

Abstract
Identification of genes responsible for embryonic induction poses a number of challenges; to name a few, secreted molecules of interest may be low in abundance, may not be secreted but tethered to the signaling cell(s), or may require the presence of binding partners or upstream regulatory molecules. Thus in a search for gene products capable of eliciting an early lens-inductive response in competent ectoderm, we utilized an expression cloning system that would allow identification of paracrine or juxtacrine factors as well as transcriptional or other regulatory proteins. Pools of mRNA were injected into Xenopus oocytes, and responding tissue placed directly on the oocytes and co-cultured. Following functional cloning of ldb1 from a neural plate stage cDNA library based on its ability to elicit the expression of the early lens placode marker foxe3 in lens-competent animal cap ectoderm, we characterized the mRNA expression pattern, and assayed developmental progression following overexpression or knockdown of ldb1. This system is suitable in a very wide variety of contexts where identification of an inducer or its upstream regulatory molecules is sought using a functional response in competent tissue.
Keywords: Developmental Biology, Issue 107, expression cloning, embryonic induction, Xenopus, oocytes, animal cap, lens, placodes, ldb1
Introduction
Forward genetic approaches to identify genes of interest through their function or loss-of-function1,2 are an integral part of understanding complex patterning events in development. Coupled with powerful reverse genetic techniques available to an ever-widening array of systems and researchers3-5, it is now possible to identify genes with a key functional role in a pathway and then elucidate that function at the cellular level and in interaction with other gene products. One approach to functionally identifying genes of interest that has yielded many key findings in the past is expression cloning6,7.
Our recent aim8 was to identify early lens-inductive factors, since it has been demonstrated that initial steps in the vertebrate lens-inductive process occur as early as gastrula stages. To that end, we used the transiently lens-competent9 animal cap ectoderm (stage 11-11.510) of Xenopus embryos as responding tissue for induction, and the stage VI Xenopus oocyte as a source of production for the inducing factors.
The following protocol builds on the expression cloning and sib selection protocols of Smith and Harland6,7, also successfully used by others11-13. In our oocyte expression system (first utilized for production of inducing factors by Lustig and Kirschner14), pools of injected transcripts capable of directly or indirectly causing the oocytes to produce factors that elicit a lens-inductive response in animal cap ectoderm are selected for and identified. Since the system is useful for expressing secreted inducing molecules directly (oocyte-injected INHBB mRNA causes mesoderm induction in mesoderm-competent animal cap ectoderm8), we originally expected the screening procedure to be useful chiefly for identification of paracrine factors. However, since we identified a nuclear factor in our screen (ldb18), it is clear that the system can be used to identify a wide variety of molecules such as transcriptional or translational regulatory factors, miRNAs, cofactors, or juxtacrine factors.
Protocol
All experimental procedures were approved by the University of Virginia Institutional Animal Care and Use Committee.
Note: Figure 1 shows a schematic overview of the experimental procedures.
1. Preparation of Oocytes
Pre-prime X. laevis females with 150 U of Pregnant Mare Serum Gonadotropin (PMSG) approximately one week in advance of oocyte isolation. Inject 1 ml 150 U/ml PMSG into dorsal lymph sac with 1 cc sterile syringe with 29 G needle.
- Prepare solutions for oocyte injection and oocyte-animal cap assay.
- Prepare Ca++/Mg++-free OR2 (OR2-): 82 mM NaCl, 2.5 mM KCl, 1.5 mM Na2HPO4, 50 mM HEPES pH 7.2.
- Prepare OR2: OR2- plus 1 mM CaCl2 and 1 mM MgCl2.
- Prepare OCM: 60% Leibovitz L15, 0.4 mg/ml BSA, 100 μg/ml Gentamycin, pH 7.8.
- Prepare 1x MBS: 88 mM NaCl, 1 mM KCl, 0.7 mM CaCl2, 1 mM MgSO4, 5 mM HEPES (pH 7.8), 2.5 mM NaHCO3.
- Prepare 3% Ficoll in 1x MBS.
- Prepare 1x NAM: 110 mM NaCl, 2 mM KCl, 1 mM Ca(NO3)2, 1 mM MgSO4, 0.1 mM EDTA, 1 mM NaHCO3, 2 mM Na3PO4, pH 7.4.
- Anesthetize the female in a 0.03% solution of Ethyl 3-aminobenzoate methanesulfonate salt (MS222) in 0.1x MBS (first dissolve 0.3 g MS222 in 10 ml 95% ethanol, then dilute in 1 L 0.1x MBS) by placing frog in solution for 10 - 15 min or until unresponsive.
- Check that anesthesia is complete by turning anesthetized frog onto its back to ensure it does not respond (incompletely anesthetized frogs move limbs or turn body over).
Surgically isolate ovarian fragments by making an abdominal incision through skin and body wall with scalpel blade, isolating ovarian tissue with forceps and scissors. Place ovarian fragments into OR2-. Close the body wall with a 3-0 silk suture on a 24 mm curved needle and close the skin separately with the same sutures. Allow recovery of female in 1 g/L aquarium salt in water.
