

Abstract
Cryo-electron tomography (Cryo-ET) is a powerful three-dimensional (3-D) imaging technique for visualizing macromolecular complexes in their native context at a molecular level. The technique involves initially preserving the sample in its native state by rapidly freezing the specimen in vitreous ice, then collecting a series of micrographs from different angles at high magnification, and finally computationally reconstructing a 3-D density map. The frozen-hydrated specimen is extremely sensitive to the electron beam and so micrographs are collected at very low electron doses to limit the radiation damage. As a result, the raw cryo-tomogram has a very low signal to noise ratio characterized by an intrinsically noisy image. To better visualize subjects of interest, conventional imaging analysis and sub-tomogram averaging in which sub-tomograms of the subject are extracted from the initial tomogram and aligned and averaged are utilized to improve both contrast and resolution. Large datasets of tilt-series are essential to understanding and resolving the complexes at different states, conditions, or mutations as well as obtaining a large enough collection of sub-tomograms for averaging and classification. Collecting and processing this data can be a major obstacle preventing further analysis. Here we describe a high-throughput cryo-ET protocol based on a computer-controlled 300kV cryo-electron microscope, a direct detection device (DDD) camera and a highly effective, semi-automated image-processing pipeline software wrapper library tomoauto developed in-house. This protocol has been effectively utilized to visualize the intact type III secretion system (T3SS) in Shigella flexneri minicells. It can be applicable to any project suitable for cryo-ET.
Keywords: Bioengineering, Issue 107, In situ structural biology, electron cryotomography. bacterial pathogen, minicell, protein secretion, injectisome, molecular machinery, high throughput image analysis
Introduction
Type III secretion systems (T3SS) are essential virulence determinants for many Gram-negative pathogens. The injectisome, also known as the needle complex, is the central T3SS machine required for direct translocation of effector proteins from the bacterium into eukaryotic host cells1, 2. The injectisome comprises an extracellular needle, a basal body, and a cytoplasmic complex also known as the sorting complex3. Previous studies have elucidated 3-D structures of purified injectisomes from Salmonella and Shigella, along with the atomic structures of major basal body proteins4, 5. Recent in situ structures of injectisomes from Salmonella, Shigella, and Yersinia were revealed by cryo-ET6, 7. However, the cytoplasmic complex, essential for effector selection and needle assembly, has not been visualized in those structures.
Cryo-ET is the most suitable technique for imaging molecular machinery at nanometer resolution within its native cellular context (in situ). Nevertheless, the achievable resolution by cryo-ET is limited by specimen thickness. To overcome the drawback, we imaged intact injectisomes in a virulent Shigella flexneri strain that was genetically modified to produce minicells thin enough for cryo-ET. Another limitation of cryo-ET is the sensitivity of the sample to the radiation induced by the electron beam, which very quickly destroys the high-resolution information in the sample. As a result, extremely low doses are used for individual tilt-images so that a suitable dose can be distributed amongst the full tilt-series. This greatly lowers the signal-to-noise ratio (SNR) in the final reconstruction, which makes it difficult to differentiate the structural features of the subject from the large amount of noise in the tomogram and limits the resolution that can be achieved by cryo-ET. Conventional image processing such as Fourier and real-space filters as well as down sampling can be used to increase contrast, but at the expense of filtering out much of the high-resolution information. Recently, sub-tomogram averaging has made it possible to greatly increase the SNR and subsequently the final resolution in some cases to sub-nanometer levels8, 9. A more detailed analysis of complexes is made possible by computationally extracting thousands of sub-tomograms containing the areas of interest from the original tomograms and then aligning and averaging the sub-tomograms to determine in situ complex structures with higher SNR and higher resolution. These methods can be integrated with genetic approaches to provide even greater insights into macromolecular assemblies and their dynamic conformations in the native cellular context.
In general, tens or even hundreds of thousand sub-tomograms need to be averaged in order to determine high-resolution structures in situ. The acquisition of a sufficient number of tilt-series needed to produce this large number of sub-tomograms quickly becomes a bottleneck. The resulting tilt-series are often affected by beam-induced shift, stage backlash, as well as magnification, rotation and skew defects, which must be solved to bring the tilt-series into alignment prior to reconstruction. The tilt series is typically aligned by tracking gold fiducial markers, which are traditionally selected manually through inspection of the tilt-series, causing yet another bottleneck. Many software packages have been developed for automated tilt-series acquisition through computer-controlled electron microscopes10, 11, 12, tilt-series alignment and reconstruction13, 14 and sub-tomogram averaging15-18. As these packages handle discrete operations in the workflow of cryo-ET, it becomes desirable to build a higher level of abstraction into the process to systematically streamline the entire scheme into a single pipeline. Therefore, we developed a software wrapper library "tomoauto" designed to organize a number of these packages into a single semi-automated unit, allowing for simple user operation while maintaining full configuration of each component in a centralized manner. The library is open-source, well documented, continually developed and freely available for use, tailored development or further integration by means of an online remote source code repository (http://github.com/DustinMorado/tomoauto).
This high-throughput cryo-ET pipeline has been utilized to visualize intact injectisomes in S. flexneri minicells. A total of 1,917 tomograms were generated using this method, revealing a high-resolution in situ structure of the intact machine including the cytoplasmic sorting platform determined by sub-tomogram averaging19. Together with molecular modeling of wild-type and mutant machines, our high-throughput pipeline provides a new avenue to understand the structure and function of the intact injectisome in the native cellular context.
Protocol
1. Minicell Preparation
To make S. flexneri minicells, transform 1 µl of plasmid pBS58, which constitutively expresses Escherichia coli cell division genes ftsQ, ftsA, and ftsZ from a low-copy spectinomycin-resistant plasmid into 5 µl electrocompetent Streptomycin-resistant serotype 5a (M90T-Sm) cells by electroporation at 2.5 kV for 5 msec in 1 mm cuvettes.
