

Abstract
To analyze gene regulatory networks active during embryonic development and organogenesis it is essential to precisely define how the different genes are expressed in spatial relation to each other in situ. Multi-target chromogenic whole-mount in situ hybridization (MC-WISH) greatly facilitates the instant comparison of gene expression patterns, as it allows distinctive visualization of different mRNA species in contrasting colors in the same sample specimen. This provides the possibility to relate gene expression domains topographically to each other with high accuracy and to define unique and overlapping expression sites. In the presented protocol, we describe a MC-WISH procedure for comparing mRNA expression patterns of different genes in Drosophila embryos. Up to three RNA probes, each specific for another gene and labeled by a different hapten, are simultaneously hybridized to the embryo samples and subsequently detected by alkaline phosphatase-based colorimetric immunohistochemistry. The described procedure is detailed here for Drosophila, but works equally well with zebrafish embryos.
Keywords: Developmental Biology, Issue 107, Digoxigenin, biotin, fluorescein, azo dye, Fast Blue, Fast Red, INT, alkaline phosphatase substrate, WISH, fruit fly
Introduction
In situ hybridization (ISH) is the standard method for detection and localization of RNA transcripts in a morphological context, within cells, tissues and organisms 1. The signals produced by the ISH procedure are commonly visualized by radioactive, fluorescent and chromogenic detection systems. In recent years significant technological advances in fluorescent ISH (FISH)2 resulted in dramatically improved sensitivity and resolution, allowing the detection and quantitation of RNA expression at single-cell and sub-cellular levels and RNA visualization up to single molecules 3,4.While sophisticated single-molecule FISH methods are used for more specialized applications, chromogenic ISH is widespread as a routine RNA in situ detection method in research and clinical diagnostics. For chromogenic detection enzyme precipitation reactions are used, which generate visible products in contrasting colors at the sites of hybridization 5. This has the advantage that RNA visualization can be combined with routine histological stains and morphological context is immediately evident by standard brightfield microscopy. Moreover, numerous of the applied color substrates produce precipitates, which are stable in organic and/or aqueous mounting media, so that permanent sample preparations can be obtained 5.
In Drosophila, initial ISH protocols applied radioisotope-labeled probes for transcript detection on sectioned material 6. While it is difficult to reconstruct from tissue sections complete transcript patterns of entire embryos or organ systems, the application of chromogenic ISH procedures with non-radioactively labeled probes makes it possible to globally detect RNA distributions in whole-mounts 7. Although many variations of chromogenic ISH exist, in typical whole-mount in situ hybridization (WISH) protocols hybridized hapten-labeled probes are detected by anti-hapten antibodies conjugated to a reporter enzyme and mRNA transcripts are visualized by a precipitating chromogen.
The palette of differently colored precipitates produced by the reporter enzymes alkaline phosphatase (AP), horseradish peroxidase (POD), and beta-galactosidase (GAL) allows for the distinctive detection of multiple targets in one and the same sample 8-14. However, POD enzymatic activity lasts only for a limited time period and the GAL colorimetric reaction is somewhat less sensitive, so that without additional (tyramide) signal amplification 15 the detection of less abundant transcripts can be challenging with these enzymes. In contrast, the enduring activity of AP allows for long-lasting substrate turnover and high signal-to-noise ratio. Therefore, sequential detection using AP reporter enzyme with differently colored substrates has proven successful in the effective and distinctive detection of up to three different transcripts in single embryos 10-12,16.
For this multi-target chromogenic WISH (MC-WISH) method (Figure 1) 14, labeled antisense RNA probes are generated by in vitro transcription and marked with one of the available hapten-labels. Embryos are formaldehyde fixed and permeabilized by methanol treatment and proteinase K digestion. Hybridization of embryos is simultaneously carried out with up to three differently labeled antisense RNA probes each specific for a different gene. After removal of unbound probe by stringency washes each hapten-label is visualized in a separate round of detection. A single detection round consists of incubation of embryos with an anti-hapten antibody coupled to AP and RNA visualization by application of an AP-substrate that produces a localized stable color precipitate. After antibody detection and staining, the applied antibody-AP conjugate is removed by a low pH wash. In multicolor experiments, each round of detection employs an antibody targeted against a different hapten-label and each transcript pattern is visualized by a different color substrate (Table 1). Embryos are mounted in glycerol and imaged under a high-resolution compound microscope using differential interference contrast (DIC) optics.
Protocol
1. Labeling of RNA probes by In Vitro Transcription
Assemble in vitro transcription reaction in a 1.5 ml microtube under RNase-free conditions: 10.5 µl DEPC-treated H2O, 4.0 µl 5x transcription buffer, 1.0 µl (1 µg) linearized, purified template DNA in DEPC-treated H2O, 1.3 µl NTP mix, 0.7 µl hapten-labeled UTP, 0.5 µl RiboLock RNA inhibitor, 2.0 µl RNA polymerase. Note: The final reaction volume is 20 µl.
Depending on the template's promoter sequence use T7, T3 or SP6 RNA polymerase for the in vitro transcription (see reference 17).
Select digoxigenin-11-UTP, biotin-16-UTP, or fluorescein-12-UTP as label for the RNA probe. For more details, see discussion of Hapten-labels and probe concentrations in the Discussion.
Mix transcription reaction and spin down shortly.
Let transcribe for 3 hr at 37 °C.
Add 1 µl RNase-free DNase I to the transcription reaction for removal of template DNA, mix well, and incubate for 15 min at 37 °C.