Using fine forceps, tear ovarian tissue into small (10 - 20 oocytes) pieces and transfer to fresh OR2-.
Defolliculate ovarian fragments in 2.0 mg/ml collagenase A in OR2- by agitating gently on shaker for 1 hr, transferring to fresh collagenase and agitating for one additional hour.
Wash oocytes 10 times in OR2 [containing Ca++/Mg++], discarding smaller oocytes.
Wash oocytes in OCM two times and transfer to fresh OCM. Isolate Stage VI oocytes by size upon visual inspection and discard immature (smaller) oocytes: Stage VI oocytes are larger than immature oocytes with even pigmentation in the animal hemisphere and are approximately 1.2-1.4 mm in diameter.
Maintain oocytes at 18 - 20 °C in OCM prior to injection. Note: Agarose-coated petri dishes may be used to minimize sticking of oocytes to dish.
2. Injection of Library Transcripts
- Prepare a directional cDNA library15 using RNA extracted at an appropriate stage of development (for example, neural plate stage 1410). Purchase a cDNA library or construct one using a commercial kit per the overview in the four steps below.
- Isolate approximately 5 μg mRNA by using a product to enrich for poly(A)+ RNA (See Table of Materials) and following manufacturer's instructions. Note: The transcripts to be used to produce the cDNA library may be restricted to an inducing tissue (such as the neural plate, following microdissection of the desired tissue), or may be from whole embryos at a particular stage.
- Produce cDNA with a commercial kit, following manufacturer's directions.
- Ligate approximately 20 ng cDNA into a vector included with the commercial kit or another suitable vector. An appropriate plasmid vector includes sequences for mRNA stability such as 5' β-globin and 3 poly(A) sequences (pCS2+, pTnT, pCS105).
- Transform ligation into competent bacterial cells using supplier's recommended protocol for heat shock transformation. Note: Alternatively, a collection of open reading frame (ORFeome) clones is available commercially and may be used to generate transcripts for injection.
- Prepare 10 pools of plasmids of 103 to 104 complexity (104 to 105 total complexity). This represents 10 plates with 1,000 - 10,000 colonies each.
- Plate library culture onto 10 15-cm LB-ampicillin plates, grow 12 - 18 hr at 37 ˚C, and collect colonies from each by gentle pressure with a glass spreader in 7 ml LB.
- Prepare a glycerol stock from 0.5 ml (add to 0.2 ml sterile glycerol and store at -20 ˚C), and use the remaining 6.5 ml to prepare DNA with a standard commercially available DNA miniprep kit following manufacturer's directions.
Linearize pooled plasmid DNA (1.0 - 2.0 μg) with appropriate restriction enzyme digest16 at 37 ˚C for 1 - 1.5 hr. Isolate the linearized DNA with phenol/chloroform extraction followed by ethanol precipitation and resuspension in water per the specifications of the RNA polymerase kit used. Synthesize sense RNA with a commercial RNA polymerase kit following manufacturer's directions.
Prepare needles for microinjection (using a needle puller with glass capillary tubing) of approximately 20 μm diameter; measure needle tips on a compound microscope with a calibrated ocular micrometer. Note: Needle puller settings must be empirically determined to produce a needle with a fine tip such that when broken with fine forceps yields the desired tip size.
Prepare (push clay into uniform layer on bottom of dish) clay-lined 35 x 10 mm petri dishes with parallel grooves to hold oocytes in place during microinjection; produce grooves with a mall probe or Pasteur pipette fused at the tip in flame. As an alternative to clay, prepare agarose dishes making indentations with an elastomer mold17. Transfer oocytes with a wide-bore pipette to 3% Ficoll in 1X MBS (approximately 2 ml) in rows in the clay-lined dishes.
Using microinjector, fill needle with 1 ng/nl RNA and adjust balance to produce slight positive pressure (to prevent drawing up of oocyte cytoplasm).
Inject oocytes with approximately 20 nl RNA in the equatorial region. Allow 1 hr for injected oocytes to remain in Ficoll-MBS, then transfer gently to 1x MBS. Incubate for 8 - 24 hr at 20 °C prior to animal cap assay.
3. Animal Cap Assay
Prepare 3/4x NAM and obtain fine forceps, hair loop, curved coverslip fragments, clay-lined dishes with cup-shaped indentations to hold oocytes. Make curved coverslip fragments by breaking apart glass coverslips into small fragments (approximately 1 - 2 mm x 2 - 4 mm) and passing through flame until edges polish and droop, producing a curved piece.
Fertilize X. laevis eggs18 through in vitro or natural mating and culture to gastrula stages (10 - 11 hr post-fertilization at RT) in 0.1x MBS prior to sorting by stage; collect mid-gastrula (stage 11 - 11.5)10 embryos.
Transfer embryos to a petri dish approximately half-full with 3/4x NAM. Using two pairs of fine forceps, remove fertilization (vitelline) membrane from gastrulae.
