

Abstract
The pea aphid Acyrthosiphon pisum, with a sequenced genome and abundant phenotypic plasticity, has become an emerging model for genomic and developmental studies. Like other aphids, A. pisum propagate rapidly via parthenogenetic viviparous reproduction, where the embryos develop within egg chambers in an assembly-line fashion in the ovariole. Previously we have established a robust platform of whole-mount in situ hybridization allowing detection of mRNA expression in the aphid embryos. For analyzing the expression of protein, though, established protocols for immunostaining the ovarioles of asexual viviparous aphids did not produce satisfactory results. Here we report conditions optimized for increasing tissue permeability and decreasing background staining, both of which were problems when applying established approaches. Optimizations include: (1) incubation of proteinase K (1 µg/ml, 10 min), which was found essential for antibody penetration in mid- and late-stage aphid embryos; (2) replacement of normal goat serum/bovine serum albumin with a blocking reagent supplied by a Digoxigenin (DIG)-based buffer set and (3) application of methanol rather hydrogen peroxide (H2O2) for bleaching endogenous peroxidase; which significantly reduced the background staining in the aphid tissues. These critical conditions optimized for immunostaining will allow effective detection of gene products in the embryos of A. pisum and other aphids.
Keywords: Developmental Biology, Issue 108, Acyrthosiphon pisum, antibody penetration, aphids, blocking reagent, embryonic development, peroxidase, hydrogen peroxide, methanol, ovary, parthenogenetic viviparous, proteinase K
Introduction
Aphids are hemipteran insects with small (1-10 mm) soft bodies. They feed on plants by sucking phloem sap with piercing mouthparts. Additionally, they rely on an obligate endosymbiotic bacterium, Buchnera aphidicola, to synthesize essential amino acids that are deficient in the phloem sap diet. Aphids have a complex life history that includes parthenogenetic viviparous reproduction during spring and summer long-day photoperiods and sexual oviparous reproduction triggered by short-day photoperiods during which they lay a limited number of overwintering eggs1,2. In spring these eggs hatch to produce the first generation of all-female aphids (fundatrices), following many rounds of parthenogenetic reproduction until autumn. The cyclical parthenogenesis in aphids, where asexual and sexual phases alternate in the annual life cycle, has been regarded as an evolutionary novelty1,2. In the parthenogenetic viviparous aphids, embryogenesis takes place within egg chambers of the ovarian tubules (ovarioles). By contrast, sexual oviparous embryos develop in the fertilized eggs. Apart from reproductive plasticity, aphids can display transgenerational wing polyphenism: in response to overcrowding signals and predator threats, the unwinged asexual females can viviparously produce winged offspring for long-distance migration. Publication of the genome sequence of the pea aphid Acyrthosiphon pisum-the first genome sequence for a basal hemimetabolous insect-allows further exploration of reproductive plasticity, wing polyphenism, and other features including insect-plant interactions, viral vectoring and symbiosis in aphids on a molecular basis3.
In addition to the sequenced genome, tools for characterizing gene expression and function are required for promoting the pea aphid as a mature model organism4. We have described robust protocols of whole-mount in situ hybridization for detecting expression of mRNA in aphid embryos5-7. RNA interference (RNAi) via double-stranded RNA injection and feeding has been used for gene silencing in aphid nymphs and adults, but stable conditions for gene knockdown in the embryos have not yet been reported8-10. Immunostaining, an antibody-based approach that can detect protein expression in samples before and after RNAi knockdown, has been performed on pea aphid embryos11-13. However, increase of tissue permeability and elimination of background staining are as yet unsatisfactory using standard protocols for immunostaining in the asexual viviparous embryos of the pea aphid. For example, we found that penetration of antibody to the tissues decreased in gastrulating embryos (stages 8-10) and that embryos with morphologically identifiable limb buds (stages 13-14) were barely permeable to antibody. In addition, background staining was visualized in the asexual viviparous pea aphid embryos stained using antibody against the germline marker Vasa as well as that against the Engrailed/Invected protein expressed in the embryonic segments12,13. Actually background staining was still clearly visible in embryos stained with the secondary antibody alone.