Adjust with DEPC-treated H2O the sample volume to 200 µl.
- Add 100 µl (0.5 vol) 7.5 M ammonium acetate and 600 µl (3 vol) ethanol to precipitate labeled RNA probe. Incubate for 30 min at RT.
- Do not use ice-cold ethanol, as this may lead to unwanted precipitation of unincorporated nucleotides.
Spin down the transcribed RNA in a temperature controlled centrifuge at maximum speed (20,800 x g) for 30 min at +20 °C. Carefully aspirate supernatant.
Wash the resulting pellet with 70% ethanol at RT and centrifuge at 20,800 x g for 10 min at +20 °C.
Remove supernatant and be careful not to accidentally aspirate the loose pellet. Let pellet air dry in the microtube with opened lid for a few min at RT.
Dissolve the obtained pellet in 100 µl of DEPC-treated H2O.
Add 300 µl pre-hybridization buffer (50% vol/vol deionized formamide, 5x SSC, 50 µg/ml heparin sodium salt, 0.1% vol/vol Tween-20, 5 mg/ml torula RNA) and store at -20 °C. Probes can be long-term stored at -20 °C. Note: For a first test of the newly transcribed probe dilute 3 µl hapten-labeled RNA probe in 100 µl hybridization buffer to obtain the final concentration for the WISH experiment.
2. Processing of Embryo Samples Using Inserts
Note: Using microtubes or multi-well plates for processing embryos through the different incubation steps bears the risk of losing significant amounts of embryos by accidentally aspirating them during exchange of solutions. To avoid this loss, it may be helpful to use baskets (such as Netwell inserts) for carrying embryo samples through the procedure. These are polystyrene inserts with a polyester mesh at the bottom (Figure 2A), on which the embryos rest during processing.
For processing embryos in the WISH procedure, assemble the inserts in a 12-well plate (Figure 2B) and add 2 ml of the appropriate solution to each well.
Pipette the embryos into the inserts using a blue tip. Make sure that all embryos are submerged in liquid.
- When incubation time is over place the embryo-containing inserts in another 12-well plate, which is filled with the solution for the next step.
- Use carriers and handles (Figure 2C) to transfer up to 12 samples at a time from one solution to another (Figure 2D). Note: To reduce the required volumes, use 2.0 ml microtubes for the pre-hybridization/hybridization steps (see STEP 4.1) and 24-well plates for the staining reactions (STEP 6.2). The hybridization and staining reactions are performed in volumes of 100 µl and 400 µl, respectively.
3. Preparation of Embryos for Hybridization
Collect, dechorionate, fix, and devitellinize embryos using standard procedures 14,18. The prepared embryos are routinely stored in methanol at -20 °C.
Transfer embryos to be used for in situ hybridization to RT and distribute into the inserts of a 12-well plate filled with 2 ml methanol per well.
Rehydrate embryos through a decreasing methanol series at RT. Incubate embryos each time for 3 min in: (i) 75% methanol in 1x PBS (ii) 50% methanol in 1x PBS, 0.1% Tween-20 (iii) 25% methanol in 1x PBS, 0.1% Tween-20 (iv) 1x PBS, 0.1% Tween-20 (v) 1x PBS, 0.1% Tween-20.
Pre-fix the embryos in 4% paraformaldehyde in 1x PBS for 20 min at RT.
Remove the fixative by 4 washes for 3 min in 1x PBS, 0.1% Tween-20 at RT. Meanwhile thaw and prepare proteinase K and glycine working solutions.
Permeabilize embryos in fresh proteinase K (10 µg/ml to 50 µg/ml) in 1x PBS, 0.1% Tween-20 for 3 min at RT. Note: The proteinase K treatment is balanced to the strength of embryo fixation. For example, if the pre-fixation step (3.4) is omitted, softer proteinase K treatment may be applied.
Stop the reaction by two 1 min rinses in 2 mg/ml glycine in 1x PBS, 0.1% Tween-20 at RT.
Post-fix the embryos in 4% paraformaldehyde in 1x PBS for 20 min at RT.
Subsequently, remove paraformaldehyde fixative by washing four times for 3 min in 1X PBS, 0.1% Tween-20 at RT.
Transfer post-fixed embryos to pre-hybridization buffer. Store embryos in pre-hybridization buffer at -20 °C or directly proceed with hybridization of probes.
4. Hybridization of Probes
Transfer embryos stored at -20 °C in pre-hybridization buffer to RT. Distribute embryos in pre-hybridization buffer into 2.0 ml microtubes.
For 30 min at a minimum, pre-hybridize embryo samples at 65 °C in pre-hybridization buffer. Note: For pre-hybridization and hybridization steps a waterbath is used. During pre-hybridization there is time to prepare the probe mix by combining up to three antisense RNA probes, each specific for a different gene transcript and labeled with a different hapten.
To obtain the probe mix, add together appropriate amounts (usually between 1 µl to 6 µl) of each desired original hapten-labeled antisense RNA probe in 100 µl hybridization buffer (50% vol/vol deionized formamide, 5x SSC, 50 µg/ml heparin sodium salt, 0.1% vol/vol Tween-20, 5 mg/ml torula RNA, 5% wt/vol dextran sulfate). Note: The addition of dextran sulfate is optional.
Denature the antisense RNA probe mix in a heat block at 80 °C for 5 min. Subsequently transfer the denatured probe mix directly to 65 °C.
Remove most of the pre-hybridization buffer from the embryo samples and add the denatured probe mix.
