Keedwell et al., 2005; Ressler and Mayberg, 2007; Seo et al., 2013). Similar findings have come from rodent studies of anxiety and depression (Covington et al., 2005; Covington et al., 2010), as well as those examining the transition to and maintenance of excessive alcohol drinking in dependence (George et al., 2012; Koob and Kreek, 2007; Koob, 2003b, 2008). Further, alcohol-dependent humans have a withdrawal-induced increase in functional connectivity between the amygdala and BNST (O'Daly et al., 2012), and disturbing the functional connection between the CeA and BNST alters anxiety and depressive-like behaviors in rodents (Tasan et al., 2010).
The PFC projects to the CeA and BNST, which have reciprocal connections with each other and project to midbrain and hindbrain regions including the periaqueductal gray, ventral tegmental area, parabrachial nucleus, and paraventricular nucleus of the thalamus to modulate stress response and alcohol-related behaviors (Flak et al., 2012; Herman, 2012; Myers et al., 2014; Stamatakis et al., 2014). The ILC has a much denser synaptic input to the BNST and CeA than the PLC (Vertes, 2004), which may explain why more robust changes in transmission and excitability were observed in the ILC than PLC. Interestingly, reciprocal projections from the extended amygdala to the PFC have not been identified (Hoover and Vertes, 2007), suggesting that the ILC exerts top-down control of the limbic extended amygdala via this glutamatergic projection (Massi et al., 2008; Radley and Sawchenko, 2011). Therefore, the robust excitatory effects of CIE on ILC function may be conferred onto the vBNST via this ILC-vBNST glutamatergic synapse. Given the density of CRF neurons in the vBNST and their role in the development and maintenance of escalated drinking in alcohol dependence, ILC hyperactivity that increases the downstream activation of CRF and other neurons in the vBNST may be a primary mechanism by which chronic alcohol exposure has its negative behavioral effects. In addition, dense GABAergic connections between the CeA and BNST (Li et al., 2012; Sun and Cassell, 1993) may contribute to the opposing effects of CIE on the excitation of these extended amygdala brain regions.
Many previous reports describe hyperactivation of the amygdala in alcohol-dependent rats and humans due to a loss of top-down modulation by the PFC (George et al., 2012; Seo et al., 2013), indicating that glutamatergic projections from the ILC to the CeA may primarily synapse onto inhibitory interneurons. Here, while we found that the PFC and CeA were oppositely affected by CIE, we found a decrease in excitation of the CeA and an increase in excitation of the PFC. This divergence between our study and previous studies in the direction of CIE effects may signify a short-term compensatory response during this 48 hr withdrawal period that is not present at later time points or during stress-evoking events as in other studies. Alternatively, it may reflect species differences in the functional connectivity of these brain regions, given that a majority of anatomical mapping studies have been performed in rats and humans. Future studies employing circuit-specific and cell type-dependent manipulations in mice will address the functional connections between these regions and the persistence of the effects of chronic alcohol exposure in limbic circuits.
Supplementary Material
Figure S1: Synaptic transmission measures in the prelimbic cortex (PLC). A-C) There were no differences in sEPSC or sIPSC frequency (A) or amplitude (B), nor a difference in sPSC synaptic drive ratio (C), of PLC neurons between CON (n=7) and EtOH (n=8) groups (p's > 0.10). D-F) Similarly, there were no differences in mEPSC or mIPSC frequency (D), amplitude (E), or mPSC synaptic drive ratio (F) between CON (n=6) and EtOH (n=8) groups (p's > 0.70).
Figure S2: Excitability measures in PLC. A&B) There were no differences in the RMP (A) or Res (B) of PLC neurons between CON (n=11) and EtOH (n=8) mice (p's > 0.35). C,D) PLC neurons from EtOH mice had a significantly lower APT than CON mice (C; t(16) = 2.56, *p = 0.021), but not a different rheobase (D; p > 0.05), at RMP. E) EtOH mouse PLC neurons had greater action potential firing at RMP than PLC neurons from CON mice, indicated by a significant interaction between CIE and current injection magnitude (F(20, 340) = 2.27, **p = 0.002).
Figure S3: Synaptic transmission measures in the dorsal BNST (dBNST). A-C) There were no differences between dBNST neurons from CON (n=12) and EtOH (n=11) mice for sPSC frequency (A), amplitude (B), or synaptic drive ratio measures (C; p's > 0.10). D,E) There were no differences in mEPSC or mIPSC frequency between CON (n=15) and EtOH (n=14) mice (D; p's > 0.10), but mEPSC amplitude was greater in EtOH than CON mice (E; t(27) = 3.265, **p = 0.003). F) mPSC synaptic drive ratio was not different between groups (p > 0.35).