In order to increase permeability without damaging integrity of aphid tissues, we carefully titrated the concentration of proteinase K and determined optimal conditions for tissue digestion on aphid embryos. In order to avoid non-specific staining in the pea aphid, we searched for compounds that could effectively block embryos and suppress activity of endogenous peroxidase (POD), an enzyme employed for amplifying signals during immunostaining. A blocking reagent provided by a Digoxigenin (DIG)-based buffer set, rather the traditionally used normal goat serum (NGS)/bovine serum albumin (BSA), significantly reduced background staining. Moreover, methanol was found to inhibit the endogenous POD activity more effectively than hydrogen peroxide (H2O2). Details regarding these aphid-specific conditions for immunostaining on embryos will be described in the following sections.
Protocol
1. Culture of Aphids
NOTE: The laboratory strain of the parthenogenetic viviparous pea aphid A. pisum was originally collected in the central Taiwan and has been reared on host plants (the garden pea Pisum sativum or broad bean Vicia faba) under long-day photoperiod for more than 300 generations (one generation: ~10 days).
- Germination of Seeds
- Soak the seeds of host plants in tap water for 3-5 days at RT. Refill with fresh water once per day. NOTE: Alternatively, putting seeds of host plants straight into moist soil can induce germination as well.
- Grow 10 germinating seeds in a small pot (9 cm diameter x 7 cm tall) with soil in the growth chamber under photoperiod 16 hr light/8 hr dark at 20 °C.
- About 10 days after growth begins, transfer aphids onto plants whose height is more than 8 cm.
- Transfer of Aphids
- Keep each pot of plants within a 1 L glass beaker, transfer 8 adult aphids onto plants using a paintbrush, and then seal the beaker with an air-permeable cover such as gauze mesh to prevent aphids from escaping.
- Incubate aphids in a growth chamber under photoperiod 16 hr light/8 hr dark at 20 °C. Water each plant pot of plants once every day.
- For obtaining the next generation of aphids, reiterate steps 1.1.3-1.2.2 ten days after the primary aphid transfer.
2. Dissection and Fixation of Ovaries
Freshly prepare 4% paraformaldehyde (PFA) in 1x phosphate-buffered saline (PBS) as the fixation buffer.
Fill one well of a spot plate with PFA (about 500 µl), place the plate under a stereo microscope at low magnification, and submerge an adult aphid within the PFA for dissection.
Dissect ovaries by holding the head and abdomen with one set of forceps, cutting open the dorsal cuticle of the abdomen, and dragging the ovaries away from the abdominal cavity.
Fix three pairs of ovaries in a 1.5 ml tube containing 1 ml of PFA at RT for 20 min.
Decant fixation buffer with a transfer pipette (either glass or disposable) and then wash ovaries with 0.2% TritonX-100 in 1x PBS (PBST) 3 times for 10 min each. Mild shaking on a mixer/rotator (rotation angle: 60° (F60)/rotation speed: 8 RPM) is recommended for both fixation and washing.
3. Treatment with Proteinase K (PK) to Increase the Permeability of Embryonic Tissues
NOTE: PK treatment is applied to embryos from germ band extension onward (stage 11 of development). For younger embryos, this step is optional.
Serially dilute the stock solution of PK (10 mg/ml) with 1x PBS to the working concentration 1 µg/ml.
Incubate ovaries with 1 µg/ml of PK (about 500 µl) for 10 mins with mild shaking.
Decant PK solution and then wash the ovaries with 700 µl of Glycine (2 mg/ml) 3 times for 5 mins each.
Wash ovaries with 0.2% PBST twice for 10 min each.
Fix ovaries again with fixation buffer for 15 min at RT with mild shaking.
Discard supernatant and wash ovaries with 0.2% PBST twice for 10 min each.
4. Methanol Incubation for Suppressing the Endogenous peroxidase (POD) activity
NOTE: Methanol incubation is not applied to embryos subjected to Phalloidin staining or antibody epitopes that are methanol sensitive.
Serially dehydrate ovaries with different percentage of methanol in 0.2% PBST (v/v: 1:3, 1:1, 3:1) by incubating ovaries at each concentration of methanol solution for 10 min with mild agitation.
Dehydrate ovaries with 100% methanol for 1 hr at RT with mild shaking. NOTE: Satisfactory results of staining could still be obtained from aphid tissues that were stored in 100% methanol at -20 °C for one month.