Store minicell samples at -80 °C in 15% glycerol in a 1.5 ml cryogenic microtube. When ready for use, scrape approximately 5 μl of cells from the unthawed microtube using a pipette tip and suspend the cells in 4 ml tryptic soy broth with spectinomycin added to 100 μg/ml concentration. Grow O/N at 37 °C.
Pipette 2 ml of the culture from 1.2 into 200 ml tryptic soy broth with spectinomycin again added to 100 μg/ml concentrations. Grow at 37 °C to late log phase.
To enrich minicells, centrifuge 200 ml of the culture from 1.3 at 1,000 x g for 5 min. Carefully pour the supernatant fraction into a new centrifuge tube and centrifuge at 20,000 x g for 10 min. Carefully pour off and discard the supernatant fraction, and gently mix the pellet with the remaining liquid using a pipette tip and transfer approximately 100 μl of the pellet mixture to a 1.5 ml microcentrifuge tube.
2. EM Grid Preparation
Place a R2/2 holey carbon film 200-mesh copper grid carbon side up on a glass slide. NOTE: A R2/2 200-mesh grid is selected to maximize the number of tilt-series that can be set to acquire in a single grid square while still supporting the specimen and placing the edge of the carbon film in the field of the camera at the desired magnification. Finer mesh grids and smaller holey carbon films such as R1.2/1.3 400-mesh can be used for samples imaged at higher magnification; larger holey carbon films such as R3.5/1 200-mesh can be used for samples imaged at lower magnification or if the micrograph should not contain the carbon edge and holey carbon films with larger spacing such as R1/4 200-mesh can be used to aid with aligning the stage to the area of interest and protecting areas from over-exposure in focusing and tracking routines.
Place the slide on the platform in a glow discharge device. NOTE: We use an in-house device in which an anode and platform has been machined into a vacuum desiccator and is powered by a high-frequency generator. After creating a vacuum, attach the high-frequency generator probe to the anode and power on the probe for 1 min to glow discharge the grid. The time necessary to glow discharge the grid can range from a few seconds to a minute. Varying the glow discharge time can be used to diagnose problems with specimen concentration and grids that appear dry with no vitreous ice.
Remove the grid with a set of forceps, and lock the forceps closed with an elastic band.
Add 100 μl of 10 nm colloidal gold solution to the microcentrifuge tube with the minicells prepared in 1.4 and mix by gently flicking the tube with a finger. With a new pipette place 4 μl of the mixture on the grid prepared in 2.2. NOTE: Colloidal gold is available in a variety of sizes and care should be taken that the size of the gold is greater than 5 pixels given the pixel size of the micrographs to be processed at the acquired magnification, while not being too large to obscure features of interest.
Prepare the plunge-freeze apparatus; fill the outer freezing container with liquid nitrogen and then fill the inner chamber with liquid ethane. Attach the forceps with the grid to the plunger rod and lock the plunger rod into the raised position. NOTE: See Iancu et al.20 for a protocol describing the use of a commercial plunge-freeze apparatus.
Blot the grid by carefully touching a piece of filter paper to the drop of sample until the meniscus between the grid and filter paper separates and the wicking on the filter paper stops, then immediately release the plunger rod, freezing the grid. Carefully remove the forceps from the plunger rod and place the grid into a grid holder.
Prepare the cryo-EM transfer station by filling the loading area and absorption pump container with liquid nitrogen. Once the loading area is at liquid nitrogen temperature place the grid holder and a microscope specimen cartridge in the loading area.
Carefully remove the lock ring, which is either a small threaded lock ring on earlier Polara microscopes or a C-style clip ring on later models; place the EM grid into the cartridge using forceps and then gently reattach the lock ring back onto the cartridge securing the grid.
Remove the multiple specimen holder from the microscope and attach it to the transfer station. Place the specimen cartridge into the holder using cartridge forceps and retract the holder from the loading area and transfer the multiple specimen holder back to the microscope. NOTE: See Chen et al.21 for a visual protocol detailing 2.1-2.9.
3. High-throughput Automated Tilt-series Collection
- Collection of Low-magnification Maps
- Open a new Navigator window by clicking 'Open' in the 'Navigator' menu of SerialEM10 (http://bio3d.colorado.edu/SerialEM)
- Find grid squares that contain acceptable imaging conditions (i.e., thin ice, no contamination, subject of interest) using the fluorescent screen at low-magnification (~2,300X for the minicell specimen). NOTE: [Optional: This step can be automated with SerialEM by montaging the entire grid, however it can be faster to simply select a few areas manually.]
- Adjust the stage to eucentric height by tilting the specimen holder to 50° then adjust the z-height until the x-y translation of the stage is minimal between the tilted and untilted views.
- Move to the center of the grid square and click the 'Add Stage Pos' button in the Navigator window to store the current stage position.
- Continue steps 3.1.1-4 above until all acceptable grid squares stage positions have been saved.
- Open a new montage MRC file by clicking 'New montage' in the 'File' menu. In the Montage Setup Dialog that opens up, select a number of pieces in X and Y that will acquire the entire grid square (e.g., 10 x 10 for a standard 200 mesh grid). Use a high binning such as 8 and select the 'Move Stage Instead of Shifting Image' and 'Skip correlations used to align pieces' radio buttons.
- In the Navigator window click the first stage position and set it to be acquired by checking the 'Acquire' checkbox. Repeat this for each stage position in the Navigator window.
- Open the Navigator Acquire Dialog by clicking 'Acquire at Points' in the 'Navigator' menu. Check the 'Acquire map image' and 'Rough eucentricity' checkbox and make sure that all other checkboxes are unchecked. Click 'Proceed' to collect a montage at each stage position.
- Tilt-series Acquisition
- In the Navigator window select one of the acquired maps and click the 'Load map' button.
