

Abstract
Endospore formation is a survival strategy found among some bacteria from the phylum Firmicutes. During endospore formation, these bacteria enter a morpho-physiological resting state that enhances survival under adverse environmental conditions. Even though endospore-forming Firmicutes are one of the most frequently enriched and isolated bacterial groups in culturing studies, they are often absent from diversity studies based on molecular methods. The resistance of the spore core is considered one of the factors limiting the recovery of DNA from endospores. We developed a method that takes advantage of the higher resistance of endospores to separate them from other cells in a complex microbial community using physical, enzymatic and chemical lysis methods. The endospore-only preparation thus obtained can be used for re-culturing or to perform downstream analysis such as tailored DNA extraction optimized for endospores and subsequent DNA sequencing. This method, applied to sediment samples, has allowed the enrichment of endospores and after sequencing, has revealed a large diversity of endospore-formers in freshwater lake sediments. We expect that the application of this method to other samples will yield a similar outcome.
Keywords: Environmental Sciences, Issue 107, Endospores, separation, vegetative cells, endospore-forming bacteria, cell lysis, diversity, microbial ecology
Introduction
The goal of this work is to provide a protocol for the separation of bacterial endospores from vegetative bacterial cells in environmental samples. The formation of bacterial endospores is a survival strategy, usually triggered by starvation, found in a number of bacterial groups belonging to the phylum Firmicutes1. Endospore-forming bacteria are well studied, mainly because a number of strains are pathogens and hence of medical importance (e.g., Bacillus anthracis or Clostridium difficile). Environmental strains of endospore-forming bacteria have been isolated from virtually every environment (soil, water, sediment, air, ice, human gut, animals gut, and more)1-3. Therefore, Firmicutes are the second most abundant phylum in culture collections4.
Because of their hardy outer cortex and protective core proteins, endospores can survive extreme environmental conditions ranging from desiccation to high radiation, extreme temperatures and harmful chemicals5. This remarkable resistance makes it a challenge to extract DNA from endospores6-8. This likely explains why they have been overlooked in environmental sequencing studies9,10. Other methods, such as targeting of endospores in environmental samples by fluorescent antibodies11, quantification of dipicolinic acid (DPA) in soil12 and sediment13, flow cytometry14 or pasteurization and subsequent cultivation15,16 have been used to retrieve or quantify endospores in environmental samples. In recent years, optimized DNA extraction methods as well as specific molecular primers to target endospore-specific gene sequences have been developed10,17-20. This has helped to reveal more biodiversity among this group of bacteria21 and has also led to applications in industry and medicine for the detection of endospores, for example in milk powder19.
The protocol presented here is based on the difference in resistance to harmful physicochemical conditions (such as heat and detergents) of bacterial endospores relative to vegetative cells. To destroy vegetative cells in a sample, we consecutively apply heat, lysozyme and low concentrations of detergents. The time and strength of these treatments have been optimized so as not to destroy spores, but to lyse all vegetative cells. Some cells in an environmental cell pool are more resistant than others, so in order to increase the probability of destroying all vegetative cells, we apply three different treatments. The advantage and novelty of this method is that the endospores after the treatment are still intact and can be used for further downstream analyses. These include DNA extraction, quantitative PCR (qPCR) and amplicon or metagenomic sequencing (targeting specifically the group of endospores and thus reducing diversity, while increasing coverage). The endospores could also be used for downstream cultivation or quantification by fluorescence microscopy, flow cytometry, or detection of DPA. An important feature of this method is that by comparing an untreated sample with a treated sample, one can deduce the quantity and diversity of endospores in an environmental sample in addition to the component corresponding to vegetative cells.
Protocol
1. Preparation of Chemicals and Equipment
Make 500 ml of a 1% sodium hexametaphosphate (SHMP) (NaPO3)n solution and sterilize by autoclaving.
Sterilize nitrocellulose (NC) filters (pore size 0.22 µm, diameter 47 mm) by autoclaving in closed glass Petri dishes.
Sterilize NC filters (pore size 0.22 µm, diameter 25 mm) by autoclaving in closed glass Petri dishes.