Serially rehydrate ovaries with different percentage of methanol in 0.2% PBST (v/v: 3:1, 1:1, 1:3) by incubating ovaries at each concentration of methanol solution for 10 min with mild agitation.
5. Antibody Staining
Dilute the 10x blocking solution from the DIG-based buffer set (DIG-B) to 1x. NOTE: The 1x DIG-B blocking solution is more effective for reducing the staining background than the standard blocking reagent composed of 5% (v/v) normal goat serum (NGS) and 0.5% (v/v) bovine serum albumin (BSA) in 0.2% PBST.
Incubate the ovaries with the 1x DIG-B blocking solution for 2.5 to 4 hr at RT or O/N at 4 °C with mild shaking. NOTE: For three pairs of ovaries in a 1.5 ml tube, 200 µl is the minimum volume for sample blocking and antibody staining.
Decant supernatant and replace with fresh 1x DIG-B blocking solution containing primary antibody at appropriate dilution ratio. Stain the ovaries for 4 hr at RT or O/N at 4 °C with mild shaking. NOTE: For the experiment described here, use the following optimal dilutions of primary antibodies: (1) ApVas1 antibody: 1:500 for chromogenic staining, 1:50 for immunofluorescence staining; (2) anti-α tubulin antibody: 1:500; (3) 4D9 monoclonal antibody: 1:25.
Wash ovaries with 0.2% PBST 4 times for 15 min each.
Incubate ovaries with 1x DIG-B blocking solution for 1 hr at RT with mild shaking.
- Decant supernatant and replace with fresh 1x DIG-B blocking solution containing secondary antibody at appropriate dilution ratio. Stain the ovaries for 4 hr at RT or O/N at 4 °C with mild shaking.
- Use the following dilution ratios of secondary antibodies: (1) For immunofluorescence staining: 1:500 for Alexa Fluor 633 goat anti-rabbit IgG or Alexa Fluor 488 goat anti-mouse IgG; (2) For chromogenic staining: 1:200 for biotinylated goat anti-rabbit IgG. NOTE: The biotinylated secondary antibody is applied for interacting with the avidin-biotin-complex (ABC) in the substrate-based (chromogenic) detection, through which the staining signals are significantly amplified.
- For immunofluorescence staining, carry out staining in the dark because the secondary antibody is light sensitive.
Wash off secondary antibody with 0.2% PBST 4 times for 10 min each.
6. Nuclear and F-actin Staining
NOTE: This is only applied for immunofluorescence staining.
Stain ovaries with 0.2% PBST containing DAPI (2 ng/µl) and Phalloidin-TRITC (100 nM) for 2 hr at RT in dark.
Wash ovaries with 0.2% PBST 4 times for 10 min each.
Incubate ovaries with the mounting medium O/N at 4 °C with mild shaking.
7. Signal Development
NOTE: This is only applied for chromogenic staining.
Prepare 100 µl of reagent avidin-biotin-complex (ABC) for enhancing the signals by adding 1 µl of Reagent A (avidin) in 98 µl of 0.2% PBST, mixing thoroughly via gentle pipetting, and adding 1 µl of Reagent B (biotin conjugated with horseradish peroxidase), followed by immediate mixing. Incubate the mixture for 30 min at RT with mild shaking.
Incubate ovaries (including the disassociated egg chambers) in the mixture of reagents A and B for 30 min at RT with mild shaking.
Wash off the mixture of reagents A and B with 0.2% PBST 4 times for 10 min each.
Transfer ovaries submerged in PBST to a well on the spot plate with a plastic dropper.
Prepare substrate solution of 3,3'-Diaminobenzidine (DAB): dissolve one DAB tablet plus another one containing urea hydrogen peroxide in 1 ml of ddH2O via vigorous vortexing for 1 min. NOTE: DAB, a precipitating substrate of peroxidase, is a popular chromogen for immunostaining. It produces a brown and insoluble precipitate after being oxidized by the peroxidase. Weak signals can be enhanced by adding nickel chloride to the substrate solution (final concentration 0.05-0.08 %).
Remove the PBST solution remaining in the well and refill with 100 µl of the DAB substrate solution for signal development.