- In the Navigator window click the 'Add Points' button and select points in the map at which to acquire a tilt-series. Then click the 'Stop Adding Points' button. Repeat for each map collected.
- In the Camera menu select 'Parameters' and define the parameters for the Focus, Trial, and Record modes. [Optional: Dose-fractionated data can be specified in parameters for Record mode.]
- Select a point in the Navigator window and check the 'Tilt-Series' check box. In the Tilt-Series Setup dialog window that opens select the parameters desired for the tilt-series collection. Repeat for the rest of the selected points in the Navigator window, but do not select the maps.
- In the Navigator menu again select the 'Acquire at Points'. In the Navigator Acquire dialog choose 'Realign to item', Autofocus' and 'Rough eucentricity' as Preliminary tasks, and select 'Acquire tilt series' as the Primary task, and select 'Close column valves at end' to close the column when all of the points have been collected. Upon proceeding a tilt-series will be collected at each point in each map.
4. High-throughput Automated Tilt-series Processing and Reconstruction Using Tomoauto
- Correction of Beam-induced Motion in Dose-fractionated data [optional] NOTE: Tomoauto uses MOTIONCORR22 (http://cryoem.ucsf.edu/software/driftcorr.html) to remove beam-induced motion from dose-fractionated micrographs. NOTIONCORR16 must be installed on the system.
- With the original tilt-series, the output log from SerialEM10 and the individual dose-fractionated images all in the current working directory, in a terminal execute the command: dose_fractioned_to_stack <filename.st> <filename.st> is the name of the tilt-series to process.
- Alignment and Reconstruction of Tilt-series NOTE: Tomoauto by default uses IMOD13 (http://bio3d.colorado.edu/imod/) to handle tilt-series automated fiducial model generation, alignment, determination of the contrast transfer function (CTF), CTF-correction23, and reconstruction. Alternatively users have the option in tomoauto to use RAPTOR24 (included in IMOD) for automated fiducial model generation, CTFFIND425 (http://grigoriefflab.janelia.org/ctf) to determine the CTF, and tomo3d26 (https://sites.google.com/site/3demimageprocessing/tomo3d) for reconstruction or in any combination of software packages by configuration. This configuration as well as the parameters available in each package is handled by a global configuration file that can be edited to suit the values most commonly used in a lab while local configuration files can also be created to detail parameters used for a specific specimen, collection set or individual tilt-series. All packages that wish to be used must be installed on the system.
- With the tilt-series in the current working directory, in a terminal execute the command tomoauto --CTF --mode=align <filename.st> <fid_diam> <filename.st> is the tilt-series to be processed and <fid_diam> is the diameter of the fiducial markers in nanometers. This command will align and estimate the CTF of the tilt-series automatically. It is possible to skip CTF processing by removing the --CTF option from the command.
- Inspect the aligned tilt-series visually for any glaring errors in the alignment processing and inspect the estimated CTF by executing the commands: 3dmod <filename>.ali submfg <filename>_ctfplotter.com respectively, where <filename> is the name of the tilt-series without suffix. Also check the output of the tomoauto command to see the mean residual error generated by the alignment, which is a quantitative statistic of alignment quality.
- Given an acceptable alignment proceed processing by executing the command: tomoauto --CTF --mode=reconstruct <filename.st> <fid_diam> with the same user substitutions as in 4.2.1. This command will correct the CTF, erase the fiducial markers from the tilt-series and compute the reconstruction. Again CTF processing can be skipped as in 4.2.1.
- [optional] To skip the visual inspection step and fully automate processing and reconstruction execute the command tomoauto --CTF <filename.st> <fid_diam>
- [optional] To use a specific local configuration, refer to the tomoauto documentation on how to generate a local configuration file, and then execute the command tomoauto [options]-L <local_config> <filename.st> <fid_diam> where <local_config> is the name of the local configuration file.
5. Sub-tomogram Averaging
NOTE: We use the i3 package15 (http://www.electrontomography.org/) to process sub-tomogram averaging experiments, however the protocol described applies generally to most available sub-tomogram averaging software packages16to process sub-tomogram averaging experiments, however the protocol described applies generally to most available sub-tomogram averaging software packages16-18.
With the reconstructed tomogram in the current working directory open up the tomogram for particle picking by executing the command: tomopick <filename>.rec where <filename> is as in 4.2.2. In the window that opens use the left mouse button to click on first the basal body and then the needle tip to select an injectisome and use the up and down arrow-keys to riff through the slices of the tomogram. Select all visible injectisomes in this manner. This stores the coordinates in a text file defining the long axis of the structure as well as estimating two of the three Euler angles describing the orientation of the structure.
Computationally extract 4003 voxel cubes from the tomogram centered at the midpoint of the defined long axis by executing the command: clip resize -cx <x> -cy <y> -cz <z> -ix 400 -iy 400 -iz 400 \ <filename>.rec <filename>_001.mrc where <x>, <y>, <z> are the midpoint coordinates of the structure and <filename> is as in 4.2.2. The size of the extracted cube should vary by the structure and the magnification used, and should be large enough to adequately enclose the structure of interest, which for this sample is 4003 voxels.
Down-sample (bin) the sub-tomogram by a factor of four to reduce the computation time for initial alignment by executing the command: binvol -b 4 <filename>_001.mrc <filename>_001.bin4.mrc where <filename> is as in 5.2.