Let embryo samples hybridize to probe mix O/N at 65 °C. Note: Temperatures ranging between 55 °C and 65 °C are routinely used for hybridization, pre- and post-hybridization steps. Hybridization at 65 °C provides most stringent conditions and is recommended in case of background problems when using lower temperatures. If there are no background problems, hybridization at 55 °C is recommended, which provides the advantage of enhanced signal strength and improved embryo integrity.
Next morning, first transfer the stringency wash solutions to 65 °C, to make sure that they are heated to the correct temperature in time.
Transfer hybridization probe mix with embryos from the 2.0 ml microtubes into inserts of a 12-well plate containing 2 ml pre-warmed hybridization wash buffer (50% formamide in 2× SSC, 0.1% Tween-20). Place the 12-well plate containing the embryo samples in an oven and incubate for 20 min at 65 °C.
Transfer embryo samples to 2 ml new hybridization wash buffer and incubate for 20 min at 65 °C. Repeat this step once more.
Remove excess probe by 20 min washes at 65 °C, once in 2× SSC, 0.1% Tween-20 and three times in 0.2× SSC, 0.1% Tween-20.
Transfer the embryo samples to 1x PBS, 0.1% Tween-20 at RT.
Incubate the embryos in blocking buffer (8% sheep serum in 1x PBS, 0.1% Tween-20) for at least 30 min at RT.
5. Antibody Detection of Hapten-labeled Probes
Note: In contrast to the simultaneous hybridization of all three differently labeled probes, each probe is detected one after another in separate rounds of antibody incubation and staining. Therefore, in each detection round only one antibody is used (either anti-digoxigenin-AP or anti-fluorescein-AP or anti-biotin-AP).
- Choose between STEP 5.1.1 to 5.1.3 and proceed with preparing the appropriate antibody dilution according to the hapten-label that is to be detected. Note: For sequential detection of the differently labeled probes, another anti-hapten-antibody is applied in each detection cycle.
- For detection of a fluorescein-labeled probe dilute anti-fluorescein-AP conjugates 1:4,000 in blocking buffer.
- For detection of a digoxigenin-labeled probe dilute anti-digoxigenin-AP conjugates 1:4,000 in blocking buffer.
- For detection of a biotin-labeled probe prepare an anti-biotin-AP conjugate solution at a 1:4,000 dilution in blocking buffer.
Incubate embryos in the selected anti-hapten-AP solution for 3 to 4 hr at RT. Alternatively, incubate O/N at 4 °C. Do not apply all three antibodies simultaneously.
In order to remove unbound antibody wash the embryos with 1x PBS, 0.1% Tween-20 for 4 times 15 min at RT.
To change buffer systems wash two times 15 min at RT in TNT (0.1 M Tris-HCl pH 8.0, 0.1 M NaCl, 0.1% Tween-20).
6. Alkaline Phosphatase Chromogenic Staining
- Subsequent to antibody detection choose between STEP 6.1.1 to 6.1.4 and proceed with one of the alkaline phosphatase chromogenic staining reactions to obtain the desired color signal (see Table 1). For each round of detection a different chromogenic substrate reaction is applied to highlight each transcript pattern in another color.
- Fast Red Staining
- Wash the embryos two times for 15 min in TT8.2 (0.1 M Tris-HCl pH 8.2, 0.1% Tween-20).
- While washing embryos, prepare the Fast Red staining solution from the Fast Red TR/NAMP Alkaline Phosphatase Substrate Tablets Set.
- Dissolve a buffer tablet from the tablet set in 1 ml distilled water and mix by vortexing to obtain 0.1 M Tris-HCl pH 8.2.
- Drop a Fast Red TR/NAMP tablet into the prepared Tris buffer and dissolve by vortexing to obtain the Fast Red staining solution.
- Clear Fast Red staining solution from non-dissolved particles through a 0.2 µm syringe filter.
- Fast Blue Staining
- Wash embryos two times for 15 min in fresh SB8.2 staining buffer (0.1 M Tris-HCl pH 8.2, 0.1 M NaCl, 0.05 M MgCl2, 0.1% Tween-20).
- While washing embryos, prepare the Fast Blue staining solution from 50 mg/ml Fast Blue BB (in DMF) and 50 mg/ml naphthol phosphate (in DMSO) stock solutions.
- Freshly prepare a Fast Blue BB solution at 0.5 mg/ml in SB8.2.
- Freshly prepare a separate naphthol phosphate solution at 0.5 mg/ml in SB8.2.
- Dropwise add the prepared naphthol phosphate solution into the Fast Blue BB solution under constant mixing with a vortex. The resulting final working concentration is 0.25 mg/ml for both substrate components. Note: Different Fast Blue / naphthol phosphate combinations result in different color shades (see Table 1).
- BCIP/NBT Staining
- Wash embryos two times for 15 min in fresh SB9.5 staining buffer (0.1 M Tris-HCl pH 9.5, 0.1 M NaCl, 0.05 M MgCl2, 0.1% Tween-20).
- For a purpleblue chromogenic signal, freshly prepare BCIP/NBT staining solution by adding 1 µl levamisole, 3.5 µl BCIP and 4.5 µl NBT per ml of SB9.5 staining buffer and mix well.
- INT Staining
- Wash the embryos two times for 15 min in fresh SB9.5 staining buffer.