Weigh and note the empty weight of sterile 50 ml tubes (with cap on) (one tube per sample).
Prepare Tris-EDTA-buffer (TE-buffer) 1x: make solution of 10 mM Tris (tris(hydroxymethyl)aminomethane) and 1 mM EDTA buffer. Adjust pH to 8 and sterilize by autoclaving.
Prepare lysozyme solution (20 mg/ml) by dissolving 0.02 g of lysozyme in 1 ml TE-buffer. Ideally, make fresh every time. Store at 4 °C for no more than 1 week.
Prepare physiological solution by dissolving 8 g/L sodium chloride (NaCl) in distilled water. Sterilize by autoclaving.
2. Separation of Biomass from Sediment
Add 3 g of sediment sample to pre-weighed sterile 50 ml tubes using ethanol-flamed metal scoops. Perform this in a clean, UV sterilized biosafety cabinet to avoid contamination.
Add 15 ml of a 1% sterile (autoclaved) SHMP solution to the sample using a sterile graduated burette. The SHMP solution can also be filtered to avoid contamination.
Homogenize the sediment and SHMP solution with a liquid disperser/homogenizer (e.g., Ultra-Turrax homogenizer). 70% ethanol-sterilize or autoclave the dispersion rotor prior to use. Run the homogenization for 1 min at 17,500 rpm. Let the sample rest for 2 min and repeat the homogenization for 1 min at the same rotor speed.
Let the sample stand for 10 min. At this step, the heaviest particles (minerals) will settle. The cells and any organic components of the sample however will remain in solution. Afterwards, transfer the supernatant solution (containing cell biomass) into a clean 50 ml tube, while taking care not to disturb the sediment pellet.
To the sediment pellet add again 15 ml of a 1% sterile (autoclaved) SHMP solution using a sterile graduated burette. Then repeat steps 2.3 and 2.4. This repetition ensures the separation of the maximum amount of cells and organic particles from the mineral component of the sediment. The supernatant of this second separation can be merged with the supernatant from the first separation. Note: The following steps are all done on the supernatant (containing cell biomass). The mineral component (sediment pellet) can be discarded.
Centrifuge the sample at 20 x g for 1 min. This step increases the g-force enough to settle small mineral particles while the biological cell material still remains in solution. After centrifugation, transfer the supernatant solution (containing cell biomass) into a clean 50 ml tube. Discard the mineral pellet.
Determine the final volume of the solution containing biomass by weighing the sample. The weight determination avoids having to transfer the sample to a graduated cylinder and reduces risk of contamination.
3. Collection of Biomass on Filter Membrane
Prepare filtration unit (for 47 mm diameter membranes) and vacuum pump. Sterilize the filtration unit either by autoclaving or (if Pyrex glass) by spraying it with 70% ethanol and flaming it with a Bunsen burner. Let it cool down before continuing with the protocol.
Add sterile NC membrane to the filtration unit using ethanol-flamed sterilized forceps.
Add half of the supernatant sample (from step 2.6) onto the membrane filtration unit and collect cells on the membrane using the vacuum pump.
- When the liquid has fully passed through the filter, stop the vacuum pump and carefully remove the membrane using ethanol-flame sterilized forceps. Place the membrane into a sterile Petri dish.
- Cut the membrane in half using ethanol-flamed sterilized scissors. Add each half of the membrane to a separate 2 ml tube. One half of the membrane will be used for DNA extraction and analysis of the entire bacterial community. The other half of filter will be stored at -80 °C and serves as a backup.
Place new NC membrane onto the filtration unit and collect biomass from the second half of sample volume (from step 2.6) using the vacuum pump.
When the liquid has fully passed through the filter, stop the vacuum pump and carefully remove the membrane using ethanol-flamed sterilized forceps. Place the entire membrane into a separate 2 ml tube. This sample will be used for the treatment to separate endospores from vegetative cells. The sample can be stored at -20 °C until use.
4. Lysis of Vegetative Cells
- Perform the treatment to separate endospores from vegetative cells on the biomass previously collected on a NC membrane (step 3.6).