Transfer oocytes to 3/4x NAM (approximately 2 ml) in clay-lined dishes and immobilize in individual impressions in the clay, producing impressions to accommodate individual embryos as grooves were described above.
Transfer embryos to the clay-lined dishes and cut animal caps off gastrulae using two pairs of fine forceps. Take care to isolate animal cap ectoderm only and not equatorial tissue18.
Place an animal cap on the animal hemisphere of each oocyte with the inner surface of the animal cap contacting the oocyte. Hold recombinants together by applying curved glass coverslip fragment and applying downward pressure to the glass, flattening the ectoderm as the coverslip contacts the clay (animal caps can remain open with the inner layer exposed to the surface of the oocyte for 6 - 8 hr or longer). Note: Alternatively, place the animal cap into a clay indentation with its inner surface facing upward and place an oocyte on the cap; secure the recombinant with small extensions of clay.
Culture at 20 °C until control embryos reach desired stage for assay.
Separate recombinant by removing coverslip/clay and isolating ectoderm with forceps and a hair loop.
Fix ectodermal fragments for 1 hr in MEMFA (3.8% formaldehyde in MEM [0.1 M MOPS pH 7.4, 2 mM EGTA, and 1 mM MgSO4]).
Transfer fragments (and control embryos) from MEMFA into ethanol and store at -20 ˚C.
4. Analysis of Response in Ectoderm by In situ Hybridization (ISH)
- Prepare solutions for ISH.
- Prepare 1x PBS: 0.01 M phosphate buffered saline, NaCl 0.138 M, pH 7.4
- Prepare PBS-Tween (PTw): 1x PBS, 0.1% Tween-20
- Prepare 100x Denhart's solution: 2% BSA, 2% Polyvinylpyrrolidone, 2% Ficoll
- Prepare Hybridization Buffer: 50% Formamide, 5x SSC, 1 mg/ml torula yeast RNA, 1 μg/mL Heparin, 1x Denhart's solution, 0.1% Tween-20, 0.1% CHAPS, 10 mM EDTA, DEPC-H2O
- Prepare Maleic Acid Buffer (MAB): 100 mM maleic acid, 150 mM NaCl, pH 7.5
- Prepare MAB + block: MAB, 2% blocking reagent (heat to 60 ˚C to dissolve)
- Prepare Alkaline Phosphatase (AP) Buffer: 100 mM Tris pH 9.5, 50 mM MgCl2, 100 mM NaCl, 0.1% Tween, dH2O
- Prepare the RNA Probe.
- Using a commercial RNA polymerase kit and dig-NTP mix, add to a 1.5 ml tube (50 μl reaction): 25.5 μl DEPC-H2O, 10 μl 5X Transcription Buffer, 2.5 μl 10x dig-NTP mix, 5 μl 100 mM DTT, 2 μl RNAsin, 2 μl linearized DNA template (~1 μg/μl), 3 μl RNA Polymerase and incubate 37 ˚C for 90 min.
- Add 2 μl RNA Polymerase and incubate 37 ˚C for 60 min.
- Check 2 μl of reaction on 1% agarose gel.
- Add 1 μl RQ1 RNase-Free DNase and incubate 37 ˚C for 20 min.
- Precipitate the probe by adding 50 μl DEPC-H2O, 25 μl 10 M Ammonium Acetate and 313 μl ethanol. Store at -20 ˚C overnight (O/N) then recover RNA by centrifugation at 13,800 x g for 20 min. Wash with 500 μl 75% ethanol, spin briefly, remove ethanol and allow pellet to air dry. Add 50 μl DEPC-H2O.
- Add Hybridization Buffer to a final concentration of ~0.5 μg/μl.
- Prepare Tissue for Hybridization. Unless otherwise noted, fill vials to the top with each solution change described (approximately 4 ml).
- Remove ethanol from vials and transfer embryos into 75% ethanol/PTw, then 50% ethanol/PTw for 10 min each, horizontally on rocker.
- Wash three times in PTw for 5 min each on rocker.
- Transfer to 10 μg/μl Proteinase K treatment in PTw; rock tubes vertically 15 min.
- Rinse twice 10 min each in 0.1 M Triethanolamine pH 7.8 - rock tubes vertically.
- Add 12.5 μl acetic anhydride to tubes and rock vertically 5 min. Repeat with additional 12.5 μl acetic anhydride for 5 min.
- Wash in PTw 5 min vertically on rocker.
- Refix in 4% paraformaldehyde 20 min on rocker. Heat Hybridization Buffer to 60 ˚C.
- Wash three times in PTw for 5 min each on rocker.
- Remove all but ~1 ml of PTw from each tube and add 250 μl Hyb Buffer; gently swirl tubes to mix. Rock tubes vertically 5 min.
- Replace with 60 ˚C Hyb Buffer (0.5 ml) and agitate gently at 60 ˚C 10 min. Replace with fresh Hyb Buffer and agitate at 60 ˚C two to 4 hr.