Apply the determined Euler angles to sub-tomograms and compute the global average to produce the initial template by executing the command. I3totsum.sh
Align and classify down-sampled sub-tomograms using a binary classification mask of the cytoplasmic area. Perform sub-tomogram averaging in Fourier space to minimize the missing wedge artifacts characteristic of tomography. For binning 4 data, please use SAMPFACT="4 4 4". i3mramsacls.sh
Repeat step 5.5 using sub-tomograms down-sampled by a factor of two (SAMPFACT="2 2 2") and once more with the original data (SAMPFACT="1 1 1"). i3mramsacls.sh
Representative Results
Samples of minicells S. flexneri were collected and processed as showed in the schematic Figure 1 using tomoauto following the pipeline detailed in Figure 2.Tilt-series were collected using SerialEM10, which allows for high-throughput tilt-series acquisition at points designated by the user on low-magnification montage maps (Figure 3). Micrographs were collected using dose-fractionation mode on a direct-detection device camera to reduce beam-induced motion22 (Figure 4). Tomoauto coordinates motion correction taking a collection of dose-fractionated micrographs processing each with MOTIONCORR22 and assembles the results into a tilt-series to be further processed (Movie 1).
The most general application of tomoauto is the automatic alignment of an initial tilt-series. Tomoauto composes sequential execution of the necessary commands in IMOD13 to coarsely align the tilt-series and generate an initial fiducial model tracking the colloidal gold particles in the sample, which are in turn used to generate the final alignment. The accuracy of this fiducial model is essential to the quality of the reconstructed tomogram, and so the user is able to visually inspect the automatically calculated fiducial model before proceeding with reconstruction or afterwards to identify tilt-series that should be processed manually. Figure 5 shows two tilt-series coarsely-aligned and the determined fiducial model as generated by tomoauto. Figures 5A, C show the untilted images and in both the fiducial model is correct with model points centered on fiducial markers. Figures 5B, D show the corresponding tilt-series at 50 degrees and while the model in Figure 5B is still tracking the gold particles correctly, several model points (red) in Figure 5D have strayed from their corresponding gold markers and the model is not suitable for fine alignment. This error can be measured quantitatively as the mean residual error between the center of the model point and the likely center of the gold marker, and tomoauto can be configured to alert the user when the measured error exceeds a user-defined threshold to expedite inspection. Tilt-series that are insufficiently aligned automatically can then be aligned manually. We find that tomoauto successfully aligns around 80%-90% of our collected tilt-series (Movie 2).
After a tilt-series has been successfully aligned, it must be reconstructed into the final tomogram. Tomoauto has been designed so that the user may use IMOD13 or tomo3d26 to generate the final reconstruction. We currently use tomo3d to take advantage of several features in modern multi-core computer processing units (CPUs) to greatly reduce the reconstruction time. The final tomogram as shown in Figure 6 and Movie 3 is a 3-D volume of the imaged sample that can then be used for cellular annotation by segmentation, or sub-tomogram averaging to obtain higher resolution information of the molecular machinery within the sample. Sub-tomogram averaging increases both the SNR and decreases the artifacts produced by the missing-wedge by averaging out the high-levels of noise in individual tomograms and utilizing the large number sub-tomograms in a well distributed set of random orientations with respect to the missing wedge to limit artifacts and improve the final resolution. The 2.7nm sub-tomogram average of the intact S. flexneri T3SS is shown in Figure 7 as deposited in the EMDB (EMD-2667), which shows the large improvement capable with this technique compared to the injectisome displayed in a single tomogram in Figure 6B.
 Figure 1. Schematic overview of high-throughput cryo-electron tomography. A liquid suspension is rapidly frozen on an EM grid and a set of tilt-series is collected by an automated computer-controlled electron microscope. The resulting micrographs are processed automatically using tomoauto to generate the tomogram. The final step here is a segmented S. flexneri minicell from a tomogram generated by this protocol from Hu et al. 201515. Please click here to view a larger version of this figure.
Figure 1. Schematic overview of high-throughput cryo-electron tomography. A liquid suspension is rapidly frozen on an EM grid and a set of tilt-series is collected by an automated computer-controlled electron microscope. The resulting micrographs are processed automatically using tomoauto to generate the tomogram. The final step here is a segmented S. flexneri minicell from a tomogram generated by this protocol from Hu et al. 201515. Please click here to view a larger version of this figure.
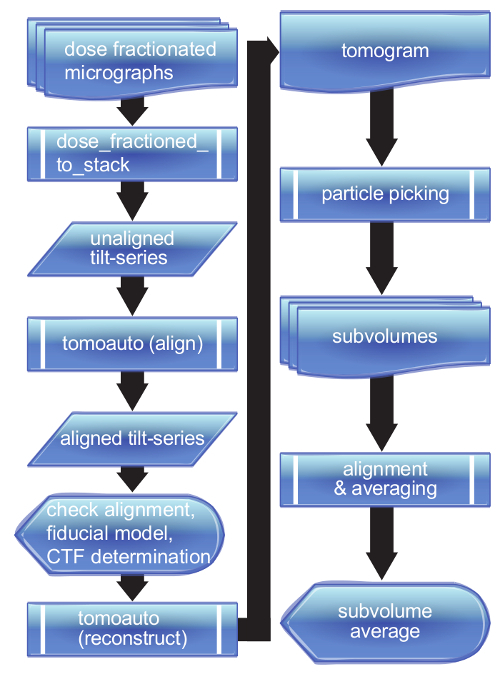 Figure 2. Flowchart of tomoauto process. A breakdown of the tomoauto workflow shows how data is processed from a collection of dose-fractionated micrographs all the way to a final sub-tomogram average. Sub-process symbols detail the tasks that tomoauto coordinates to process input by running the configured appropriate software. Data symbols show output generally not used by the user, while document and multi-document symbols show the output actually handled by the user. Finally display symbols show where user intervention occurs in the workflow. Please click here to view a larger version of this figure.
Figure 2. Flowchart of tomoauto process. A breakdown of the tomoauto workflow shows how data is processed from a collection of dose-fractionated micrographs all the way to a final sub-tomogram average. Sub-process symbols detail the tasks that tomoauto coordinates to process input by running the configured appropriate software. Data symbols show output generally not used by the user, while document and multi-document symbols show the output actually handled by the user. Finally display symbols show where user intervention occurs in the workflow. Please click here to view a larger version of this figure.