- If the membrane was frozen, leave it at RT for 10 min to thaw. Then place the membrane in a sterile Petri dish and cut it (approximately 4 times) into smaller pieces using ethanol-flamed sterilized scissors. Then place all membrane filter pieces into a sterile 2 ml tube.
Add 900 µl of 1x TE (Tris-EDTA) buffer (see 1.5) to the tube containing the sample membrane and mix thoroughly by vortex. At this step, the biomass is removed from the membrane into the TE-buffer solution.
Place the tube in an incubator at 65 °C for 10 min and 80 rpm. Afterwards remove the tube from the incubator and let it cool down for 15 min.
Add 100 µl of freshly prepared lysozyme (see 1.5) to reach a final concentration of 2 mg/ml. Do not add the lysozyme before the sample has cooled down to 37 °C, as this could degrade the enzyme.
Incubate the sample at 37 °C for 60 min and 80 rpm, the optimal conditions for the lysozyme to lyse vegetative cells.
After lysis is complete, add 250 µl of 3 N sodium hydroxide (NaOH) and 250 µl of 6% sodium dodecyl sulfate (SDS) solution to the sample. By adding this, the sample volume reaches 1.5 ml and there is a final concentration of 0.5 N NaOH and final concentration of 1% SDS.
Incubate this mix at RT for 60 min and 80 rpm. Adding the base and detergents will help in final cell lysis. The concentration of these detergents has been optimized as to not harm the endospores, while lysing vegetative cells.
Prepare a sterile filtration unit that holds 25 mm diameter membranes by autoclaving, or (if Pyrex glass) spraying it with 70% ethanol and flaming it with a Bunsen burner. Let it cool down.
Place a 0.2 µm NC membrane (25 mm diameter) on the filtration unit using ethanol-flame sterilized forceps.
Add the sample from step 4.7 onto the membrane and filter the liquid using the vacuum pump. When liquid has passed through, turn off vacuum pump. At this step, the lysed vegetative cell material is removed, as it is not retained on the membrane. Only endospores will remain on the membrane.
Add 2 ml of sterile physiological solution to wash off residual detergents and filter the liquid using the vacuum pump.
When liquid has fully filtered, turn off the vacuum pump. Leave the membrane on the filtration unit.
5. DNase Treatment
Note: Perform the DNase treatment directly on the filter membrane. It is important that the filtration unit does not leak and the vacuum pump is turned off.
Add 450 µl of sterile water, 50 µl of DNase reaction buffer (1x) and 0.5 µl DNase enzyme directly onto the filter membrane and let it stand for 15 min. If possible, do this digestion in a room that is slightly warmer than average RT, since the enzyme works better at temperatures of 25 °C and above. Note: Keeping the Bunsen burner aside the filtration unit also increases the temperature and has the added benefit of keeping the surroundings sterile, therefore reduced risk of contamination of the samples.
When the DNase digestion is finished, turn on vacuum pump to remove the enzyme from the sample.
Wash off residual enzyme by adding and filtering 1 ml of physiological solution.
If liquid has fully passed, turn off vacuum pump and remove the filter membrane containing endospores using sterile forceps and place it into a sterile Petri dish. Note: The sample of separated endospores is now ready for downstream analysis. Store at -20 °C if used for DNA extraction or alternatively at 4°C if used for germination and cultivation.
Representative Results
The results presented here have been published earlier10,21. Please refer to those articles for the environmental interpretation and discussion of the data.