- Heat probe (1 ml at 0.5 μg/μl in Hyb Buffer) to 60 ˚C (3 min). Remove Hyb Buffer and add probe to tubes. Agitate gently O/N at 60 ˚C.
- Prepare Tissue for Antibody.
- Warm Hyb Buffer and 2x SSC + 0.1% Tween-20 solutions to 60 ˚C.
- Replace probe solution with Hyb Buffer (save probes at -20 ˚C for 2 - 3x reuse). Wash at 60 ˚C for 10 min.
- Wash three times at 60 ˚C in 2x SSC-Tween (20 min each with agitation).
- Wash three times at 60 ˚C in 0.2x SSC-Tween (20 min each with agitation).
- Wash two times in MAB (RT) for 15 min each horizontally on rocker.
- Add 1 ml MAB + block. Wash 2 hr vertically on rocker.
- Transfer to 1 ml MAB + block containing a 1/2,000 Anti-digoxygenin-AP. Rock vertically at 4 ˚C O/N.
- Prepare Tissue for Color Reagent.
- Replace antibody solution with MAB; wash three times in MAB 5 min each horizontally on rocker.
- Wash three times in MAB 1 hr each horizontally on rocker.
- Wash two times in AP Buffer 10 min each horizontally on rocker.
- Transfer tissue to multiple-well plastic tray, remove AP Buffer and add BM Purple reagent (~1 ml). Allow reaction to proceed in dark 5 min to O/N.
- When appropriate staining is achieved, transfer tissue to vials of PTw. Wash two times 10 min each in PTw on rocker.
- Fix in Bouin's solution at 4 ˚C O/N on rocker.
- Wash out Bouin's with three 70% ethanol/30% PTw washes at RT. Proceed to image capture using epi-illumination and a microscope-mounted camera, bleaching if necessary18.
5. Sib Selection and Cloning
Select pool with the highest activity (greatest response in ectodermal tissue).
Titrate (dilute with LB to appropriate density) the glycerol stock for the pool with the highest activity and plate out ten new plates with approximately one-tenth the colonies from the previous step (using section 2 in protocol above).
Reduce pool size until activity is traced to single colony.
Obtain DNA sequence using standard primers in the vector.
Representative Results
In response to expression of mRNA injected into oocytes, responding animal cap tissue was assayed for expression of otx2 by in situ hybridization (Figure 2 and Table 1); otx2 is expressed in the presumptive lens ectoderm (PLE) from neural tube closure through lens placode thickening19. However, since otx2 is also expressed in the anterior neural ectoderm as well as non-neural head ectoderm outside the PLE, it is associated with both neural and placodal responses. The use of foxe3 to screen the library for a gene product capable of producing a lens-inductive response in lens-competent animal cap ectoderm allowed a more specific approach to the goal of the expression cloning, since foxe3 is expressed in the PLE from neural plate stages and throughout lens vesicle formation20, present in adjacent placodal regions but absent from neuroectoderm. Using the expression cloning and sib selection protocol above and injecting pools of library transcripts, a gene capable of producing foxe3 expression in the animal caps was isolated (Table 2). Following isolation of the clone, 179 additional animal cap assays using oocytes injected with library transcripts were screened for expression of foxe3; 50 were positive (28%). Of 140 animal cap pieces placed on uninjected oocytes, 0 were positive (Figure 3).
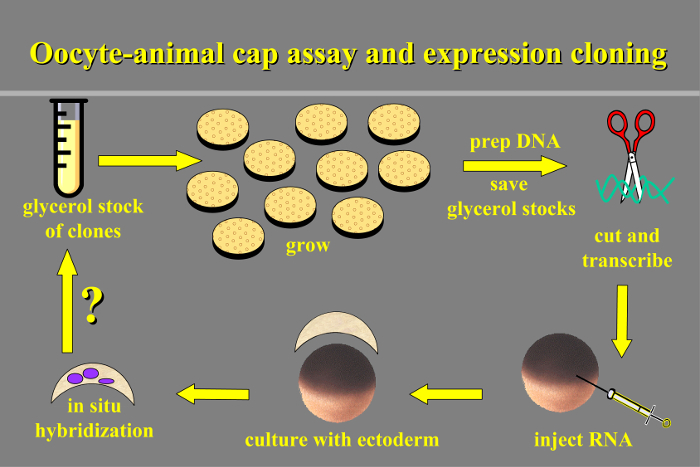 Figure 1. Oocyte-animal Cap Assay and Expression Cloning. Schematic overview of the protocol: transcripts are prepared from the clone library and injected into oocytes, animal cap ectoderm is cultured with oocytes and then assayed for induced gene expression by in situ hybridization. Please click here to view a larger version of this figure.
Figure 1. Oocyte-animal Cap Assay and Expression Cloning. Schematic overview of the protocol: transcripts are prepared from the clone library and injected into oocytes, animal cap ectoderm is cultured with oocytes and then assayed for induced gene expression by in situ hybridization. Please click here to view a larger version of this figure.