- For a yellowbrown chromogenic signal, freshly prepare INT staining solution by adding 1 µl levamisole, 5 µl Magenta-Phos and 5 µl INT per ml of SB9.5 staining buffer and mix well. Note: Alternatively use 7.5 µl/ml BCIP/INT ready-to-use staining solution.
For the staining reaction transfer embryo samples to a 24-well plate. Incubate each sample in 400 µl of the chosen staining solution in the dark.
Monitor occasionally signal development under a stereomicroscope. Stop the staining reaction when the desired signal strength is obtained and before significant background signal is observed.
To stop the chromogenic reaction, transfer stained embryos into inserts of a 12-well plate containing 2 ml TNT per well. Rinse the stained embryos two times in TNT.
Wash the embryos two times for 10 min in 1× PBS, 0.1% Tween-20 at RT to remove residual substrate.
7. Antibody Removal/Alkaline Phosphatase Inactivation
Remove antibody and inactivate alkaline phosphatase by incubation in 2 ml of low pH stop solution (0.1 M glycine-HCl pH 2.2, 0.1% Tween-20) for 10 min at RT.
Rinse for 1 min with 2 ml of 1× PBS, 0.1% Tween-20 at RT.
Wash 3 times for 3 min in 1× PBS, 0.1% Tween-20 at RT.
8. Second Detection Round
To detect the distribution pattern of a second transcript proceed with antibody detection of hapten-labeled probes (STEP 5.1 to 5.4) using antibody-AP conjugates targeted against the second hapten-label.
Perform alkaline phosphatase chromogenic staining (STEP 6.1 to 6.5) applying a substrate combination that produces a contrasting color to the one used in the first detection round (for available colors see Table 1).
Remove second antibody-AP conjugate as described in STEP 7.1 to 7.3.
9. Third Detection Round
In order to detect the distribution pattern of a third transcript, proceed with antibody detection of hapten-labeled probes (STEP 5.1 to 5.4) using antibody-AP conjugates specific for the third hapten-label.
Perform alkaline phosphatase chromogenic staining (STEP 6.1 to 6.5) applying a substrate combination that was not applied in the previous detection rounds. Choose a chromogenic reaction that produces a color precipitate easy to distinguish from those of the previous two detection rounds (for color choice see Table 1).
10. Mounting and Imaging
Transfer properly stained embryos to 30% glycerol in PBS and incubate until embryos are fully soaked in the glycerol solution. They are well equilibrated when they sink to the bottom.
Transfer embryos to 70% glycerol in H2O and let equilibrate at RT. The stained embryos can be stored in 70% glycerol at 4 °C.
For mounting, pipette stained embryos in a 70% glycerol droplet onto an object slide and without touching the spacers glued to the slide (Figure 3). Apply the cover slip.
View mounted embryos under a dissecting microscope. Move the applied cover slip, so that the embryos rotate to the desired orientation.
Observe properly oriented embryos at high resolution (20X, 40X objectives) under a compound microscope with Differential Interference Contrast optics.
Capture images with a digital color camera and save on digital storage device.
Representative Results
The described protocol allows for the simultaneous visualization of multiple transcript patterns in different colors. MC-WISH provides the advantage to directly compare the expression patterns of different genes in the same embryo. As an example, the expression patterns of empty spiracles (ems) 19, fushi tarazu (ftz) 20 and sloppy paired 1 (slp1) 21 are directly compared in Drosophila melanogaster blastoderm stage embryos and visualized in a variety of color shades (Figure 4).
Different AP substrate combinations are available to obtain a panel of contrasting color precipitates (Table 1). The red color precipitate produced by Fast Red and the purple precipitate produced by BCIP/NBT can be easily distinguished and are therefore used most commonly in two-color experiments (Figure 4A). However, the BCIP/NBT stain can quickly turn into a rather dark color, so that the Fast Red color is disguised in case of partial co-distribution of transcripts. Therefore, the application of Fast Blue can be of advantage, which generates blue, green and violet products in combination with NAMP, NAGP and NABP, respectively (Table 1). The combination of Fast Blue with NAMP gives a light blue color that is easily discernible from the Fast Red product in adjacent or overlapping expression domains as shown here for ems and slp1 expression sites visualized in red and blue, respectively (Figure 4B). The Fast Blue NAGP combination provides a similar strong contrast to the Fast Red stain, so that the green ems domain is clearly distinctive from the red slp1 expression site (Figure 4C). However, if ems and slp1 expression sites are detected by Fast Blue NABP and Fast Red, respectively, the resulting violet and red color precipitates are less discernible (Figure 4D). As mentioned above, Fast Red and Fast Blue NAGP are easy to distinguish as shown by slp1 and ftz expression in red and green, respectively (Figure 4D).
MagentaPhos in combination with INT produces a yellow precipitate, which is used to reveal the segmental expression of ftz (Figure 4C, E). The yellow colored precipitate generates good contrast to the blue colored substrates as exemplified by the detection of slp1 and ftz with Fast Blue NAMP and MAG/INT, respectively (Figure 4E). However, the yellow precipitate is less easy discernible from the Fast Red product. The yellow stained expression domain of ems can be hardly distinguished from the rostral expression of slp1 shown in red (Figure 4F). Therefore, the combination of color substrates has to be carefully chosen for each experiment.