The overall procedure is summarized in Figure 1 and corresponds to three main steps: first, the separation of biomass from sediment or any other environmental matrix; second, the destruction of vegetative cells; and third, the downstream analysis of the separated endospores. Downstream analysis could consist, for example, of DNA extraction and amplicon sequencing to determine diversity. The DNA extraction needs to be optimized as to guarantee lysis of endospores. In sediment samples, we have achieved this by using a forceful sequential DNA extraction procedure10. The application of the method in sediment demonstrates a significant increase in the fraction of endospore-forming Firmicutes after separation. In amplicon sequencing of the 16S rRNA gene, Firmicutes in the untreated samples (whole community) corresponded only to 8.0 and 19.0% of the sequences (Table 1). In contrast, after the treatment Firmicutes represented 90.6% and 83.9% of the endospore-enriched sample. The two major orders of endospore-forming Firmicutes, Bacilliales and Clostridiales, were enriched with the method, contrasting with the absence of orders like Lactobacilliales that are non endospore-forming Firmicutes. In order to demonstrate that the method is particularly suitable for the enrichment of true endospores, other groups capable of producing spore-like structures were also analyzed. Accordingly, the frequency of Actinobacteria, Cyanobacteria and Myxococcales decreased after the treatment to lyse cells.
The efficiency of the treatment was also demonstrated using pure cultures. An endospore preparation (> 95% endospores) of Paenibacillus alvei, Bacillus subtilis and Bacillus megaterium, as well as a vegetative cell culture of Escherichia coli, were treated as described in the protocol for the lysis of vegetative cells. All cultures were then incubated in nutrient broth at 37 °C and growth was measured using optical density at 600 nm wavelength. Growth was observed for the endospore cultures while no growth was observed in the treated E. coli culture, proving that vegetative cells are irreversibly damaged (Figure 2).
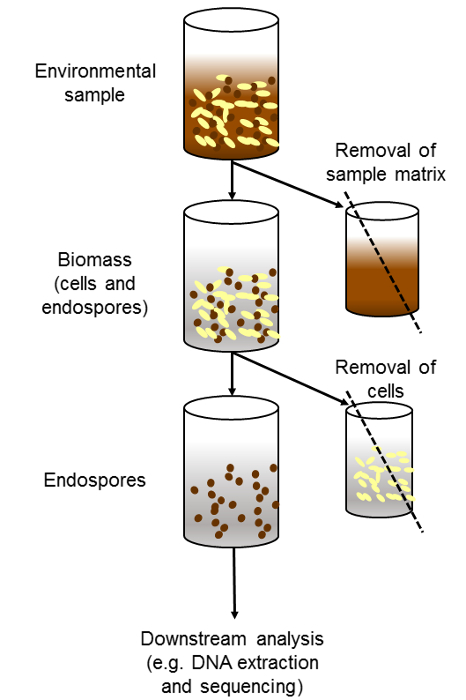 Figure 1. Overview of the experimental procedure. Procedure used to enrich endospore-forming bacteria in environmental samples. The step of separating cells and endospores from the environmental matrix can be omitted depending on the type of sample (i.e., water sample). In the figure the downstream methods applied on the spore-enriched fraction correspond to DNA extraction and high throughput sequencing. These steps can be replaced by culturing. Please click here to view a larger version of this figure.
Figure 1. Overview of the experimental procedure. Procedure used to enrich endospore-forming bacteria in environmental samples. The step of separating cells and endospores from the environmental matrix can be omitted depending on the type of sample (i.e., water sample). In the figure the downstream methods applied on the spore-enriched fraction correspond to DNA extraction and high throughput sequencing. These steps can be replaced by culturing. Please click here to view a larger version of this figure.
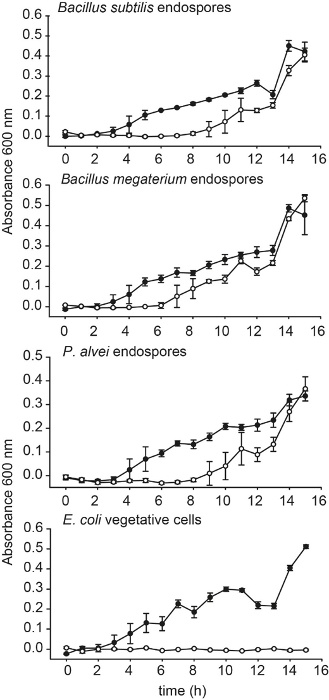 Figure 2. Verification of the treatment on pure cultures. Growth curves verifying the growth of endospore-forming bacteria from a suspension of endospores of Bacillus subtilis, Bacillus megaterium, and Paenibacillus alvei and a cell culture of Escherichia coli treated with the method for the lysis of cells. Treated = ○. Untreated = ●. Error bars from three independent cultures. Growth of control cultures and re-growth of treated cultures was measured as optical density at 600 nm wavelength. This figure has been re-printed from Wunderlin et al. 2014 with permission. Please click here to view a larger version of this figure.