Figure S4: Excitability measures in the dBNST. There were no differences in the RMP (A) or Res (B) of dBNST neurons between CON (n=18) and EtOH (n=12) mice (p's > 0.35). There were also no differences between groups in measures of current-injected firing of action potentials, including the threshold for firing (C), the rheobase (D), or the relationship between the number of action potentials and current injection magnitude (E), when neurons were held at a common potential of -70 mV (p's > 0.15).
Figure S5: Synaptic transmission measures in the lateral CeA (lCeA). A) sEPSC frequency was lower in neurons from EtOH (n=10) than CON (n=10) mice (t(17) = 2.20, *p = 0.042), but sIPSC frequency was not different (p > 0.85). B) There were no differences between lCeA neurons from CON and EtOH mice in sEPSC or sIPSC amplitude (p's > 0.20). C) The sPSC synaptic drive ratio in the lCeA of EtOH mice was significantly lower than that in CON mice (t(15) = 3.19, **p = 0.006). D-F) There were no differences between CON (n=9) and EtOH (n=12) mice in any mPSC measures in the lCeA (p's > 0.05).
Figure S6: Excitability measures in the lCeA. A&B) There were no differences in the RMP (A) or Res (B) of lateral CeA neurons between CON (n=13) and EtOH (n=11) mice (p's > 0.35). C-E) There were no differences between groups in the APT (C), rheobase (D), or the number of action potentials per current injection step (E) when lCeA neurons were held at -70 mV (p's > 0.25).
Highlights.
Ethanol-exposed mice displayed increased anxiety compared to air-exposed controls
CIE induced hyperexcitability of the infralimbic cortex
CIE induced a shift toward excitation in synaptic drive and hyperexcitability of vBNST neurons
CIE exposure induced a net inhibition of the CeA.
Acknowledgments
Support for this research was provided by NIH grants F32 AA021043 and K99 AA023599 (KEP), U01 AA020911 and R01 AA019454 (TLK), U01 AA020935 (ALM), F31 AA022280 (NAC), F32 AA022549 (EGLG), F32 AA021319 (CAM), T32 ES007126 (AMM), F31 DA03558 (JHR), P01 AA017056 and U01 AA014091 (SRJ), and the Bowles Center for Alcohol Studies (KEP, EGLG, NAC, CAM, CL, NMM, AMM, ALM, and TLK).
Footnotes
Author contributions: TLK, KEP, ALM, and SRJ were responsible for the study concept. KEP, NAC, EGLG, JHR, CL, CAM, NMM, and AMM performed the in vivo ethanol exposure paradigm. KEP, NAC, EGLG, CL, and CAM performed ex vivo electrophysiological recordings. JHR conducted the MBT, measured BECs, and analyzed the behavioral data. KEP analyzed the electrophysiology data, interpreted the results, and drafted the manuscript. All authors critically reviewed the content of the manuscript and approve the final version for publication.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amodeo DA, Jones JH, Sweeney JA, Ragozzino ME. Differences in BTBR T+ tf/J and C57BL/6J mice on probabilistic reversal learning and stereotyped behaviors. Behav Brain Res. 2012;227:64–72. doi: 10.1016/j.bbr.2011.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badanich KA, Becker HC, Woodward JJ. Effects of chronic intermittent ethanol exposure on orbitofrontal and medial prefrontal cortex-dependent behaviors in mice. Behav Neurosci. 2011;125:879–891. doi: 10.1037/a0025922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker HC. Animal models of excessive alcohol consumption in rodents. Curr Top Behav Neurosci. 2013;13:355–377. doi: 10.1007/7854_2012_203. [DOI] [PubMed] [Google Scholar]
- Becker HC, Lopez MF. Increased ethanol drinking after repeated chronic ethanol exposure and withdrawal experience in C57BL/6 mice. Alcohol Clin Exp Res. 2004;28:1829–1838. doi: 10.1097/01.alc.0000149977.95306.3a. [DOI] [PubMed] [Google Scholar]
- Becker HC, Ron D. Animal models of excessive alcohol consumption: recent advances and future challenges. Alcohol. 2014;48:205–208. doi: 10.1016/j.alcohol.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogg T, Fukunaga R, Finn PR, Brown JW. Cognitive control links alcohol use, trait disinhibition, and reduced cognitive capacity: Evidence for medial prefrontal cortex dysregulation during reward-seeking behavior. Drug Alcohol Depend. 2012;122:112–118. doi: 10.1016/j.drugalcdep.2011.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremner JD. Traumatic stress: effects on the brain. Dialogues Clin Neurosci. 2006;8:445–461. doi: 10.31887/DCNS.2006.8.4/jbremner. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgos-Robles A, Bravo-Rivera H, Quirk GJ. Prelimbic and infralimbic neurons signal distinct aspects of appetitive instrumental behavior. PLoS One. 2013;8:e57575. doi: 10.1371/journal.pone.0057575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrara-Nascimento PF, Lopez MF, Becker HC, Olive MF, Camarini R. Similar ethanol drinking in adolescent and adult C57BL/6J mice after chronic ethanol exposure and withdrawal. Alcohol Clin Exp Res. 2013;37:961–968. doi: 10.1111/acer.12056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covington HE, 3rd, Kikusui T, Goodhue J, Nikulina EM, Hammer RP, Jr, Miczek KA. Brief social defeat stress: long lasting effects on cocaine taking during a binge and zif268 mRNA expression in the amygdala and prefrontal cortex. Neuropsychopharmacology. 2005;30:310–321. doi: 10.1038/sj.npp.1300587. [DOI] [PubMed] [Google Scholar]
- Covington HE, 3rd, Lobo MK, Maze I, Vialou V, Hyman JM, Zaman S, LaPlant Q, Mouzon E, Ghose S, Tamminga CA, Neve RL, Deisseroth K, Nestler EJ. Antidepressant effect of optogenetic stimulation of the medial prefrontal cortex. J Neurosci. 2010;30:16082–16090. doi: 10.1523/JNEUROSCI.1731-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabbe JC, Metten P, Belknap JK, Spence SE, Cameron AJ, Schlumbohm JP, Huang LC, Barkley-Levenson AM, Ford MM, Phillips TJ. Progress in a replicated selection for elevated blood ethanol concentrations in HDID mice. Genes Brain Behav. 2014;13:236–246. doi: 10.1111/gbb.12105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crestani CC, Alves FH, Gomes FV, Resstel LB, Correa FM, Herman JP. Mechanisms in the bed nucleus of the stria terminalis involved in control of autonomic and neuroendocrine functions: a review. Curr Neuropharmacol. 2013;11:141–159. doi: 10.2174/1570159X11311020002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M, Walker DL, Lee Y. Amygdala and bed nucleus of the stria terminalis: differential roles in fear and anxiety measured with the acoustic startle reflex. Philos Trans R Soc Lond B Biol Sci. 1997;352:1675–1687. doi: 10.1098/rstb.1997.0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePoy L, Daut R, Brigman JL, MacPherson K, Crowley N, Gunduz-Cinar O, Pickens CL, Cinar R, Saksida LM, Kunos G, Lovinger DM, Bussey TJ, Camp MC, Holmes A. Chronic alcohol produces neuroadaptations to prime dorsal striatal learning. Proc Natl Acad Sci U S A. 2013;110:14783–14788. doi: 10.1073/pnas.1308198110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhaher R, Finn D, Snelling C, Hitzemann R. Lesions of the extended amygdala in C57BL/6J mice do not block the intermittent ethanol vapor-induced increase in ethanol consumption. Alcohol Clin Exp Res. 2008;32:197–208. doi: 10.1111/j.1530-0277.2007.00566.x. [DOI] [PubMed] [Google Scholar]
- Dong HW, Petrovich GD, Watts AG, Swanson LW. Basic organization of projections from the oval and fusiform nuclei of the bed nuclei of the stria terminalis in adult rat brain. J Comp Neurol. 2001;436:430–455. doi: 10.1002/cne.1079. [DOI] [PubMed] [Google Scholar]
- Dong HW, Swanson LW. Organization of axonal projections from the anterolateral area of the bed nuclei of the stria terminalis. J Comp Neurol. 2004;468:277–298. doi: 10.1002/cne.10949. [DOI] [PubMed] [Google Scholar]
- Drevets WC. Functional anatomical abnormalities in limbic and prefrontal cortical structures in major depression. Prog Brain Res. 2000a;126:413–431. doi: 10.1016/S0079-6123(00)26027-5. [DOI] [PubMed] [Google Scholar]
- Drevets WC. Neuroimaging studies of mood disorders. Biol Psychiatry. 2000b;48:813–829. doi: 10.1016/s0006-3223(00)01020-9. [DOI] [PubMed] [Google Scholar]
- Drevets WC. Neuroimaging and neuropathological studies of depression: implications for the cognitive-emotional features of mood disorders. Curr Opin Neurobiol. 2001;11:240–249. doi: 10.1016/s0959-4388(00)00203-8. [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Cardinal RN, Parkinson JA, Robbins TW. Appetitive behavior: impact of amygdala-dependent mechanisms of emotional learning. Ann N Y Acad Sci. 2003;985:233–250. [PubMed] [Google Scholar]
- Fatseas M, Serre F, Alexandre JM, Debrabant R, Auriacombe M, Swendsen J. Craving and substance use among patients with alcohol, tobacco, cannabis or heroin addiction: a comparison of substance- and person-specific cues. Addiction. 2015 doi: 10.1111/add.12882. [DOI] [PubMed] [Google Scholar]