Monitor intensity of signals under a stereo microscope at low magnification.
Stop reactions by removing the DAB solution, followed by refilling with 1x PBS immediately. Repeat the wash twice.
Transfer ovaries back to the 1.5 ml tube and then wash with 1x PBS or PBST twice for 15 min each.
Incubate ovaries in a glycerol-based mounting medium (70% glycerol) O/N at 4 °C with mild shaking.
8. Mounting the Aphid Embryos
NOTE: The thickness of aphid embryos varies between stages of development. Mounting strategies are thus modified to fit early (germaria and stages 0-10), mid (stages 11-18), and late embryos (stages 19-20), which are demonstrated in Figure 2B-D. Embryonic staging followed Miura et al.12
Transfer ovaries together with mounting medium to the cell tray with a plastic dropper and observe samples under a stereo microscope at low magnification.
Cut the calyces associated with the lateral oviduct using insect pins and then transfer an isolated ovariole to an empty glass well with a dropper. Make up the final volume to 50-100 µl using mounting medium.
Transfer an ovariole onto the slide with a glass dropper and then dissect egg chambers using insect pins. NOTE: For embryos older than stage 6 of development, separation of egg chambers is suggested; for germaria and the first two egg chambers younger than stage 6, separation is optional.
Relocate a dissected egg chamber to a clean slide using a glass dropper.
- Put a coverslip (size: 22 x 22 mm) over the dissected germaria or egg chambers (containing embryos at stages 1-10 of development) slowly to avoid bubbles.
- Mount egg chambers (containing embryos at stages 11-18 of development) on a slide with one-sided coverslip bridge and place another coverslip (size: 18 x 18 mm) on top of the sample.
- Mount egg chambers (containing embryos older than stage 19 of development) on a slide with double-sided coverslip bridge and place another coverslip (size: 18 x 18 mm) on top of the sample.
Fill the space beneath the top coverslip with mounting media to avoid drying the sample.
Mildly roll the embryo by sliding the coverslip to obtain the right orientation for observation.
Seal around the edge of coverslips (including the bridge coverslips) with nail polish.
9. Imaging Analysis
Photograph differential interference contrast (DIC) images of whole-mount embryos with a compound microscope equipped with DIC optics and a dry objective lens (10X, 20X, 40X) connected to a camera. Install the software for image transfer between the camera and computer using manufacturer’s instructions.
- Acquire projections of the fluorescently labeled embryos with a laser-scanning confocal microscope14. Follow the manufacturer's instructions for photographing, z-stacking, and 3D projecting with imaging software.
- Turn the slide upside down, find the mounted sample with 10X objective, and circle the sample area with a fine oiled-based pen. NOTE: This makes identifying the sample easier when searching for it through objectives of a confocal microscope.
- Add a drop of oil on the top of the coverslip area whose opposite side is labeled as described in 9.2.1.
Find the focal plane at 40X oil-immersion objective then change to a 63X oil-immersion objective. Manually move the fine focus control up and down to capture the best focal plane. NOTE: For observing structural details of germaria and early embryos, we suggest using 40X objectives or those with higher magnification.
Scan the embryo in different excitation channels and obtain a z-stack image. NOTE: For pea aphid tissues, reduce thickness of each optical section down to 1.5 µm or less.
Representative Results
In this study, we performed whole-mount immunostaining on embryos of asexual pea aphids (Figure 1A). These females produce offspring parthenogenetically and viviparously. These female embryos develop within egg chambers of the ovarian tubules (ovarioles) (Figure 1B and Figure 2A). Before microscopy, the dissected ovarioles are the staining targets; however, separation of egg chambers is required for observation of embryos under a microscope (Figure 2B-D).
Increase of tissue permeability
Proteinase K (PK) treatment is a standard approach for enhancing tissue permeability, but for embryos of some model organisms-such as Caenorhabditis elegans (nematode), Drosophila melanogaster (fly), and Danio rerio (zebrafish)-this step is optional. In the pea aphid, the requirement for PK treatment is stage-dependent: for germaria and embryos prior to gastrulation (stages 0-7), PK treatment can be omitted; but for embryos under germband extension (stage 11) or in later stages this step is highly recommended. For example, during mid embryogenesis signals were barely detected in embryos without PK treatment (Figure 3A-C). By contrast, signal intensity was significantly enhanced in embryos subjected to PK digestion (Figure 3A'-C', A"-C").