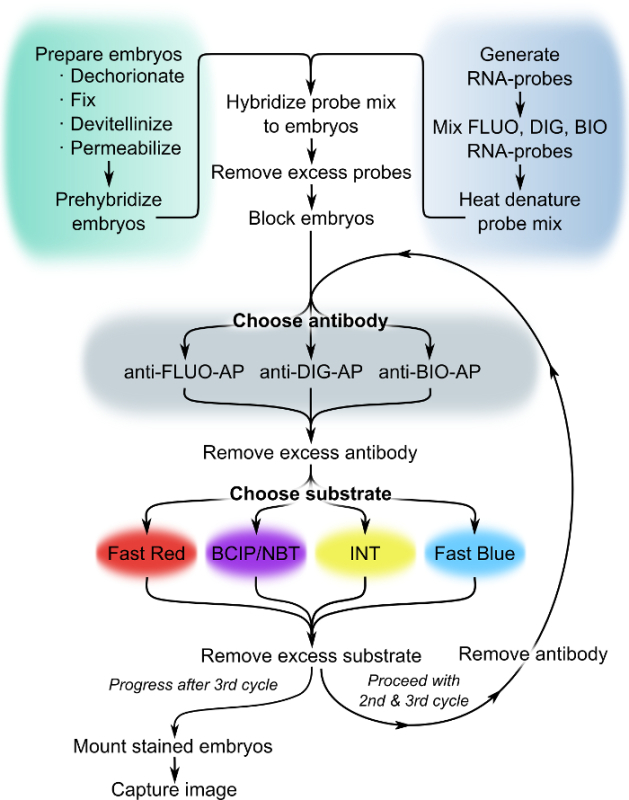 Figure 1. Flowchart of MC-WISH. For multicolored visualization of three unique transcript patterns, choose between different antibody-AP conjugates and color substrate combinations in each detection cycle. Abbreviations: AP, alkaline phosphatase; BIO, biotin; DIG, digoxigenin; FLUO, fluorescein. For other abbreviations see legends to Figure 4 and Table 1. Please click here to view a larger version of this figure.
Figure 1. Flowchart of MC-WISH. For multicolored visualization of three unique transcript patterns, choose between different antibody-AP conjugates and color substrate combinations in each detection cycle. Abbreviations: AP, alkaline phosphatase; BIO, biotin; DIG, digoxigenin; FLUO, fluorescein. For other abbreviations see legends to Figure 4 and Table 1. Please click here to view a larger version of this figure.
 Figure 2. Inserts as embryo carriers. (A) Inserts fitted at the bottom with polyester membranes of 15 mm diameter and 74 µm mesh size are used as Drosophila embryo carriers. (B) The inserts are assembled in 12-well plates, which function as reservoir trays for the various solutions. (C) Available carriers and handles allow processing of (D) 12 inserts/embryo samples simultaneously. Please click here to view a larger version of this figure.
Figure 2. Inserts as embryo carriers. (A) Inserts fitted at the bottom with polyester membranes of 15 mm diameter and 74 µm mesh size are used as Drosophila embryo carriers. (B) The inserts are assembled in 12-well plates, which function as reservoir trays for the various solutions. (C) Available carriers and handles allow processing of (D) 12 inserts/embryo samples simultaneously. Please click here to view a larger version of this figure.
 Figure 3. Mounting of stained embryos. Embryos are mounted in a drop of 70% glycerol between two stacks of coverslips, which function as spacers. Gentle moving of the applied large coverslip helps to rotate the mounted embryos into the desired orientation for observation and photography. Please click here to view a larger version of this figure.
Figure 3. Mounting of stained embryos. Embryos are mounted in a drop of 70% glycerol between two stacks of coverslips, which function as spacers. Gentle moving of the applied large coverslip helps to rotate the mounted embryos into the desired orientation for observation and photography. Please click here to view a larger version of this figure.
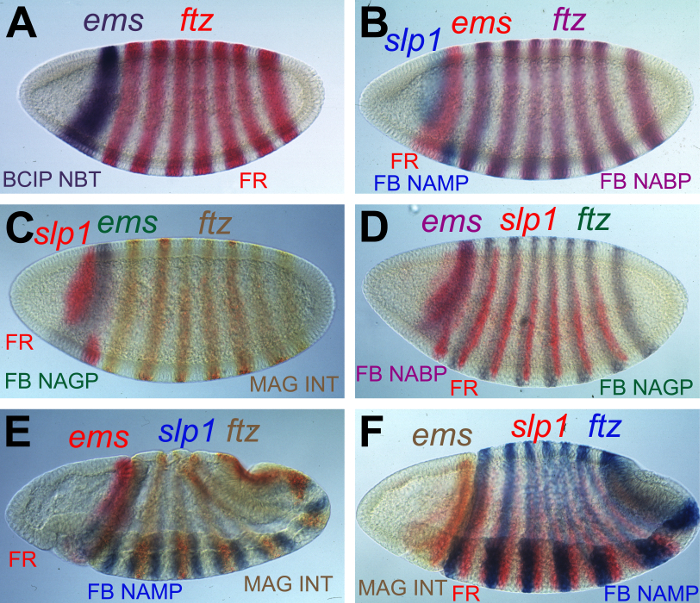 Figure 4. Multicolor visualization of mRNA transcripts in Drosophila embryos. Examples of MC-WISH experiments in Drosophila embryos are shown from lateral views. The mRNA expression patterns of empty spiracles (ems), fushi tarazu (ftz) and sloppy paired 1 (slp1) are visualized by different AP color substrate combinations, which are indicated on each panel. Transcripts were detected by (A) ftz FLUO and ems DIG, (B) slp1 FLUO, ems BIO, and ftz DIG, (C) ems BIO, slp1 FLUO, and ftz DIG, (D) ftz BIO, slp1 FLUO, and ems DIG, (E) slp1 FLUO, ems DIG, and ftz BIO, (F) ftz BIO, slp1 FLUO, and ems DIG labeled probes. The hapten-labeled probes were immunohistochemically detected one after another in the listed orders. Abbreviations: FB Fast Blue; FR, Fast Red tablet. For other abbreviations see legends to Figure 1 and Table 1. Please click here to view a larger version of this figure.