- Feder A, Nestler EJ, Charney DS. Psychobiology and molecular genetics of resilience. Nat Rev Neurosci. 2009;10:446–457. doi: 10.1038/nrn2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald PJ, Whittle N, Flynn SM, Graybeal C, Pinard CR, Gunduz-Cinar O, Kravitz AV, Singewald N, Holmes A. Prefrontal single-unit firing associated with deficient extinction in mice. Neurobiol Learn Mem. 2014;113:69–81. doi: 10.1016/j.nlm.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flak JN, Solomon MB, Jankord R, Krause EG, Herman JP. Identification of chronic stress-activated regions reveals a potential recruited circuit in rat brain. Eur J Neurosci. 2012;36:2547–2555. doi: 10.1111/j.1460-9568.2012.08161.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk CK, Koob GF. A CRF(2) agonist administered into the central nucleus of the amygdala decreases ethanol self-administration in ethanol-dependent rats. Brain Res. 2007;1155:172–178. doi: 10.1016/j.brainres.2007.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk CK, O'Dell LE, Crawford EF, Koob GF. Corticotropin-releasing factor within the central nucleus of the amygdala mediates enhanced ethanol self-administration in withdrawn, ethanol-dependent rats. J Neurosci. 2006;26:11324–11332. doi: 10.1523/JNEUROSCI.3096-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gass JT, Trantham-Davidson H, Kassab AS, Glen WB, Jr, Olive MF, Chandler LJ. Enhancement of extinction learning attenuates ethanol-seeking behavior and alters plasticity in the prefrontal cortex. J Neurosci. 2014;34:7562–7574. doi: 10.1523/JNEUROSCI.5616-12.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George O, Sanders C, Freiling J, Grigoryan E, Vu S, Allen CD, Crawford E, Mandyam CD, Koob GF. Recruitment of medial prefrontal cortex neurons during alcohol withdrawal predicts cognitive impairment and excessive alcohol drinking. Proc Natl Acad Sci U S A. 2012;109:18156–18161. doi: 10.1073/pnas.1116523109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilpin NW, Roberto M. Neuropeptide modulation of central amygdala neuroplasticity is a key mediator of alcohol dependence. Neurosci Biobehav Rev. 2012;36:873–888. doi: 10.1016/j.neubiorev.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin WC., 3rd Alcohol dependence and free-choice drinking in mice. Alcohol. 2014;48:287–293. doi: 10.1016/j.alcohol.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin WC, 3rd, Lopez MF, Yanke AB, Middaugh LD, Becker HC. Repeated cycles of chronic intermittent ethanol exposure in mice increases voluntary ethanol drinking and ethanol concentrations in the nucleus accumbens. Psychopharmacology (Berl) 2009;201:569–580. doi: 10.1007/s00213-008-1324-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilig M, Koob GF. A key role for corticotropin-releasing factor in alcohol dependence. Trends Neurosci. 2007;30:399–406. doi: 10.1016/j.tins.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman JP. Neural pathways of stress integration: relevance to alcohol abuse. Alcohol Res. 2012;34:441–447. [PMC free article] [PubMed] [Google Scholar]
- Herman JP, Cullinan WE, Watson SJ. Involvement of the bed nucleus of the stria terminalis in tonic regulation of paraventricular hypothalamic CRH and AVP mRNA expression. J Neuroendocrinol. 1994;6:433–442. doi: 10.1111/j.1365-2826.1994.tb00604.x. [DOI] [PubMed] [Google Scholar]
- Holmes A, Fitzgerald PJ, MacPherson KP, DeBrouse L, Colacicco G, Flynn SM, Masneuf S, Pleil KE, Li C, Marcinkiewcz CA, Kash TL, Gunduz-Cinar O, Camp M. Chronic alcohol remodels prefrontal neurons and disrupts NMDAR-mediated fear extinction encoding. Nat Neurosci. 2012;15:1359–1361. doi: 10.1038/nn.3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover WB, Vertes RP. Anatomical analysis of afferent projections to the medial prefrontal cortex in the rat. Brain Struct Funct. 2007;212:149–179. doi: 10.1007/s00429-007-0150-4. [DOI] [PubMed] [Google Scholar]