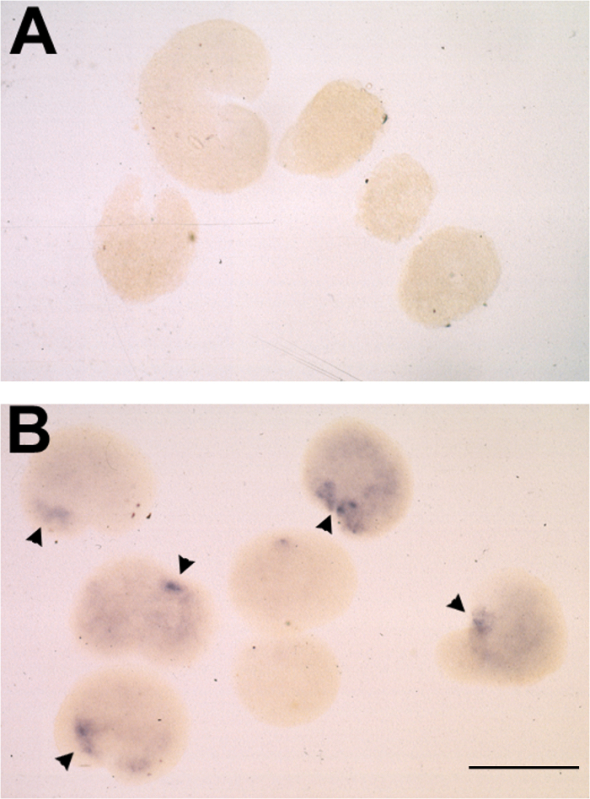 Figure 2. Typical Results of Animal Cap Assay following mRNA Injection and in situ Hybridization for Otx2. (A-B) Representative result of inductive response to dorsalized stage 14 poly(A)+ RNA in animal caps, assayed for otx2 expression by whole mount in situ hybridization. (A) Animal caps placed on uninjected oocytes at stage 10.5 and cultured to stage 25. (B) Stage 10.5 animal caps placed on oocytes injected with 10 ng RNA and cultured to stage 25. otx2 expression observed in 6/7 cases and indicated by arrowheads. Bar = 500 μm. Please click here to view a larger version of this figure.
Figure 2. Typical Results of Animal Cap Assay following mRNA Injection and in situ Hybridization for Otx2. (A-B) Representative result of inductive response to dorsalized stage 14 poly(A)+ RNA in animal caps, assayed for otx2 expression by whole mount in situ hybridization. (A) Animal caps placed on uninjected oocytes at stage 10.5 and cultured to stage 25. (B) Stage 10.5 animal caps placed on oocytes injected with 10 ng RNA and cultured to stage 25. otx2 expression observed in 6/7 cases and indicated by arrowheads. Bar = 500 μm. Please click here to view a larger version of this figure.
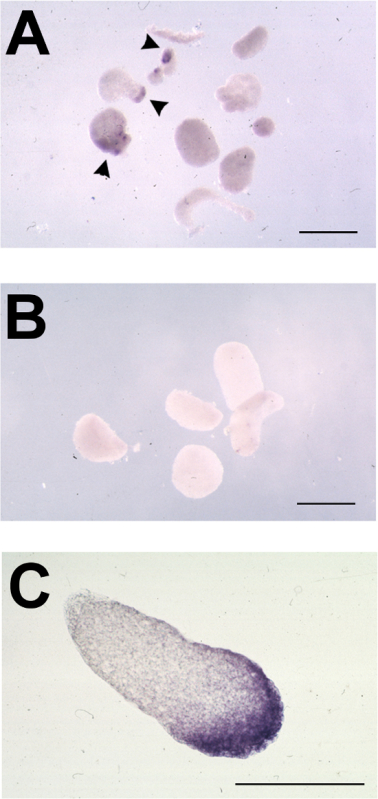 Figure 3. Typical Results of Animal Cap Assay following in situ Hybridization for Foxe3. (A-C) Representative animal caps from oocyte-animal cap assays, tested for expression of foxe3 by in situ hybridization. (A) Stage 11-11.5 animal caps placed on ldb1-injected oocytes and cultured to stage 23. foxe3 expression is indicated by arrowheads. (B) Stage 11-11.5 animal caps placed on uninjected oocytes and cultured to stage 23; no foxe3 expression detected. (C) Section through foxe3-positive induced animal cap from A showing expression in inner and outer layers of ectoderm. Bars = 500 μm. Please click here to view a larger version of this figure.