Reduction of background staining
A high level of the endogenous peroxidase (POD) activity was identified in the embryonic tissues of aphids. To suppress this enzyme activity, the paraformaldehyde-fixed embryos were incubated in the presence of hydrogen peroxide (H2O2), a common reagent for the oxidation of POD. Our results showed that H2O2 treatment did not suppress the activity of endogenous POD effectively in the late-stage embryos (Figure 4A, D). By contrast, background staining was largely reduced in embryos subjected to methanol incubation (Figure 4B, C, E, F). Before application of the primary antibody embryos were blocked in solution containing NGS and BSA as described in standard protocols for antibody staining. However, residual background staining in the aphid embryos was detected (Figure 3A'-C'). This problem was resolved after replacing NGS/BSA with the blocking reagent supplied by a buffer set for DIG-labeling experiments such as in situ hybridization (Figure 3A"-C"). We thus conclude that post-fixation with methanol and incubation with the DIG-B blocking reagent are both essential for reducing background staining in the aphid embryos.
PK treatment and the DIG-B blocking reagent are required not only for effective signal detection using the chromogenic but also fluorescent approaches. Actin staining with phalloidin or staining for methanol-sensitive epitope, however, does not work in embryos subjected to pre-fixation with methanol, which can destroy the native form of actin or antibodies. Accordingly, this treatment should be avoided when performing fluorescence immunostaining. Apart from eliminating the methanol treatment steps immunofluorescence allows multi-labeling experiments in the aphid embryos. Using confocal microscopy, for example, pea aphid Vasa1 protein (ApVas1) in germ cells, α-tubulin in microtubules, F-actin in microfilaments, and DNA in nuclei could be concurrently marked and visualized (Figure 5A, C). In comparison with the chromogenic method (Figure 5B), confocal sectioning of fluorescently labeled samples provides better resolution of localized signals such as ApVas1 in the germ plasm and cellularizing germ cells in early aphid embryos (Figure 5C', D-D"). Adopting the conditions for labeling germ cells described above, the Engrailed/Invected protein in segments of the extending germ band was also stained with the antibody 4D9 (Figure 6). This shows that our protocol for immunostaining-including chromogenic and fluorescent methods-can be effectively applied to signal detection in both germline and somatic cell lineages in aphids.
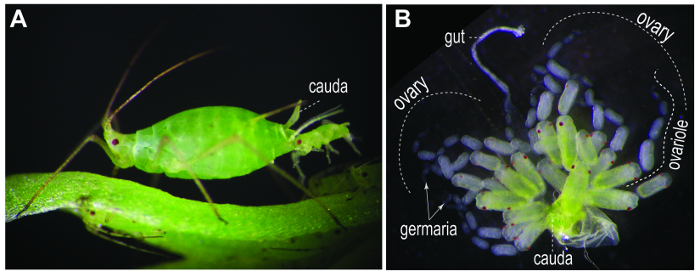 Figure 1. Parthenogenetic viviparous pea aphids. (A) A first-instar nymph emerging from an asexual viviparous female adult. (B) A pair of dissected ovaries. Each ovary is composed of 7 ovarian tubules (ovarioles). Nonetheless, the number of ovarioles within an ovary may be variable between strains12. An ovariole contains a germarium in the tip (arrows), 1-2 oocytes, and 5-7 embryonic chambers. Gut and cauda are usually associated with the dissected ovaries. Please click here to view a larger version of this figure.
Figure 1. Parthenogenetic viviparous pea aphids. (A) A first-instar nymph emerging from an asexual viviparous female adult. (B) A pair of dissected ovaries. Each ovary is composed of 7 ovarian tubules (ovarioles). Nonetheless, the number of ovarioles within an ovary may be variable between strains12. An ovariole contains a germarium in the tip (arrows), 1-2 oocytes, and 5-7 embryonic chambers. Gut and cauda are usually associated with the dissected ovaries. Please click here to view a larger version of this figure.