- Jennings JH, Sparta DR, Stamatakis AM, Ung RL, Pleil KE, Kash TL, Stuber GD. Distinct extended amygdala circuits for divergent motivational states. Nature. 2013;496:224–228. doi: 10.1038/nature12041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone T, van Reekum CM, Urry HL, Kalin NH, Davidson RJ. Failure to regulate: counterproductive recruitment of top-down prefrontal-subcortical circuitry in major depression. J Neurosci. 2007;27:8877–8884. doi: 10.1523/JNEUROSCI.2063-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kash TL. The role of biogenic amine signaling in the bed nucleus of the stria terminals in alcohol abuse. Alcohol. 2012;46:303–308. doi: 10.1016/j.alcohol.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kash TL, Baucum AJ, 2nd, Conrad KL, Colbran RJ, Winder DG. Alcohol exposure alters NMDAR function in the bed nucleus of the stria terminalis. Neuropsychopharmacology. 2009;34:2420–2429. doi: 10.1038/npp.2009.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kash TL, Pleil KE, Marcinkiewcz CA, Lowery-Gionta EG, Crowley N, Mazzone C, Sugam J, Hardaway JA, McElligott ZA. Neuropeptide regulation of signaling and behavior in the BNST. Mol Cells. 2015;38:1–13. doi: 10.14348/molcells.2015.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keedwell PA, Andrew C, Williams SC, Brammer MJ, Phillips ML. The neural correlates of anhedonia in major depressive disorder. Biol Psychiatry. 2005;58:843–853. doi: 10.1016/j.biopsych.2005.05.019. [DOI] [PubMed] [Google Scholar]
- Kissler JL, Sirohi S, Reis DJ, Jansen HT, Quock RM, Smith DG, Walker BM. The one-two punch of alcoholism: role of central amygdala dynorphins/kappa-opioid receptors. Biol Psychiatry. 2014;75:774–782. doi: 10.1016/j.biopsych.2013.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob G, Kreek MJ. Stress, dysregulation of drug reward pathways, and the transition to drug dependence. Am J Psychiatry. 2007;164:1149–1159. doi: 10.1176/appi.ajp.2007.05030503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF. Alcoholism: allostasis and beyond. Alcohol Clin Exp Res. 2003a;27:232–243. doi: 10.1097/01.ALC.0000057122.36127.C2. [DOI] [PubMed] [Google Scholar]
- Koob GF. Neuroadaptive mechanisms of addiction: studies on the extended amygdala. Eur Neuropsychopharmacol. 2003b;13:442–452. doi: 10.1016/j.euroneuro.2003.08.005. [DOI] [PubMed] [Google Scholar]
- Koob GF. A role for brain stress systems in addiction. Neuron. 2008;59:11–34. doi: 10.1016/j.neuron.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreifeldt M, Le D, Treistman SN, Koob GF, Contet C. BK channel beta1 and beta4 auxiliary subunits exert opposite influences on escalated ethanol drinking in dependent mice. Front Integr Neurosci. 2013;7:105. doi: 10.3389/fnint.2013.00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroener S, Mulholland PJ, New NN, Gass JT, Becker HC, Chandler LJ. Chronic alcohol exposure alters behavioral and synaptic plasticity of the rodent prefrontal cortex. PLoS One. 2012;7:e37541. doi: 10.1371/journal.pone.0037541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Pleil KE, Stamatakis AM, Busan S, Vong L, Lowell BB, Stuber GD, Kash TL. Presynaptic inhibition of gamma-aminobutyric acid release in the bed nucleus of the stria terminalis by kappa opioid receptor signaling. Biol Psychiatry. 2012;71:725–732. doi: 10.1016/j.biopsych.2011.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little HJ, Stephens DN, Ripley TL, Borlikova G, Duka T, Schubert M, Albrecht D, Becker HC, Lopez MF, Weiss F, Drummond C, Peoples M, Cunningham C. Alcohol withdrawal and conditioning. Alcohol Clin Exp Res. 2005;29:453–464. doi: 10.1097/01.alc.0000156737.56425.e3. [DOI] [PubMed] [Google Scholar]
- Loeber S, Duka T, Welzel H, Nakovics H, Heinz A, Flor H, Mann K. Impairment of cognitive abilities and decision making after chronic use of alcohol: the impact of multiple detoxifications. Alcohol Alcohol. 2009;44:372–381. doi: 10.1093/alcalc/agp030. [DOI] [PubMed] [Google Scholar]
- Loeber S, Duka T, Welzel Marquez H, Nakovics H, Heinz A, Mann K, Flor H. Effects of repeated withdrawal from alcohol on recovery of cognitive impairment under abstinence and rate of relapse. Alcohol Alcohol. 2010;45:541–547. doi: 10.1093/alcalc/agq065. [DOI] [PubMed] [Google Scholar]
- Lopez MF, Becker HC. Effect of pattern and number of chronic ethanol exposures on subsequent voluntary ethanol intake in C57BL/6J mice. Psychopharmacology (Berl) 2005;181:688–696. doi: 10.1007/s00213-005-0026-3. [DOI] [PubMed] [Google Scholar]