Figure 3. Typical Results of Animal Cap Assay following in situ Hybridization for Foxe3. (A-C) Representative animal caps from oocyte-animal cap assays, tested for expression of foxe3 by in situ hybridization. (A) Stage 11-11.5 animal caps placed on ldb1-injected oocytes and cultured to stage 23. foxe3 expression is indicated by arrowheads. (B) Stage 11-11.5 animal caps placed on uninjected oocytes and cultured to stage 23; no foxe3 expression detected. (C) Section through foxe3-positive induced animal cap from A showing expression in inner and outer layers of ectoderm. Bars = 500 μm. Please click here to view a larger version of this figure.
| Injected RNA | Positive cases | Gene | % |
| Stage 14 dorsalized mRNA, 10 ng | 12/24 | otx2 | 50 |
| Library pools of 105 clones, 20 ng | 3/9 | otx2 | 33 |
| Library pools of 105 to 100 clones, 20 ng | 50/179 | foxe3 | 28 |
| None | 0/140 | foxe3 | 0 |
Table 1. Oocyte-Animal Cap Assay Results. Results of animal cap assay assessed by in situ hybridization with otx2 and foxe3, using oocytes injected with mRNA or with transcripts synthesized from cDNA library pools; or uninjected oocytes.
| Injected RNA | Pool designation/selection | Positive foxe3 expression |
| Library pools of 105 clones, 20 ng | A | 2/4 |
| B* | 4/28 | |
| C | 4/44 | |
| Library pools of 104 clones, 20 ng | 1 | 0/11 |
| 2 | 0/10 | |
| 3 | 3/14 | |
| 4 | 0/12 | |
| 5 | 0/15 | |
| 6 | 1/20 | |
| 7 | 3/23 | |
| 8 | 0/16 | |
| 9 | 0/16 | |
| 10* | 5/20 | |
| Library pools of 5,000 clones, 20 ng | 1 | 0/7 |
| 2* | 5/16 | |
| 3 | 1/7 | |
| 4 | 0/7 | |
| 5 | 0/7 | |
| Library pools of 400 clones, 20 ng | 1 | 0/10 |
| 2 | 0/10 | |
| 3 | 0/10 | |
| 4 | 0/10 | |
| 5* | 8/26 | |
| 6 | 0/11 | |
| 7 | 0/10 | |
| 8 | 0/10 | |
| 9 | 0/10 | |
| 10 | 0/8 | |
| Library pools of 70-200 colonies, 20 ng | 1 | 0/8 |
| 2 | 0/10 | |
| 3* | 1/10 | |
| 4* | 1/10 | |
| 5 | 0/10 | |
| 6 | 0/9 | |
| 7 | 0/10 | |
| 8 | 0/10 | |
| Library pools of 20 colonies, 20 ng | 1 | 0/10 |
| 2 | 0/10 | |
| 3 | 0/10 | |
| 4* | 7/21 | |
| 5 | 0/10 | |
| 6 | 0/10 | |
| 7 | 0/10 | |
| 8 | 0/10 | |
| 9 | 0/10 | |
| 10 | 0/10 | |
| Library pools of 6 - 7 colonies, 20 ng | K2-6, L1 | 0/10 |
| L2-6, M7 | 0/10 | |
| M8-12, N7-8 | 0/10 | |
| K5-6, L1-4 | 1/10 | |
| L5-6, M7-10 | 0/10 | |
| M11-12, N7-8, K2-4 | 0/10 | |
| K2, K5, L2, L5, M8, M11 | 0/10 | |
| K3, K6, L3, L6, M9, M12 | 0/9 | |
| K4, L1, L4, M7, M10, N7, N8 | 1/9 | |
| Library RNAs, 20 ng | K6 | 0/10 |
| L1* | 3/10 | |
| L3 | 0/10 | |
| L4 | 0/10 | |
| Library RNA, 20 ng | L1 (ldb1) confirmation | 50/179 |
| * indicates selected pool |
Table 2. Sib Selection and Expression Cloning Results. The pool with the highest level of response in each animal cap assay (ranging between 10% and 36% positive for foxe3 expression) was selected for use in the next experiment to narrow down activity to one clone. Asterisk indicates selected pool.
Discussion
The method described here for the functional cloning of genes capable of inducing a response in competent ectoderm can be used to identify a wide range of gene products. This method expands upon past work by combining tissue-inducing assays with expression cloning techniques. We utilize the metabolic pathways of the Xenopus oocyte as a source of production of inducing factors, directly or indirectly, following RNA injection. This, in combination with the use of established methods for cloning a gene of interest6,7 using expression of transcripts generated from a cDNA library or other collection of clones, provides a valuable approach for those seeking to identify genes of new interest involved in embryonic induction. The broad applicability of this method to functional identification of new genes is a useful complement to exciting new reverse genetic approaches, and could also be used to functionally test transcripts identified using high-throughput methods (such as RNA-seq)21.
Controls are critical in monitoring the metabolic function of oocytes used in the cap assay. Both to ensure the health of each batch of oocytes and to establish the usefulness of this system to detect low-abundance transcripts, varying concentrations of mRNA of the mesoderm inducer INHBB were tested; this gene is unrelated to the lens-induction pathway and is a well-documented independent control14. Muscle-inducing activity, assayed by the expression of muscle-specific antigens in animal cap ectoderm with the 12/101 antibody, was observed with 2 - 200 pg INHBB mRNA, even in the presence of 500-fold excess stage 14 poly(A)+ RNA. The system is thus useful over a very wide range of injected RNA quantity, and oocytes are able to stably produce protein and remain viable following cytoplasmic injection of volumes of 50 nl and higher.