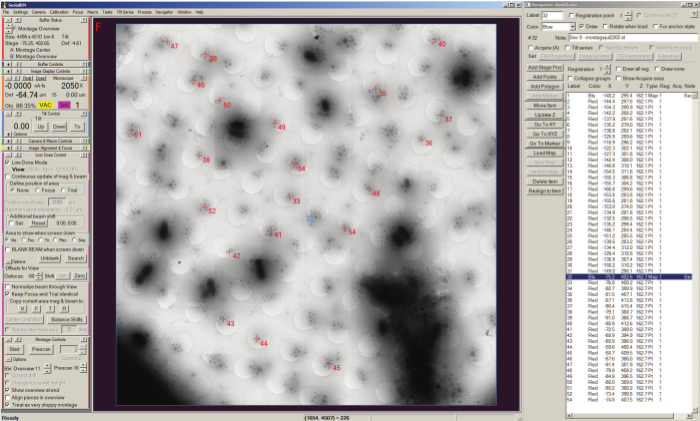 Figure 3. Batch tilt-series acquisition with SerialEM Navigator. Positions of montage maps are stored as stage positions (shown selection) in the Navigator window list shown on the left side of the screen, and the currently loaded map is displayed in the buffer window along with the selected points labeled numerically with a red cross added to the map for acquisition. Acquisition points are listed by label in the Navigator window and can be set to acquire using the "Tilt series" checkbox. Please click here to view a larger version of this figure.
Figure 3. Batch tilt-series acquisition with SerialEM Navigator. Positions of montage maps are stored as stage positions (shown selection) in the Navigator window list shown on the left side of the screen, and the currently loaded map is displayed in the buffer window along with the selected points labeled numerically with a red cross added to the map for acquisition. Acquisition points are listed by label in the Navigator window and can be set to acquire using the "Tilt series" checkbox. Please click here to view a larger version of this figure.
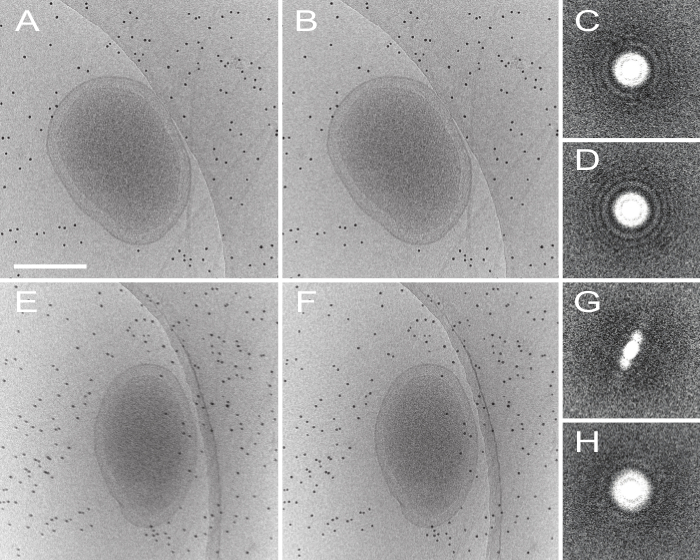 Figure 4. Effect of motion-correction on dose-fractionated data. (A) Shows an untilted and uncorrected micrograph, and the motion-corrected image (processed by MOTIONCORR) is shown in (B), the contrast is slightly improved after correction. Improvement can be seen more apparently by looking at the Fourier transform of the micrograph before (C) and after (D) motion correction. Images E-H show the same information but with a micrograph tilted at 60 degrees, where contrast is diminished and Thon rings visible in C and D are no longer visible at high tilt. Scale bar 250 nm. Please click here to view a larger version of this figure.
Figure 4. Effect of motion-correction on dose-fractionated data. (A) Shows an untilted and uncorrected micrograph, and the motion-corrected image (processed by MOTIONCORR) is shown in (B), the contrast is slightly improved after correction. Improvement can be seen more apparently by looking at the Fourier transform of the micrograph before (C) and after (D) motion correction. Images E-H show the same information but with a micrograph tilted at 60 degrees, where contrast is diminished and Thon rings visible in C and D are no longer visible at high tilt. Scale bar 250 nm. Please click here to view a larger version of this figure.
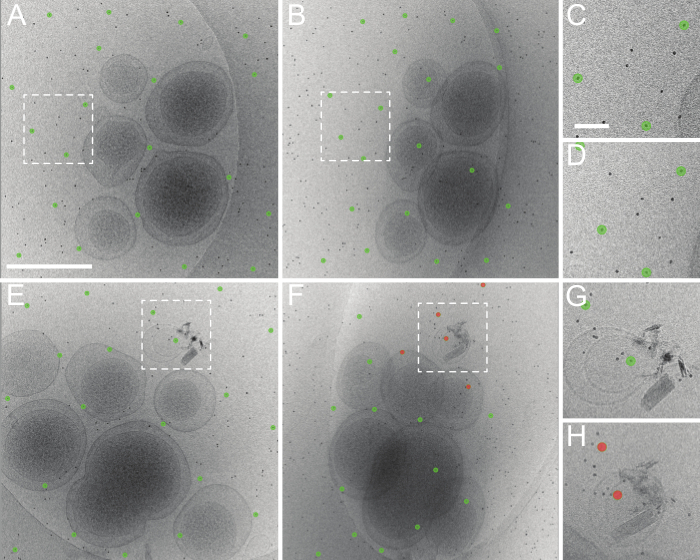 Figure 5. Good and bad results of tomoauto automated tilt-series alignment. (A) An untilted roughly aligned micrograph and the fiducial model produced automatically using tomoauto. (B) The determined fiducial model at 50 degrees tilt. The model still fits well and is centered on the appropriate fiducial markers. (C, D) Shows the model in (A, B) respectively, zoomed in at the boxed area. This tilt-series was aligned with a mean residual error of 1.06 pixels. (E) An untilted micrograph and fiducial model from another tilt-series and (F) the series at 50 degrees tilt. Here we see that model has lost track of several fiducial markers (shown in red) and this is representative of a bad automated tracking. (G, H) Shows the model in (E, F) respectively, zoomed in at the boxed area. This tilt-series was aligned with a mean residual error of 3.51 pixels and had to be processed by manual alignment of the series. (A) Scale bar 500 nm (C) Scale bar 50 nm. Please click here to view a larger version of this figure.