Figure 4. Multicolor visualization of mRNA transcripts in Drosophila embryos. Examples of MC-WISH experiments in Drosophila embryos are shown from lateral views. The mRNA expression patterns of empty spiracles (ems), fushi tarazu (ftz) and sloppy paired 1 (slp1) are visualized by different AP color substrate combinations, which are indicated on each panel. Transcripts were detected by (A) ftz FLUO and ems DIG, (B) slp1 FLUO, ems BIO, and ftz DIG, (C) ems BIO, slp1 FLUO, and ftz DIG, (D) ftz BIO, slp1 FLUO, and ems DIG, (E) slp1 FLUO, ems DIG, and ftz BIO, (F) ftz BIO, slp1 FLUO, and ems DIG labeled probes. The hapten-labeled probes were immunohistochemically detected one after another in the listed orders. Abbreviations: FB Fast Blue; FR, Fast Red tablet. For other abbreviations see legends to Figure 1 and Table 1. Please click here to view a larger version of this figure.
| Substrate combination | Add to 1 ml AP buffer | Concentration [µg/ml] | AP buffer | Color | Signal sensitivity |
| BCIP NBT | 3.5 µl 4.5 µl | 175 337.5 | SB9.5 | purple | +++ |
| MAG INT | 5 µl 5 µl | 250 250 | SB9.5 | yellow | + |
| BCIP/INT stock solution | 7.5 µl | 250 250 | SB9.5 | yellow | + |
| Fast Blue NAMP | 5 µl 5 µl | 250 250 | SB8.2 | blue | ++ |
| Fast Blue NABP | 20 µl 10 µl | 1,000 500 | SB8.2 | violet | ++ |
| Fast Blue NAGP | 10 µl 10 µl | 500 500 | TT8.2 | green | + |
| Fast Red NAMP | 1 tablet | 1,000 400 | TT8.2 | red | ++ |
Table 1. Substrate combinations. Abbreviations: +++, strong; ++, medium; +, weak NBT, 4-nitro-blue-tetrazolium chloride; BCIP, 5-bromo-4-chloro-3-indolyl-phosphate; INT, 2-(4-iodophenyl)-3-(4-nitrophenyl)-5-phenyl-tetrazolium chloride; MAG, Magenta-Phos, 5-bromo-6-chloro-3-indolyl-phosphate, NAMP, Naphthol-AS-MX-phosphate; NABP, Naphthol-AS-BI-phosphate; NAGP, Naphthol-AS-GR-phosphate; SB9.5, staining buffer at pH 9.5; SB8.2, staining buffer at pH 8.2; TT8.2, Tris-Tween buffer at pH 8.2.
Discussion
In situ hybridization (ISH) with radioactively labeled nucleic acid probes is often used to detect the localization of RNA on tissue sections. The radioactive ISH method, however, is time consuming, less sensitive, and does not allow appreciation of complete transcript distribution patterns in whole-mounts. In contrast, the herein described MC-WISH method permits the direct comparison of multiple gene expression domains in contrasting colors within intact embryos. MC-WISH has the advantage that histological context is immediately evident and visualized simply by standard brightfield microscopy. This provides the possibility to relate gene expression domains topographically to each other with high accuracy and define unique and overlapping expression sites in a fast and reliable way. On the other hand, multiplexed fluorescent ISH (FISH) is more powerful with respect to sensitivity and resolution and is preferably used, if cellular or even sub-cellular resolution of mRNA detection is required.
The wide spectrum of transcripts that has been mapped by MC-WISH suggests that virtually any transcript can be analyzed not only in embryonic but also in larval and adult tissues and in species other than Drosophila. Indeed, the described procedure detailed here for Drosophila, works similarly efficient with zebrafish embryos 11,16,22. Moreover, the MC-WISH method can be adapted to a wide variety of invertebrate and vertebrate embryos and tissue specimen.
Hapten-labels and probe concentrations
We routinely use fluorescein, digoxigenin, and biotin as hapten labels of RNA probes. Moreover, dinitrophenol-labeled probes may be applied, which have been introduced in zebrafish WISH experiments 23-25. Fluorescein-labeled probes display a lesser sensitivity in comparison to the other hapten-labels, so that fluorescein is best used for detection of abundant transcripts. Each newly transcribed probe is first tested in a single WISH experiment, where its performance is evaluated either using BCIP/NBT or Fast Red as substrate. A probe concentration is considered as optimal when a strong signal without background development is achieved within min to few hr.
To make sure that the expression domains of interest have been detected in their full extent by MC-WISH, it is essential to visualize each transcript pattern by single-label WISH experiments using the most sensitive substrate combination, BCIP/NBT. This ensures that weak expression domains are not overlooked in the multi-target experiment. To compensate for the lesser sensitivity of the other substrate combinations it is essential to use doubled (to tripled) probe concentrations as compared to standard BCIP/NBT staining.