- Lopez MF, Becker HC, Chandler LJ. Repeated episodes of chronic intermittent ethanol promote insensitivity to devaluation of the reinforcing effect of ethanol. Alcohol. 2014;48:639–645. doi: 10.1016/j.alcohol.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowery-Gionta EG, Marcinkiewcz CA, Kash TL. Functional Alterations in the Dorsal Raphe Nucleus Following Acute and Chronic Ethanol Exposure. Neuropsychopharmacology. 2014 doi: 10.1038/npp.2014.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowery-Gionta EG, Marcinkiewcz CA, Kash TL. Functional alterations in the dorsal raphe nucleus following acute and chronic ethanol exposure. Neuropsychopharmacology. 2015;40:590–600. doi: 10.1038/npp.2014.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowery-Gionta EG, Navarro M, Li C, Pleil KE, Rinker JA, Cox BR, Sprow GM, Kash TL, Thiele TE. Corticotropin releasing factor signaling in the central amygdala is recruited during binge-like ethanol consumption in C57BL/6J mice. J Neurosci. 2012;32:3405–3413. doi: 10.1523/JNEUROSCI.6256-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magableh A, Lundy R. Somatostatin and Corticotrophin Releasing Hormone Cell Types Are a Major Source of Descending Input From the Forebrain to the Parabrachial Nucleus in Mice. Chem Senses. 2014 doi: 10.1093/chemse/bju038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado-Devincci AM, Cook JB, O'Buckley TK, Morrow DH, McKinley RE, Lopez MF, Becker HC, Morrow AL. Chronic Intermittent Ethanol Exposure and Withdrawal Alters (3alpha,5alpha)-3-Hydroxy-Pregnan-20-One Immunostaining in Cortical and Limbic Brain Regions of C57BL/6J Mice. Alcohol Clin Exp Res. 2014;38:2561–2571. doi: 10.1111/acer.12530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massi L, Elezgarai I, Puente N, Reguero L, Grandes P, Manzoni OJ, Georges F. Cannabinoid receptors in the bed nucleus of the stria terminalis control cortical excitation of midbrain dopamine cells in vivo. J Neurosci. 2008;28:10496–10508. doi: 10.1523/JNEUROSCI.2291-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall NM, Sprow GM, Delpire E, Thiele TE, Kash TL, Pleil KE. Effects of sex and deletion of neuropeptide Y2 receptors from GABAergic neurons on affective and alcohol drinking behaviors in mice. Front Integr Neurosci. 2013;7:100. doi: 10.3389/fnint.2013.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinhardt MW, Sommer WH. Postdependent state in rats as a model for medication development in alcoholism. Addict Biol. 2015;20:1–21. doi: 10.1111/adb.12187. [DOI] [PubMed] [Google Scholar]
- Myers B, Mark Dolgas C, Kasckow J, Cullinan WE, Herman JP. Central stress-integrative circuits: forebrain glutamatergic and GABAergic projections to the dorsomedial hypothalamus, medial preoptic area, and bed nucleus of the stria terminalis. Brain Struct Funct. 2014;219:1287–1303. doi: 10.1007/s00429-013-0566-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Daly OG, Trick L, Scaife J, Marshall J, Ball D, Phillips ML, Williams SS, Stephens DN, Duka T. Withdrawal-associated increases and decreases in functional neural connectivity associated with altered emotional regulation in alcoholism. Neuropsychopharmacology. 2012;37:2267–2276. doi: 10.1038/npp.2012.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochsner KN, Ray RD, Cooper JC, Robertson ER, Chopra S, Gabrieli JD, Gross JJ. For better or for worse: neural systems supporting the cognitive down- and up-regulation of negative emotion. Neuroimage. 2004;23:483–499. doi: 10.1016/j.neuroimage.2004.06.030. [DOI] [PubMed] [Google Scholar]
- Perez EE, De Biasi M. Assessment of affective and somatic signs of ethanol withdrawal in C57BL/6J mice using a short-term ethanol treatment. Alcohol. 2015;49:237–243. doi: 10.1016/j.alcohol.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips ML, Drevets WC, Rauch SL, Lane R. Neurobiology of emotion perception I: The neural basis of normal emotion perception. Biol Psychiatry. 2003;54:504–514. doi: 10.1016/s0006-3223(03)00168-9. [DOI] [PubMed] [Google Scholar]
- Pleil KE, Lopez A, McCall N, Jijon AM, Bravo JP, Kash TL. Chronic stress alters neuropeptide Y signaling in the bed nucleus of the stria terminalis in DBA/2J but not C57BL/6J mice. Neuropharmacology. 2012;62:1777–1786. doi: 10.1016/j.neuropharm.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleil KE, Rinker JA, Lowery-Gionta EG, Mazzone CM, McCall NM, Kendra AM, Olson DP, Lowell BB, Grant KA, Thiele TE, Kash TL. NPY signaling inhibits extended amygdala CRF neurons to suppress binge alcohol drinking. Nat Neurosci. 2015;18:545–552. doi: 10.1038/nn.3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radley JJ, Sawchenko PE. A common substrate for prefrontal and hippocampal inhibition of the neuroendocrine stress response. J Neurosci. 2011;31:9683–9695. doi: 10.1523/JNEUROSCI.6040-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repunte-Canonigo V, Herman MA, Kawamura T, Kranzler HR, Sherva R, Gelernter J, Farrer LA, Roberto M, Paolo Sanna P. Nf1 regulates alcohol dependence-associated excessive drinking and gamma-aminobutyric Acid release in the central amygdala in mice and is associated with alcohol dependence in humans. Biol Psychiatry. 