Figure 2. Verification of the treatment on pure cultures. Growth curves verifying the growth of endospore-forming bacteria from a suspension of endospores of Bacillus subtilis, Bacillus megaterium, and Paenibacillus alvei and a cell culture of Escherichia coli treated with the method for the lysis of cells. Treated = ○. Untreated = ●. Error bars from three independent cultures. Growth of control cultures and re-growth of treated cultures was measured as optical density at 600 nm wavelength. This figure has been re-printed from Wunderlin et al. 2014 with permission. Please click here to view a larger version of this figure.
| Sediment 1 | Sediment 2 | |||
| whole community | endospore-enriched | whole community | endospore-enriched | |
| Firmicutes | 8.0 | 90.6 | 19.0 | 83.9 |
| Bacilli | 0.5 | 10.0 | 5.7 | 15.1 |
| Clostridia | 7.4 | 76.9 | 12.5 | 63.2 |
| Actinobacteria | 2.4 | 1.0 | 4.4 | 2.1 |
| Cyanobacteria | 1.1 | 0.1 | 0.7 | 0.1 |
| Myxococcales | 0.7 | 0.0 | 0.2 | 0.0 |
| Other bacteria | 87.8 | 8.3 | 75.7 | 13.9 |
Table 1. Abundance of endospores and other spore-forming bacterial groups. Relative frequency of Firmicutes (endospore-formers) and other bacterial groups producing spore-like structures in two sediment samples corresponding to the whole (untreated) and endospore-enriched (treated) communities.
Discussion
The resistance of endospores to external aggressive physicochemical factors (e.g., temperature or detergents) was used to devise a method to separate bacterial endospores from vegetative cells in environmental samples. This is the first comprehensive method to isolate endospores from environmental samples in a non-destructive manner. Previous methods to quantify, detect or analyze endospores in samples were based on the measurement of specific proxies for endospores such as dipicolinic acid or specific marker genes. In contrast, with the protocol presented here, sample components other than endospores are removed and so the endospores eventually remain as the sole and purified remnants of the sample. Although other methods such as pasteurization or density gradient centrifugation have been proposed to enrich for endospores22, our tests showed that density gradient centrifugation is not appropriate as a general method for environmental samples, due to physiological differences and different densities of various species of endospore-formers (data not shown). By contrast, our protocol can easily be applied to all types of environmental samples and is suitable for downstream applications including molecular methods or culturing.
There are two critical steps in the protocol. The first is the separation of biomass from sample matrix (i.e., sediment particles). It is essential that the number of cells separated from the matrix be maximized, so as not to overlook part of the diversity. For sediment samples, as used here, one of the principal limitations of the protocol is the fact that some cells or spores may not have been detached from the sediment matrix and therefore not included in the downstream analysis, which is a potential limitation of the method. Depending on the sample matrix (for example if there are humic acids), cells may be tightly bound to it and therefore difficult to release. The use of ortho-phosphate buffer as well as good homogenization with a homogenizer or blender are important at this step. To boost recovery, the procedure of homogenization and removal of biomass is repeated twice as written in the protocol. The protocol described here has been developed and optimized for samples of freshwater lake sediment. Some parameters may not be optimal for other types of samples. One way to analyze the community fraction that was potentially overlooked in the process would be to subject the sediment matrix to DNA extraction and amplicon sequencing. As possible modifications to the technique, the step of separation of biomass from sample matrix may be omitted. Particularly if the environmental sample does not contain organic material or mineral particles that could interfere with the downstream protocol (e.g., water, other liquids or purified cultures), prior separation may not be needed. For liquid samples the protocol can start directly at chapter 3 (Collection of biomass on filter membrane).