Low pH inactivation
The low pH inactivation step can lead to partial disintegration of the antisense hapten-labeled RNA probe/sense mRNA hybrids resulting in reduced signal detection in the second and third staining rounds. To minimize loss in sensitivity, the inactivation steps are as short as possible. We experienced that a 10 min incubation time at low pH is sufficient for elimination of AP-activity. The subsequent quick washes in large volumes ensure rapid dilution of unbound antibody-AP conjugates and prevent re-binding to the haptenized probes. Alternative procedures include paraformaldehyde fixation and heat inactivation. However, some of the color precipitates are not heat stable and paraformaldehyde fixation may not be sufficient for complete inactivation of AP. Incomplete inactivation of the first applied antibody-AP conjugate can lead to re-visualization of the mRNA pattern in the following detection round leading to false positive overlap in expression with the second mRNA species to be detected. Therefore, in a control experiment the embryos are split into two fractions after inactivation of the first applied antibody-AP conjugate. The second antibody-AP conjugate is omitted from the control fraction and should not produce a signal in the second color reaction. However, if the second color reaction generates a signal distribution pattern corresponding to the first one, then the inactivation procedure was not efficient. In this case, the incubation time in low pH stop solution should be prolonged for the next experiment.
Orders of AP substrate application
Since sensitivity drops with each subsequent round of detection, it is advisable to detect less abundant mRNAs prior to more abundant transcripts. In addition, the Fast Red and Fast Blue substrate combinations are significantly less sensitive than the purpleblue BCIP/NBT stain, so that Fast dyes are preferably applied in the first staining round for detection of the stronger expressed transcript. This also has the advantage that subsequent BCIP/NBT staining can be monitored and stopped in time before the purpleblue signal gets too dark in relation to the lighter Fast dyes. Thus in a standard two-color experiment, first the stronger expressed mRNA is detected by Fast Red and second the weaker one by BCIP/NBT. As an alternative to the Fast dyes the yellow INT substrate combinations may be applied, which however produce precipitates that become diffuse after some time. Therefore BCIP/INT is exclusively applied in the last staining round. Consequently in a three-color experiment we often use first Fast Red, second BCIP/NBT (or Fast Blue) and third an INT substrate combination. Note that it is advisable to photograph stained embryos as soon as possible when applying INT.
Fluorescent detection of azo dyes
It can be sometimes difficult to recognize overlapping expression patterns when a darker color precipitate is shadowing a lighter one. For example, a strongly developed BCIP/NBT precipitate can mask lighter Fast dye signals. One way to get around this problem is to capture images immediately after each staining and remove the applied color precipitate before the next detection round. In this case, alcohol-soluble azo dye (Fast Red) and INT precipitates are removed by ethanol washes after the first and second detection rounds, respectively, and BCIP/NBT staining is applied as the last AP-substrate 26. Another possibility is to take advantage of the fluorescent properties of azo dyes. Fast Red can be visualized using rhodamine filter sets 27, while Fast Blue can be observed with far-red filters 28,29. Comparison or overlay of chromogenic and fluorescent images can reveal the site of co-distribution.Using this approach, BCIP/NBT signal development has to be stopped in time, so that it does not become too dense and quench the fluorescent signal of the azo dye reaction product.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank our colleagues for cDNA plasmids for template generation. The authors' work is supported by the Swedish Cancer Foundation (CAN 2010/553), the Swedish Foundation for International Cooperation in Research and Higher Education (IG2011-2042) and the Knut and Alice Wallenberg Foundation (KAW2012.0058).
References
- Hauptmann G. In Situ Hybridization Methods. 1. NY: Humana Press, Springer; 2015. [Google Scholar]
- Kosman D, et al. Multiplex detection of RNA expression in Drosophila embryos. Science. 2004;305(5685):846. doi: 10.1126/science.1099247. [DOI] [PubMed] [Google Scholar]
- Itzkovitz S, van Oudenaarden A. Validating transcripts with probes and imaging technology. Nature Methods. 2011;8(4):S12–S19. doi: 10.1038/nmeth.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon S. Single-molecule fluorescence in situ hybridization: Quantitative imaging of single RNA molecules. Bmb Reports. 2013;46(2):65–72. doi: 10.5483/BMBRep.2013.46.2.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speel EJ. Robert Feulgen Prize Lecture 1999. Detection and amplification systems for sensitive, multiple-target DNA and RNA in situ hybridization: looking inside cells with a spectrum of colors. Histochem Cell Biol. 1999;112(2):89–113. doi: 10.1007/s004180050396. [DOI] [PubMed] [Google Scholar]
- Hafen E, Levine M, Garber RL, Gehring WJ. An improved in situ hybridization method for the detection of cellular RNAs in Drosophila tissue sections and its application for localizing transcripts of the homeotic Antennapedia gene complex. EMBO J. 1983;2(4):617–623. doi: 10.1002/j.1460-2075.1983.tb01472.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tautz D, Pfeifle C. A non-radioactive in situ hybridization method for the localization of specific RNAs in Drosophila embryos reveals translational control of the segmentation gene hunchback. Chromosoma. 1989;98(2):81–85. doi: 10.1007/BF00291041. [DOI] [PubMed] [Google Scholar]
- Hartmann C, Jäckle H. Spatiotemporal relationships between a novel Drosophila stripe expressing gene and known segmentation genes by simultaneous visualization of transcript patterns. Chromosoma. 1995;104(2):84–91. doi: 10.1007/BF00347690. [DOI] [PubMed] [Google Scholar]
- Hauptmann G. Two-color detection of mRNA transcript localizations in fish and fly embryos using alkaline phosphatase and beta-galactosidase conjugated antibodies. Dev Genes Evol. 1999;209(5):317–321. doi: 10.1007/s004270050258. [DOI] [PubMed] [Google Scholar]
- Hauptmann G. One-, two-, and three-color whole-mount in situ hybridization to Drosophila embryos. Methods. 2001;23(4):359–372. doi: 10.1006/meth.2000.1148. [DOI] [PubMed] [Google Scholar]
- Hauptmann G, Gerster T. Two-color whole-mount in situ hybridization to vertebrate and Drosophila embryos. Trends Genet. 1994;10(8):266. doi: 10.1016/0168-9525(90)90008-t. [DOI] [PubMed] [Google Scholar]
- Hauptmann G, Gerster T. Multicolour whole-mount in situ hybridization to Drosophila embryos. Development Genes and Evolution. 1996;206(4):292–295. doi: 10.1007/s004270050055. [DOI] [PubMed] [Google Scholar]
- O'Neill JW, Bier E. Double-label in situ hybridization using biotin and digoxigenin-tagged RNA probes. Biotechniques. 1994;17(5):870–875. [PubMed] [Google Scholar]
- Söll I, Hauptmann G. G Hauptmann., editor. Multicolored visualization of transcript distributions in Drosophila embryos. Neuromethods. 2015. pp. 45–59.