Figure 5. Good and bad results of tomoauto automated tilt-series alignment. (A) An untilted roughly aligned micrograph and the fiducial model produced automatically using tomoauto. (B) The determined fiducial model at 50 degrees tilt. The model still fits well and is centered on the appropriate fiducial markers. (C, D) Shows the model in (A, B) respectively, zoomed in at the boxed area. This tilt-series was aligned with a mean residual error of 1.06 pixels. (E) An untilted micrograph and fiducial model from another tilt-series and (F) the series at 50 degrees tilt. Here we see that model has lost track of several fiducial markers (shown in red) and this is representative of a bad automated tracking. (G, H) Shows the model in (E, F) respectively, zoomed in at the boxed area. This tilt-series was aligned with a mean residual error of 3.51 pixels and had to be processed by manual alignment of the series. (A) Scale bar 500 nm (C) Scale bar 50 nm. Please click here to view a larger version of this figure.
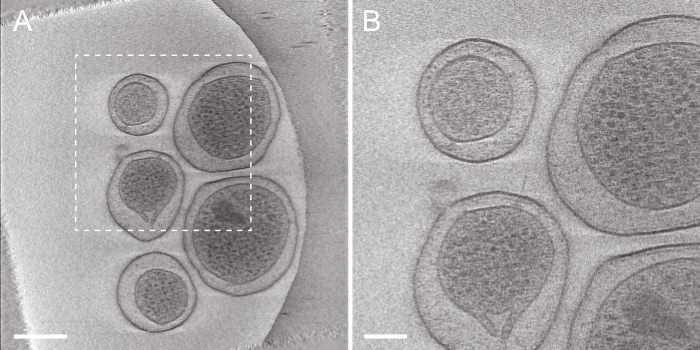 Figure 6. Tomogram generated automatically by tomoauto. (A) This displays a projection of seven slices from the center of the reconstruction of the tilt-series displayed in Figure 4A. Scale bar 250 nm. (B) A zoomed in view of the boxed area in (A) displaying an intact injectisome. Scale bar 100 nm. Please click here to view a larger version of this figure.
Figure 6. Tomogram generated automatically by tomoauto. (A) This displays a projection of seven slices from the center of the reconstruction of the tilt-series displayed in Figure 4A. Scale bar 250 nm. (B) A zoomed in view of the boxed area in (A) displaying an intact injectisome. Scale bar 100 nm. Please click here to view a larger version of this figure.
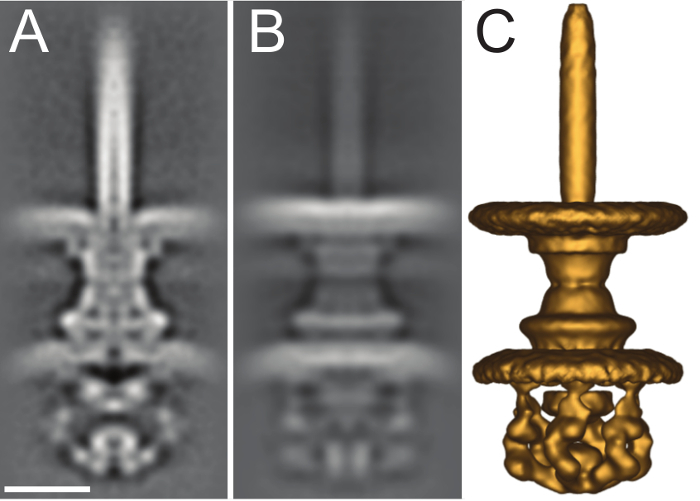 Figure 7. Sub-tomogram average of intact S. flexneri type III secretion system. (A) Central slice of the 2.7 nm sub-tomogram average of the intact S. flexneri T3SS from EMDB (EMD-2667). (B) Full projection along the X-axis of the volume. (C) Isosurface rendering of the volume viewed at a contour threshold of 130 in IMOD. Scale bar 5 nm. Please click here to view a larger version of this figure.
Figure 7. Sub-tomogram average of intact S. flexneri type III secretion system. (A) Central slice of the 2.7 nm sub-tomogram average of the intact S. flexneri T3SS from EMDB (EMD-2667). (B) Full projection along the X-axis of the volume. (C) Isosurface rendering of the volume viewed at a contour threshold of 130 in IMOD. Scale bar 5 nm. Please click here to view a larger version of this figure.
 Movie 1: Animation of unaligned tilt-series (Right click to download). This animation runs through the tilt-series as initially collected by SerialEM. Translational shifts are easily identified by the erratic path of individual fiducial markers from image to image, and these shifts along with less noticeable defects must be corrected before the tilt-series can be reconstructed.
Movie 1: Animation of unaligned tilt-series (Right click to download). This animation runs through the tilt-series as initially collected by SerialEM. Translational shifts are easily identified by the erratic path of individual fiducial markers from image to image, and these shifts along with less noticeable defects must be corrected before the tilt-series can be reconstructed.
 Movie 2: Animation of aligned tilt-series (Right click to download). This animation runs through the same micrographs displayed in Movie 1 after automated alignment by tomoauto. The erratic paths of fiducial markers now follow a smooth trajectory through the tilt-series, and the tilt-axis is aligned vertically with respect to the viewer.