2014;77:870–879. doi: 10.1016/j.biopsych.2014.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repunte-Canonigo V, Herman MA, Kawamura T, Kranzler HR, Sherva R, Gelernter J, Farrer LA, Roberto M, Paolo Sanna P. Nf1 regulates alcohol dependence-associated excessive drinking and gamma-aminobutyric Acid release in the central amygdala in mice and is associated with alcohol dependence in humans. Biol Psychiatry. 2015;77:870–879. doi: 10.1016/j.biopsych.2014.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ressler KJ, Mayberg HS. Targeting abnormal neural circuits in mood and anxiety disorders: from the laboratory to the clinic. Nat Neurosci. 2007;10:1116–1124. doi: 10.1038/nn1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Cruz MT, Gilpin NW, Sabino V, Schweitzer P, Bajo M, Cottone P, Madamba SG, Stouffer DG, Zorrilla EP, Koob GF, Siggins GR, Parsons LH. Corticotropin Releasing Factor-Induced Amygdala Gamma-Aminobutyric Acid Release Plays a Key Role in Alcohol Dependence. Biol Psychiatry. 2010 doi: 10.1016/j.biopsych.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Gilpin NW, Siggins GR. The central amygdala and alcohol: role of gamma-aminobutyric acid, glutamate, and neuropeptides. Cold Spring Harb Perspect Med. 2012;2:a012195. doi: 10.1101/cshperspect.a012195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Madamba SG, Stouffer DG, Parsons LH, Siggins GR. Increased GABA release in the central amygdala of ethanol-dependent rats. J Neurosci. 2004;24:10159–10166. doi: 10.1523/JNEUROSCI.3004-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AJ, Heyser CJ, Cole M, Griffin P, Koob GF. Excessive ethanol drinking following a history of dependence: animal model of allostasis. Neuropsychopharmacology. 2000;22:581–594. doi: 10.1016/S0893-133X(99)00167-0. [DOI] [PubMed] [Google Scholar]
- Sailer U, Robinson S, Fischmeister FP, Konig D, Oppenauer C, Lueger-Schuster B, Moser E, Kryspin-Exner I, Bauer H. Altered reward processing in the nucleus accumbens and mesial prefrontal cortex of patients with posttraumatic stress disorder. Neuropsychologia. 2008;46:2836–2844. doi: 10.1016/j.neuropsychologia.2008.05.022. [DOI] [PubMed] [Google Scholar]
- Seo D, Lacadie CM, Tuit K, Hong KI, Constable RT, Sinha R. Disrupted ventromedial prefrontal function, alcohol craving, and subsequent relapse risk. JAMA Psychiatry. 2013;70:727–739. doi: 10.1001/jamapsychiatry.2013.762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberman Y, Matthews RT, Winder DG. A corticotropin releasing factor pathway for ethanol regulation of the ventral tegmental area in the bed nucleus of the stria terminalis. J Neurosci. 2013;33:950–960. doi: 10.1523/JNEUROSCI.2949-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrow AM, Lowery-Gionta EG, Pleil KE, Li C, Sprow GM, Cox BR, Rinker JA, Jijon AM, Pena J, Navarro M, Kash TL, Thiele TE. Central neuropeptide Y modulates binge-like ethanol drinking in C57BL/6J mice via Y1 and Y2 receptors. Neuropsychopharmacology. 2012;37:1409–1421. doi: 10.1038/npp.2011.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis AM, Sparta DR, Jennings JH, McElligott ZA, Decot H, Stuber GD. Amygdala and bed nucleus of the stria terminalis circuitry: Implications for addiction-related behaviors. Neuropharmacology. 2014;76 Pt B:320–328. doi: 10.1016/j.neuropharm.2013.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens DN, Duka T. Review. Cognitive and emotional consequences of binge drinking: role of amygdala and prefrontal cortex. Philos Trans R Soc Lond B Biol Sci. 2008;363:3169–3179. doi: 10.1098/rstb.2008.0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun N, Cassell MD. Intrinsic GABAergic neurons in the rat central extended amygdala. J Comp Neurol. 1993;330:381–404. doi: 10.1002/cne.903300308. [DOI] [PubMed] [Google Scholar]