- Bobrow MN, Harris TD, Shaughnessy KJ, Litt GJ. Catalyzed reporter deposition, a novel method of signal amplification. Application to immunoassays. J Immunol Methods. 1989;125(1-2):279–285. doi: 10.1016/0022-1759(89)90104-x. [DOI] [PubMed] [Google Scholar]
- Hauptmann G, Gerster T. Multicolor whole-mount in situ hybridization. Methods Mol Biol. 2000;137:139–148. doi: 10.1385/1-59259-066-7:139. [DOI] [PubMed] [Google Scholar]
- Söll I, Hauptmann G. Hauptmann G, editor. Manual and automated whole-mount in situ hybridization for systematic gene expression analysis in embryonic zebrafish forebrain. Neuromethods. 2015. pp. 171–206.
- Bergalet J, et al. Hauptmann G, et al., editors. Subcellular transcript localization in Drosophila embryos and tissues visualized by multiplex FISH. Neuromethods. 2015. pp. 369–391.
- Walldorf U, Gehring WJ. Empty spiracles, a gap gene containing a homeobox involved in Drosophila head development. Embo J. 1992;11(6):2247–2259. doi: 10.1002/j.1460-2075.1992.tb05284.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafen E, Kuroiwa A, Gehring WJ. Spatial distribution of transcripts from the segmentation gene fushi tarazu during Drosophila embryonic development. Cell. 1984;37(3):833–841. doi: 10.1016/0092-8674(84)90418-5. [DOI] [PubMed] [Google Scholar]
- Grossniklaus U, Pearson RK, Gehring WJ. The Drosophila sloppy paired locus encodes two proteins involved in segmentation that show homology to mammalian transcription factors. Genes Dev. 1992;6(6):1030–1051. doi: 10.1101/gad.6.6.1030. [DOI] [PubMed] [Google Scholar]
- Hauptmann G, Gerster T. Regulatory gene expression patterns reveal transverse and longitudinal subdivisions of the embryonic zebrafish forebrain. Mech Dev. 2000;91(1-2):108–118. doi: 10.1016/s0925-4773(99)00277-4. [DOI] [PubMed] [Google Scholar]
- Long S, Rebagliati M. Sensitive two-color whole-mount in situ hybridizations using digoxygenin- and dinitrophenol-labeled RNA probes. Biotechniques. 2002;32(3):494–500. doi: 10.2144/02323bm04. [DOI] [PubMed] [Google Scholar]
- Lauter G, Söll I, Hauptmann G. Multicolor fluorescent in situ hybridization to define abutting and overlapping gene expression in the embryonic zebrafish brain. Neural Dev. 2011;6(1):10. doi: 10.1186/1749-8104-6-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauter G, Söll I, Hauptmann G. Sensitive whole-mount fluorescent in situ hybridization in zebrafish using enhanced tyramide signal amplification. Methods Mol Biol. 2014;1082:175–185. doi: 10.1007/978-1-62703-655-9_12. [DOI] [PubMed] [Google Scholar]
- Chen J, Laramore C, Shifman M. Triple-labeling whole-mount in situ hybridization method for analysis of overlapping gene expression in brain tissue with high level of autofluorescence. J Cytol Histol. 2015. pp. S3–S011.
- Murdoch A, Jenkinson EJ, Johnson GD, Owen JJ. Alkaline phosphatase-fast red, a new fluorescent label. Application in double labelling for cell cycle analysis. J Immunol Methods. 1990;132(1):45–49. doi: 10.1016/0022-1759(90)90396-d. [DOI] [PubMed] [Google Scholar]
- Hauptmann G, Lauter G, Söll I. Hauptmann G, editor. Application of alkaline phosphatase-mediated azo dye staining for dual fluorescent in situ hybridization in zebrafish. Neuromethods. 2015. pp. 393–404.
- Lauter G, Söll I, Hauptmann G. Two-color fluorescent in situ hybridization in the embryonic zebrafish brain using differential detection systems. BMC Dev Biol. 2011;11:43. doi: 10.1186/1471-213X-11-43. [DOI] [PMC free article] [PubMed] [Google Scholar]