Movie 2: Animation of aligned tilt-series (Right click to download). This animation runs through the same micrographs displayed in Movie 1 after automated alignment by tomoauto. The erratic paths of fiducial markers now follow a smooth trajectory through the tilt-series, and the tilt-axis is aligned vertically with respect to the viewer.
 Movie 3: Animation of reconstructed tilt-series (Right click to download). This animation runs through the tomogram shown in Figure 6 generated after automated reconstruction of the tilt-series displayed in Movie 2 by tomoauto.
Movie 3: Animation of reconstructed tilt-series (Right click to download). This animation runs through the tomogram shown in Figure 6 generated after automated reconstruction of the tilt-series displayed in Movie 2 by tomoauto.
Discussion
The high-throughput method described here enabled us to process 1,917 cryo tilt-series and produce over 4,500 sub-tomograms of the intact S. flexneri injectisome19. The collected data led to the detailed characterization of in situ injectisome, including the cytoplasmic sorting complex. The method was also utilized to visualize several mutant cells with specific deletion of putative protein components, which helped elucidate the composition of the sorting platform of the injectisome. Our method provided new avenues to investigate the structure-function relationship of the injectisome. As a result, the new stage was set for further dissection of the mechanisms underlying T3SS-mediated secretion and pathogenesis.
The protocol presented here describes high-throughput cryo-ET of intact S. flexneri, but is applicable to any project suitable for cryo-ET. This method has been used in the structural characterization of the flagellar motor of Borrelia burgdorferi27, the infection of E. coli minicells by bacteriophage T728, and chemoreceptor arrays in E. coli29. By facilitating the collection of massive datasets, it is possible to screen multiple mutants as well as image a large number of conditions that permit inferences of dynamic processes such as machine assembly27 and the progress of phage infection28. By collecting multiple software packages and allowing for full control over processing execution, users are able to customize different combinations of packages for optimal results. Chen et al.21 previously published a similar protocol describing collecting data using the software package Leginon12 and automating tilt-series processing using IMOD13 and RAPTOR24. The current protocol complements this method detailing an alternative method to collecting and processing data while also showing how much the technology and procedure has advanced, driven by the increased focus on high-resolution through sub-tomogram averaging, with dose-fractionated data, automated CTF estimation and correction, and more robust automated alignment routines within IMOD13. While the previous protocol goes into visual detail with emphasis on data collection, this method focuses on the details of processing the collected data.
High-throughput methods allow for massive data collection that maximize usage of microscope and computer resources, while limiting the amount of tedious user manual interventions that slow down the project and act as a major bottleneck. The wrapper library tomoauto has been designed to allow full configuration of all parameters used in each software package in a simple and centralized manner. Once a suitable configuration has been determined, it is then easy to apply the settings to whole dataset. A majority of the tilt-series can be processed with acceptable results (Figure 4A), while a minor subset is required to be processed manually. These tilt-series are usually less ideal acquisitions plagued by excessive image shift, poor contrast, or a lack of sufficient fiducial markers which causes the automated fiducial tracking routine to fail (Figure 4B). To obtain the best possible tomograms, extensive care must be taken at every crucial step from sample preparation, image acquisition, to image processing.
Further developments in high-throughput cryo-ET such as automated sub-tomogram extraction by template matching and the integration of modern sub-tomogram averaging software packages such as Dynamo into existing workflow pipelines like tomoauto are now being investigated. The recent advent of new-generation direct detection device cameras has made major improvements in increasing tilt-series SNR and enabling more consistent CTF determination due to the higher efficiency of the detector. The use of new full gold grid-types may decrease tilt-series collection defects, improving the success rate for automated tilt-series processing and reconstruction with less need for manual intervention30. Finally with the now ubiquitous use of computer clusters and graphic processing units (GPUs) to parallelize and speed up large dataset processing, development of pipelines that can utilize these systems will soon hopefully be able to shorten execution time from days to hours, providing users with still even less downtime in between experiment design and meaningful data analysis while still increasing dataset size and achieving higher resolutions.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
We thank Dr. William Margolin for comments. We are grateful for the support on SerialEM from Drs. David Mastronarde and Chen Xu. D.M., B.H. and J.L. were supported by Grant R01AI087946 from the National Institute of Allergy and Infectious Diseases, Grants R01GM110243 and R01GM107629 from the National Institute of General Medical Sciences (NIGMS), and Grant AU-1714 from the Welch Foundation. The direct electron detector was funded by National Institutes of Health Award S10OD016279.