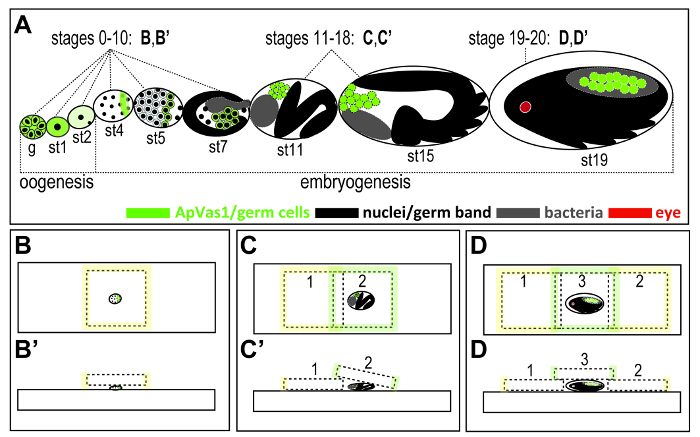 Figure 2. Illustration of strategies for mounting aphid embryos at different stages of development. (A) Outline of embryonic development in the ovariole dissected from a parthenogenetic viviparous female adult. Brief oogenesis (germarial stage and stages 0-2) is followed by embryogenesis (stages 3-20). Outline of embryogenesis: formation of syncytial blastoderm (stages 3-5); blastulation (stages 6-7); gastrulation (stages 8-10); elongation of germ band (stages 11-14); katatrepsis (stages 15); post katatrepsis and germ band retraction (stages 16-17); organogenesis (stages 18-20). Color keys are at the bottom of the figure. (B, B') Germarium and stages 0-10 embryos. No coverslip bridge is required. (C, C') Stages 11-18 embryos. A coverslip bridge is required. (D, D') Stages 19-20 embryos. Double coverslip bridges are required. Rolling the embryos by sliding the coverslip can create different angles of observation. Sizes of coverslips: 22 x 22 mm in (B), 18 x 18 mm in (C) and (D). The thickness of coverslips: 0.13 to 0.16 mm. Embryonic staging followed Miura et al.12 Abbreviations: g: germarium; st: stage. Please click here to view a larger version of this figure.
Figure 2. Illustration of strategies for mounting aphid embryos at different stages of development. (A) Outline of embryonic development in the ovariole dissected from a parthenogenetic viviparous female adult. Brief oogenesis (germarial stage and stages 0-2) is followed by embryogenesis (stages 3-20). Outline of embryogenesis: formation of syncytial blastoderm (stages 3-5); blastulation (stages 6-7); gastrulation (stages 8-10); elongation of germ band (stages 11-14); katatrepsis (stages 15); post katatrepsis and germ band retraction (stages 16-17); organogenesis (stages 18-20). Color keys are at the bottom of the figure. (B, B') Germarium and stages 0-10 embryos. No coverslip bridge is required. (C, C') Stages 11-18 embryos. A coverslip bridge is required. (D, D') Stages 19-20 embryos. Double coverslip bridges are required. Rolling the embryos by sliding the coverslip can create different angles of observation. Sizes of coverslips: 22 x 22 mm in (B), 18 x 18 mm in (C) and (D). The thickness of coverslips: 0.13 to 0.16 mm. Embryonic staging followed Miura et al.12 Abbreviations: g: germarium; st: stage. Please click here to view a larger version of this figure.