One consideration for the use of a cDNA library in this protocol is the necessity for full-length or nearly full-length clones. Prior to use in the protocol, the library should be examined by Northern analysis to determine the size of known transcripts in the library and therefore reduce the possibility that dominant-negative effects are obtained from potentially truncated injected transcripts. Since a full-length cDNA pool is important, the use of EST clones22 or optimally, the Xenopus ORFeome23 (which provides a complete set of validated full-length clones) are preferred to a traditional cDNA library unless a stage- and tissue-specific source of cDNA are sought and used for the library construction. Another consideration is the presence of β-globin and poly(A) sequences in the library vector intended to augment mRNA stability in injected transcripts. While this is desirable to increase the quantity of protein produced from injected synthetic mRNA, it also raises the possibility of producing a dominant-negative effect from mRNA that is higher in stability and activity than in vivo. A low-copy number mRNA may not exert the same effects as its endogenous counterpart in the background of a large pool of transcripts; one that is stabilized in the cloning and transcription process may persist to allow detection in the cap assay. A third issue is pool size; considerable reduction of pool size (10 clones or fewer) has been demonstrated to be advantageous in recent gain-of-function screening projects in embryos24 and is likely to provide clearer results and easier identification of candidate genes. A smaller pool size than the 103 - 104 recommended in this protocol is achievable if the total complexity of the collection of clones is less than 104, as is the case with the ORFeome23; one could screen the ~9,000 clones beginning with 10 pools of 900, though it may be prohibitively labor-intensive to process more than 10 pools in an experiment.
Determination of the type of gene product (by sequence analysis) made by the gene identified in the screen determines the nature of tests of its function. Since a nuclear transcriptional cofactor was identified in our screen8, it was necessary to confirm the role of the nucleus in the inductive process triggered by introduction of ldb1. Enucleated oocytes have fully functioning protein synthetic machinery and are viable and capable of producing secreted factors from injected transcripts. However, the absence of the nucleus will abolish functioning of transcription factors, cofactors, or other indirectly-acting gene products. Subsequent analyses of genes identified in the screen include determination of developmental expression pattern by in situ hybridization and gain-of-function and loss-of-function tests. Overexpression by injection of RNA into zygotes or by limiting injection to specific blastomeres to restrict its effects to particular regions of the embryo can provide insights, as well as knockdown of gene function through injection of morpholino oligonucleotides directed against the in vivo transcript.
Direct visualization of an inductive effect in the responding animal cap ectoderm using a transgenic line with a reporter gene (such as GFP) may greatly expedite the screening process and eliminate the need for the analysis of RNA expression by in situ hybridization. Similarly, the use of antibodies as quicker means of screening is desirable if an appropriate antibody (such as the 12/101 for muscle response discussed above) is available. Albino embryos may be used for the animal caps, which although more difficult to stage accurately at gastrula stages, streamline the in situ hybridization process by eliminating the need for any bleaching of wild-type pigmentation to better visualize the color reaction product.
Finally, it is important to consider that for several reasons, a large number of trials may need to be conducted to identify a given gene. For one, the number of cases one may reasonably process in a given trial is limited by the development (and eventual loss of competence) that occurs over time even given a large batch of embryos and a range of temperatures at which to culture them, as well as the loss that may occur of small pieces of ectoderm in processing. For another, success rates of expression of a chosen marker in the ectoderm may be very low and require many cases to observe a statistically significant effect.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
This work was supported by a Professional Development Grant to C.Z.P. from the Shepherd University Foundation. The authors wish to thank Brett Zirkle and Malia Deshotel for helpful discussions on the protocols, and Dr. Carol Hurney for generous assistance.