References
- Cornelis GR. The type III secretion injectisome. Nat. Rev. Microbiol. 2006;4(11):811–825. doi: 10.1038/nrmicro1526. [DOI] [PubMed] [Google Scholar]
- Galan JE, Wolf-Watz H. Protein delivery into eukaryotic cells by type III secretion machines. Nature. 2006;444(7119):567–573. doi: 10.1038/nature05272. [DOI] [PubMed] [Google Scholar]
- Kubori T, et al. Supramolecular structure of the Salmonella typhimurium type III protein secretion system. Science. 1998;280(5363):602–605. doi: 10.1126/science.280.5363.602. [DOI] [PubMed] [Google Scholar]
- Schraidt O, Marlovits TC. Three-dimensional model of Salmonella's needle complex at subnanometer resolution. Science. 2011;331(6021):1192–1195. doi: 10.1126/science.1199358. [DOI] [PubMed] [Google Scholar]
- Hodgkinson JL, et al. Three-dimensional reconstruction of the Shigella T3SS transmembrane regions reveals 12-fold symmetry and novel features throughout. Nat. Struct. Mol. Biol. 2009;16(5):477–485. doi: 10.1038/nsmb.1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudryashev M, et al. In situ structural analysis of the Yersinia enterocolitica injectisome. eLife. 2013;2:e00792. doi: 10.7554/eLife.00792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamoto A, et al. Common and distinct structural features of Salmonella injectisome and flagellar basal body. Scientific Reports. 2013;3:3369–3369. doi: 10.1038/srep03369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs JA. Structural biology in situ-the potential of subtomogram averaging. Curr. Opin. Struct. Biol. 2013;23(2):261–267. doi: 10.1016/j.sbi.2013.02.003. [DOI] [PubMed] [Google Scholar]
- Schur FK, Hagen WJ, de Marco A, Briggs JA. Determination of protein structure at 8.5Å resolution using cryo-electron tomography and sub-tomogram averaging. J. Struct. Biol. 2013;184(3):394–400. doi: 10.1016/j.jsb.2013.10.015. [DOI] [PubMed] [Google Scholar]
- Mastronarde DN. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 2005;152(1):36–51. doi: 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Zheng SQ, et al. UCSF tomography: an integrated software suite for real-time electron microscopic tomographic data collection, alignment and reconstruction. J. Struct. Biol. 2007;157(1):138–147. doi: 10.1016/j.jsb.2006.06.005. [DOI] [PubMed] [Google Scholar]
- Suloway C, et al. Fully automated, sequential tilt-series acquisition with Leginon. J. Struct. Biol. 2009;167(1):11–18. doi: 10.1016/j.jsb.2009.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, McIntosh JR. Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 1996;116(1):71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- Winkler H, Taylor KA. Accurate marker-free alignment with simultaneous geometry determination and reconstruction of tilt-series in electron tomography. Ultramicroscopy. 2006;106(3):240–254. doi: 10.1016/j.ultramic.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Winkler H, Zhu P, Liu J, Ye F, Roux KH, Taylor KA. Tomographic subvolume alignment and classification applied to myosin V and SIV envelope spikes. J. Struct. Biol. 2009;165(2):64–77. doi: 10.1016/j.jsb.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicastro D, Schwartz CL, Pierson J, Gaudette R, Porter ME, McIntosh JR. The Molecular Architecture of Axonemes Revealed by Cryoelectron Tomography. Science. 2006;313(5789):944–948. doi: 10.1126/science.1128618. [DOI] [PubMed] [Google Scholar]
- Castaño-Díez D, Kudryashev M, Arheit M, Stahlberg H. Dynamo: a flexible, user-friendly development tool for subtomogram averaging of cryo-EM data in high-performance computing environments. J. Struct. Biol. 2012;178(2):139–151. doi: 10.1016/j.jsb.2011.12.017. [DOI] [PubMed] [Google Scholar]
- Hrabe T, Chen Y, Pfeffer S, Cuellar LK, Mangold AV, Förster F. PyTom: a python-based toolbox for localization of macromolecules in cryo-electron tomograms and subtomogram analysis. J. Struct. Biol. 2012;178(2):177–188. doi: 10.1016/j.jsb.2011.12.003. [DOI] [PubMed] [Google Scholar]
- Hu B, et al. Visualization of the type III secretion sorting platform of Shigella flexneri. Proc. Natl. Acad. Sci. 2015;112(4):1047–1052. doi: 10.1073/pnas.1411610112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iancu CV, et al. Electron cryotomography sample preparation using the Vitrobot. Nat. Protoc. 2007;1(6):2813–2819. doi: 10.1038/nprot.2006.432. [DOI] [PubMed] [Google Scholar]
- Chen S, et al. Electron Cryoelectrontomography of Bacterial Cells. J. Vis. Exp. 2010. p. e1943.
- Li X, et al. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods. 2013;10(6):584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Q, Morphew MK, Schwartz CL, Hoenger AH, Mastronarde DM. CTF determination and correction for low dose tomographic tilt series. J. Struct. Biol. 2009;168(3):378–387. doi: 10.1016/j.jsb.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amat F, Moussavi F, Comolli LR, Elidan G, Downing KH, Horowitz M. Markov random field based automatic image alignment for electron tomography. J. Struct. Biol. 2008;161(3):260–275. doi: 10.1016/j.jsb.2007.07.007. [DOI] [PubMed] [Google Scholar]
- Rouhou A, Grigorieff N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. bioRxiv. 2015. [DOI] [PMC free article] [PubMed]
- Agulleiro JI, Fernandez JJ. Tomo3D 2.0 – Exploitation of Advanced Vector eXtensions (AVX) for 3D reconstruction. J. Struct. Biol. 2015;189(2):147–152. doi: 10.1016/j.jsb.2014.11.009. [DOI] [PubMed] [Google Scholar]
- Zhao X, Zhang K, Boquoi T, Hu B, Motaleb MA, Miller K, James M, Charon NW, Manon MD, Norris SJ, Li C, Liu J. Cryo-Electron Tomography Reveals the Sequential Assembly of Bacterial Flagella in Borrelia burgdorferi. Proc Natl Acad Sci U S A. 2013;110(35):14390–14395. doi: 10.1073/pnas.1308306110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B, Margolin W, Molineux IJ, Liu J. The Bacteriophage T7 Virion Undergoes Extensive Structural Remodeling during infection. Science. 2013;339(6119):576–579. doi: 10.1126/science.1231887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Hu B, Morado DR, Jani S, Manson MD, Margolin W. W: Molecular architecture of chemoreceptor arrays revealed by cryoelectron tomography of Escherichia coli minicells. Proc Natl Acad Sci USA. 2012;109(23):e1481–e1488. doi: 10.1073/pnas.1200781109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo CJ, Passmore LA. Electron microscopy: Ultrastable gold substrates for electron cryomicroscopy. Science. 2014;346(6215):1377–1380. doi: 10.1126/science.1259530. [DOI] [PMC free article] [PubMed] [Google Scholar]