The second important step of the protocol is the treatment with lysozyme, heat, NaOH and SDS to destroy vegetative cells. Temperature, duration as well as concentration of chemicals have all been optimized as not to harm endospores, while destroying the cells. These parameters should therefore be strictly kept as they are. The use of a first step of heating the sample at 65 °C was very effective to specifically select for endospores compared to other types of bacterial spores or spore-like structures occurring among the bacterial phyla of Actinobacteria, Myxobacteria, Cyanobacteria, that are generally not heat-resistant. As seen in Table 1, non-spore forming bacteria are still present after the treatment (8.3% to 14%). Some explanations for this are that sediment samples are highly complex and can also harbor resistant non-spore cells. In addition, leftover organic material may allow certain attached cells to survive the treatment. In order to further reduce the percentage of non-spore-forming cells in the treated samples, the method should be optimized based on individual sample type.
The method represents a novel tool for the targeted study of endospore-formers in environmental samples. The treatment to destroy vegetative cells can be tested on pure cultures of cells and endospores and adapted to suit individual resistance of cells. The destruction of cells can be seen in the curves of treated cultures shown in Figure 2. The delayed growth of the treated cultures can be due to the time that endospores need to re-germinate and move into the exponential growth phase. Other possible explanations for this delay could also be a weakening of the endospore culture or lower cell numbers after treatment. If the separated endospores are not used for DNA extraction, the last step of DNase treatment can be omitted.
Future applications of this technique could be envisaged in industries where pathogenic endospores need to be detected. One example is in the food industry and clinical research, where the detection and analysis (for identification) of endospores, and its differentiation from vegetative cells (for example, from foods products such as milk or milk-based products) is vital. This might be particular relevant considering that many standard procedures consider direct DNA extraction from a sample using lytic enzymes that are not adapted to lyse endospores, which will thus be overlooked. With the method presented here, cells can be removed and subsequent analysis will provide answers to the presence or absence of endospores as opposed to cells. Other applications include the analysis of endospore communities in different environmental samples (for example ice or sediment cores) that are historical archives of environmental conditions. In many environments with relatively stable environmental conditions (for example lake sediment surface) endospore-forming Firmicutes can thrive in the form of vegetative cells. When conditions deteriorate (for example by nutrient depletion during sediment burial or a shift towards anaerobic conditions) the cells move into endospore state. Therefore, the retrieved endospore communities from sediment cores can reflect the environmental conditions at time of burial and thus be used as a paleoecological indicator of the conditions prior to burial. However, we are still gathering information to support this claim.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors acknowledge the Swiss National Science Foundation for Grant No. 31003A-132358/1, 31003A_152972 and No. 151948, and Fundation Pierre Mercier pour la Science.
References
- Nicholson WL. Roles of Bacillus endospores in the environment. Cell Mol Life Sci. 2002;59(3):410–416. doi: 10.1007/s00018-002-8433-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester ED, Satomi M, Ponce A. Microflora of extreme arid Atacama Desert soils. Soil Biol Biochem. 2007;39(2):704–708. [Google Scholar]
- Wilson MS, Siering PL, White CL, Hauser ME, Bartles AN. Novel archaea and bacteria dominate stable microbial communities in North America's Largest Hot Spring. Microb Ecol. 2008;56(2):292–305. doi: 10.1007/s00248-007-9347-6. [DOI] [PubMed] [Google Scholar]