References
- Yergeau D, Kelley C, Zhu H, Kuliyev E, Mead P. Forward Genetic Screens in Xenopus. Using Transposon-Mediated Insertional Mutagenesis. Methods in Molecular Biology. 2012;917:111–127. doi: 10.1007/978-1-61779-992-1_6. [DOI] [PubMed] [Google Scholar]
- Grainger R. Xenopus tropicalis. as a Model Organism for Genetics and Genomics: Past, Present and Future. Methods in Molecular Biology. 2012;917:3–15. doi: 10.1007/978-1-61779-992-1_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama T, Fish M, Fisher M, Oomen-Hajagos J, Thomsen G, Grainger R. Simple and Efficient CRISPR/Cas9-Mediated Targeted Mutagenesis in Xenopus tropicalis. Genesis. 2013;51:835–843. doi: 10.1002/dvg.22720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi S, Cliffe R, Amaya E. Highly efficient bi-allelic mutation rates using TALENs in Xenopus tropicalis. Biology Open. 2012;1(12):1273–1276. doi: 10.1242/bio.20123228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Ishibashi S, Amaya E. Reverse Genetic Studies Using Antisense Morpholino Oligonucleotides. Methods in Molecular Biology. 2012;917:143–154. doi: 10.1007/978-1-61779-992-1_8. [DOI] [PubMed] [Google Scholar]
- Smith W, Harland R. Injected Xwnt-8. RNA acts early in Xenopus. embryos to promote formation of a vegetal dorsalizing center. Cell. 1991;67:753–765. doi: 10.1016/0092-8674(91)90070-f. [DOI] [PubMed] [Google Scholar]
- Smith W, Harland R. Expression cloning of noggin., a new dorsalizing factor localized in the Spemann organizer in Xenopus embryos. Cell. 1992;70:829–840. doi: 10.1016/0092-8674(92)90316-5. [DOI] [PubMed] [Google Scholar]
- Plautz C, Zirkle B, Deshote M, Grainger R. Early stages of induction of anterior head ectodermal properties in Xenopus. embryos are mediated by transcriptional cofactor ldb1. Developmental Dynamics. 2014;243(12):1606–1618. doi: 10.1002/dvdy.24193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servetnick M, Grainger R. Changes in neural and lens competence in Xenopus. ectoderm: evidence for an autonomous developmental timer. Development. 1991;112:177–188. doi: 10.1242/dev.112.1.177. [DOI] [PubMed] [Google Scholar]
- Nieuwkoop P, Faber J. Normal Table of Xenopus laevis (Daudin) New York and London: Garland Publishing, Inc; 1994. [Google Scholar]
- Lemaire P, Garrett N, Gurdon J. Expression cloning of Siamois., a Xenopus. homeobox gene expressed in dorsal-vegetal cells of blastulae and able to induce a complete secondary axis. Cell. 1995;81:85–94. doi: 10.1016/0092-8674(95)90373-9. [DOI] [PubMed] [Google Scholar]
- Lustig K, Kroll K, Sun E, Kirschner M. Expression cloning of a Xenopus T.-related gene (Xombi.) involved in mesodermal patterning and blastopore lip formation. Development. 1996;122:4001–4012. doi: 10.1242/dev.122.12.4001. [DOI] [PubMed] [Google Scholar]
- Hsu D, Economides A, Wang X, Eimon P, Harland R. The Xenopus. dorsalizing factor Gremlin identifies a novel family of secreted proteins that antagonize BMP activities. Molecular Cell. 1998;1(5):673–683. doi: 10.1016/s1097-2765(00)80067-2. [DOI] [PubMed] [Google Scholar]
- Lustig K, Kirschner M. Use of an oocyte expression assay to reconstitute inductive signaling. Proc. Natl. Acad. Sci. USA. 1995;92:6234–6238. doi: 10.1073/pnas.92.14.6234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basic Methods in Cellular and Molecular Biology. Molecular Cloning. JoVE Science Education Database. 2015.
- Basic Methods in Cellular and Molecular Biology. Restriction Enzyme Digests. JoVE Science Education Database. 2015.
- Viczian A, Zuber M. Tissue Determination Using the Animal Cap Transplant (ACT) Assay in Xenopus laevis. J. Vis. Exp. 2010. p. e1932. [DOI] [PMC free article] [PubMed]
- Sive H, Grainger R, Harland R. Early development of.Xenopus laevis.: A laboratory manual. New York: Cold Spring Harbor Laboratory Press Cold Spring Harbor; 2000. [Google Scholar]
- Zygar C, Cook T, Grainger R. Gene activation during early stages of lens induction in Xenopus. Development. 1998;125:3509–3519. doi: 10.1242/dev.125.17.3509. [DOI] [PubMed] [Google Scholar]
- Medina-Martinez O, Jamrich M. Foxe view of lens development and disease. Development. 2007;134:1455–1463. doi: 10.1242/dev.000117. [DOI] [PubMed] [Google Scholar]
- Amin N, Tandon P, Osborne NE, Conlon F. RNA-seq in the tetraploid Xenopus.laevis. enables genome-wide insight in a classic developmental biology model organism. Methods. 2014;66:398–409. doi: 10.1016/j.ymeth.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilchrist M, Zorn A, Voigt J, Smith J, Papalopulu N, Amaya E. Defining a large set of full-length clones from a Xenopus tropicalis EST project. Developmental Biology. 2004;271:498–516. doi: 10.1016/j.ydbio.2004.04.023. [DOI] [PubMed] [Google Scholar]
- Karpinka J, et al. Xenbase, the Xenopus. model organism database; new virtualized system, data types and genomes. Nucleic Acids Research. 2015;43:D756–D763. doi: 10.1093/nar/gku956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Li J, Lea R, Amaya E, Dorey K. A Functional Genome-Wide In Vivo Screen Identifies New Regulators of Signaling Pathways during Early Xenopus Embryogenesis. PLoS ONE. 2013. p. e79469. [DOI] [PMC free article] [PubMed]