- Tasan RO, Nguyen NK, Weger S, Sartori SB, Singewald N, Heilbronn R, Herzog H, Sperk G. The central and basolateral amygdala are critical sites of neuropeptide Y/Y2 receptor-mediated regulation of anxiety and depression. J Neurosci. 2010;30:6282–6290. doi: 10.1523/JNEUROSCI.0430-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas A, Burant A, Bui N, Graham D, Yuva-Paylor LA, Paylor R. Marble burying reflects a repetitive and perseverative behavior more than novelty-induced anxiety. Psychopharmacology (Berl) 2009;204:361–373. doi: 10.1007/s00213-009-1466-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vendruscolo LF, Barbier E, Schlosburg JE, Misra KK, Whitfield TW, Jr, Logrip ML, Rivier C, Repunte-Canonigo V, Zorrilla EP, Sanna PP, Heilig M, Koob GF. Corticosteroid-dependent plasticity mediates compulsive alcohol drinking in rats. J Neurosci. 2012;32:7563–7571. doi: 10.1523/JNEUROSCI.0069-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vertes RP. Differential projections of the infralimbic and prelimbic cortex in the rat. Synapse. 2004;51:32–58. doi: 10.1002/syn.10279. [DOI] [PubMed] [Google Scholar]
- Wager TD, Davidson ML, Hughes BL, Lindquist MA, Ochsner KN. Prefrontal-subcortical pathways mediating successful emotion regulation. Neuron. 2008;59:1037–1050. doi: 10.1016/j.neuron.2008.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills TA, Klug JR, Silberman Y, Baucum AJ, Weitlauf C, Colbran RJ, Delpire E, Winder DG. GluN2B subunit deletion reveals key role in acute and chronic ethanol sensitivity of glutamate synapses in bed nucleus of the stria terminalis. Proc Natl Acad Sci U S A. 2012;109:E278–287. doi: 10.1073/pnas.1113820109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M, Atallah BV, Scanziani M. Equalizing excitation-inhibition ratios across visual cortical neurons. Nature. 2014;511:596–600. doi: 10.1038/nature13321. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Synaptic transmission measures in the prelimbic cortex (PLC). A-C) There were no differences in sEPSC or sIPSC frequency (A) or amplitude (B), nor a difference in sPSC synaptic drive ratio (C), of PLC neurons between CON (n=7) and EtOH (n=8) groups (p's > 0.10). D-F) Similarly, there were no differences in mEPSC or mIPSC frequency (D), amplitude (E), or mPSC synaptic drive ratio (F) between CON (n=6) and EtOH (n=8) groups (p's > 0.70).
Figure S2: Excitability measures in PLC. A&B) There were no differences in the RMP (A) or Res (B) of PLC neurons between CON (n=11) and EtOH (n=8) mice (p's > 0.35). C,D) PLC neurons from EtOH mice had a significantly lower APT than CON mice (C; t(16) = 2.56, *p = 0.021), but not a different rheobase (D; p > 0.05), at RMP. E) EtOH mouse PLC neurons had greater action potential firing at RMP than PLC neurons from CON mice, indicated by a significant interaction between CIE and current injection magnitude (F(20, 340) = 2.27, **p = 0.002).
Figure S3: Synaptic transmission measures in the dorsal BNST (dBNST). A-C) There were no differences between dBNST neurons from CON (n=12) and EtOH (n=11) mice for sPSC frequency (A), amplitude (B), or synaptic drive ratio measures (C; p's > 0.10). D,E) There were no differences in mEPSC or mIPSC frequency between CON (n=15) and EtOH (n=14) mice (D; p's > 0.10), but mEPSC amplitude was greater in EtOH than CON mice (E; t(27) = 3.265, **p = 0.003). F) mPSC synaptic drive ratio was not different between groups (p > 0.35).
Figure S4: Excitability measures in the dBNST. There were no differences in the RMP (A) or Res (B) of dBNST neurons between CON (n=18) and EtOH (n=12) mice (p's > 0.35). There were also no differences between groups in measures of current-injected firing of action potentials, including the threshold for firing (C), the rheobase (D), or the relationship between the number of action potentials and current injection magnitude (E), when neurons were held at a common potential of -70 mV (p's > 0.15).
Figure S5: Synaptic transmission measures in the lateral CeA (lCeA). A) sEPSC frequency was lower in neurons from EtOH (n=10) than CON (n=10) mice (t(17) = 2.20, *p = 0.042), but sIPSC frequency was not different (p > 0.85). B) There were no differences between lCeA neurons from CON and EtOH mice in sEPSC or sIPSC amplitude (p's > 0.20). C) The sPSC synaptic drive ratio in the lCeA of EtOH mice was significantly lower than that in CON mice (t(15) = 3.19, **p = 0.006). D-F) There were no differences between CON (n=9) and EtOH (n=12) mice in any mPSC measures in the lCeA (p's > 0.05).
Figure S6: Excitability measures in the lCeA. A&B) There were no differences in the RMP (A) or Res (B) of lateral CeA neurons between CON (n=13) and EtOH (n=11) mice (p's > 0.35). C-E) There were no differences between groups in the APT (C), rheobase (D), or the number of action potentials per current injection step (E) when lCeA neurons were held at -70 mV (p's > 0.25).