- Hugenholtz P. Exploring prokaryotic diversity in the genomic era. Genome Biol. 2002;3(2) doi: 10.1186/gb-2002-3-2-reviews0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onyenwoke RU, Brill JA, Farahi K, Wiegel J. Sporulation genes in members of the low G+C Gram-type-positive phylogenetic branch (Firmicutes) Arch Microbiol. 2004;182(2-3):182–192. doi: 10.1007/s00203-004-0696-y. [DOI] [PubMed] [Google Scholar]
- Delmont TO, et al. Accessing the soil metagenome for studies of microbial diversity. Appl Environ Microbiol. 2011;77(4):1315–1324. doi: 10.1128/AEM.01526-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricca E, Henriques AO, Cutting SM. Bacterial spore formers: probiotics and emerging applications. Horizon Bioscience; 2004. [Google Scholar]
- Kuske CR, et al. Small-Scale DNA Sample Preparation Method for Field PCR Detection of Microbial Cells and Spores in Soil. Appl Environ Microbiol. 1998;64(7):2463–2472. doi: 10.1128/aem.64.7.2463-2472.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Mering C, et al. Quantitative phylogenetic assessment of microbial communities in diverse environments. Science. 2007;315(8515):1126–1130. doi: 10.1126/science.1133420. [DOI] [PubMed] [Google Scholar]
- Wunderlin T, Junier T, Roussel-Delif L, Jeanneret N, Junier P. Stage 0 sporulation gene A as a molecular marker to study diversity of endospore-forming Firmicutes. Environ Microbiol Rep. 2013;5(6):911–924. doi: 10.1111/1758-2229.12094. [DOI] [PubMed] [Google Scholar]
- Siala A, Hill IR, Gray TRG. Populations of spore-forming bacteria in an acid forest soil, with special reference to Bacillus subtilis. J General Microbiol. 1974;81(1):183–190. [Google Scholar]
- Brandes Ammann A, Kolle L, Brandl H. Detection of bacterial endospores in soil by terbium fluorescence. Int J Microbiol. 2011. p. 435281. [DOI] [PMC free article] [PubMed]
- Fichtel J, Koster J, Rullkotter J, Saas H. High variations in endospore numbers within tidal flat sediments revealed by quantification of dipicolinic acid. Geomicrobiol J. 2008;25(7-8) [Google Scholar]
- Cronin UP, Wilkinson MG. The potential of flow cytometry in the study of Bacillus cereus. J Appl Microbiol. 2010;108(1):1–16. doi: 10.1111/j.1365-2672.2009.04370.x. [DOI] [PubMed] [Google Scholar]
- de Rezende JR, et al. Dispersal of thermophilic Desulfotomaculum endospores into Baltic Sea sediments over thousands of years. ISME J. 2013;7(1):72–84. doi: 10.1038/ismej.2012.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubert C, et al. Thermophilic anaerobes in Arctic marine sediments induced to mineralize complex organic matter at high temperature. Environ Microbiol. 2010;12(4):1089–1104. doi: 10.1111/j.1462-2920.2010.02161.x. [DOI] [PubMed] [Google Scholar]
- Garbeva P, van Veen JA, van Elsas JD. Predominant Bacillus spp. in agricultural soil under different management regimes detected via PCR-DGGE. Microb Ecol. 2003;45(3):302–316. doi: 10.1007/s00248-002-2034-8. [DOI] [PubMed] [Google Scholar]
- Brill JA, Wiegel J. Differentiation between spore-forming and asporogenic bacteria using a PCR and southern hybridization based method. J Microbiol Methods. 1997;31(1-2):29–36. [Google Scholar]
- Rueckert A, Ronimus RS, Morgan HW. Development of a real-time PCR assay targeting the sporulation gene, spo0A., for the enumeration of thermophilic bacilli in milk powder. Food Microbiol. 2006;23(3):220–230. doi: 10.1016/j.fm.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Bueche M, et al. Quantification of endospore-forming firmicutes by quantitative PCR with the functional gene spo0A. Appl Environ Microbiol. 2013;79(17):5302–5312. doi: 10.1128/AEM.01376-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wunderlin T, Junier T, Roussel-Delif L, Jeanneret N, Junier P. Endospore-enriched sequencing approach reveals unprecedented diversity of Firmicutes in sediments. Environ Microbiol Rep. 2014;6(6):631–639. doi: 10.1111/1758-2229.12179. [DOI] [PubMed] [Google Scholar]
- Nicholson WL, Law JF. Method for purification of bacterial endospores from soils: UV resistance of natural Sonoran desert soil populations of Bacillus spp. with reference to B. subtilis strain 168. J Microbiol Methods. 1999;35(1):13–21. doi: 10.1016/s0167-7012(98)00097-9. [DOI] [PubMed] [Google Scholar]