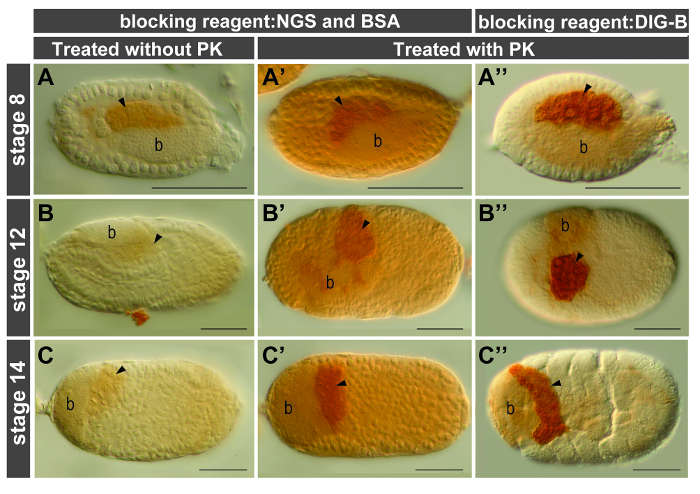 Figure 3. Proteinase K treatment and comparison of reagents with different blocking effects. Anterior of egg chambers is to the left; all views are lateral except embryos shown in (B", C', C"), which are dorsal. Embryos are all stained using ApVas1 antibody (dilution 1:500) and signals of ApVas1 are developed within 10-20 sec. Arrowheads indicate location of germ cells. (A-C) Embryos without treatment of proteinase K (PK). ApVas1 signals in germ cells are barely detected. (A'-C', A"-C") Comparison of background staining in embryos blocked with NGS and BSA (A'-C') and of the commercial blocking reagent in the DIG Wash and Block Buffer Set (DIG-B) (A"-C"). Background is significantly reduced in embryos shown in (A"-C"). Abbreviation: b: bacteria. Scale bars: 100 µm. Please click here to view a larger version of this figure.
Figure 3. Proteinase K treatment and comparison of reagents with different blocking effects. Anterior of egg chambers is to the left; all views are lateral except embryos shown in (B", C', C"), which are dorsal. Embryos are all stained using ApVas1 antibody (dilution 1:500) and signals of ApVas1 are developed within 10-20 sec. Arrowheads indicate location of germ cells. (A-C) Embryos without treatment of proteinase K (PK). ApVas1 signals in germ cells are barely detected. (A'-C', A"-C") Comparison of background staining in embryos blocked with NGS and BSA (A'-C') and of the commercial blocking reagent in the DIG Wash and Block Buffer Set (DIG-B) (A"-C"). Background is significantly reduced in embryos shown in (A"-C"). Abbreviation: b: bacteria. Scale bars: 100 µm. Please click here to view a larger version of this figure.
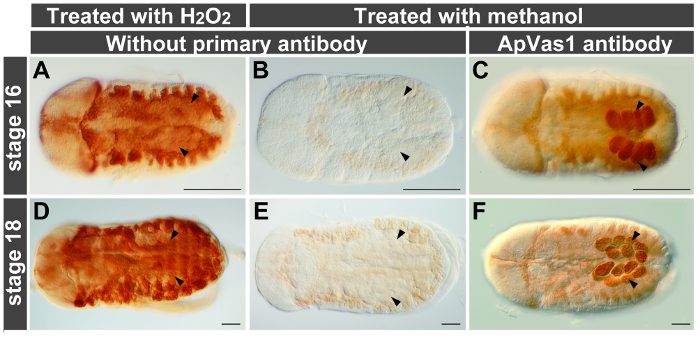 Figure 4. Minimizing background staining with methanol. Anterior of embryos is to the left; all views are dorsal. Embryos are treated with PK for increasing permeability of antibody. Arrowheads indicate location of germ cells. (A, B, D, E) Comparison of treatments with hydrogen peroxide (H2O2) and methanol. Embryos are only stained with secondary antibody. H2O2 treatment (0.3% w/v, 10 min): high background (A, D); methanol treatment (100%, 60 min): low background (B, E). (C, F) Primary antibody staining on embryos treated with methanol. Conditions of methanol treatment are identical to those used for embryos shown in (B, E). The ApVas1 antibody preferentially labels the embryonic germ cells. Scale bars: 100 µm. Please click here to view a larger version of this figure.
Figure 4. Minimizing background staining with methanol. Anterior of embryos is to the left; all views are dorsal. Embryos are treated with PK for increasing permeability of antibody. Arrowheads indicate location of germ cells. (A, B, D, E) Comparison of treatments with hydrogen peroxide (H2O2) and methanol. Embryos are only stained with secondary antibody. H2O2 treatment (0.3% w/v, 10 min): high background (A, D); methanol treatment (100%, 60 min): low background (B, E). (C, F) Primary antibody staining on embryos treated with methanol. Conditions of methanol treatment are identical to those used for embryos shown in (B, E). The ApVas1 antibody preferentially labels the embryonic germ cells. Scale bars: 100 µm. Please click here to view a larger version of this figure.
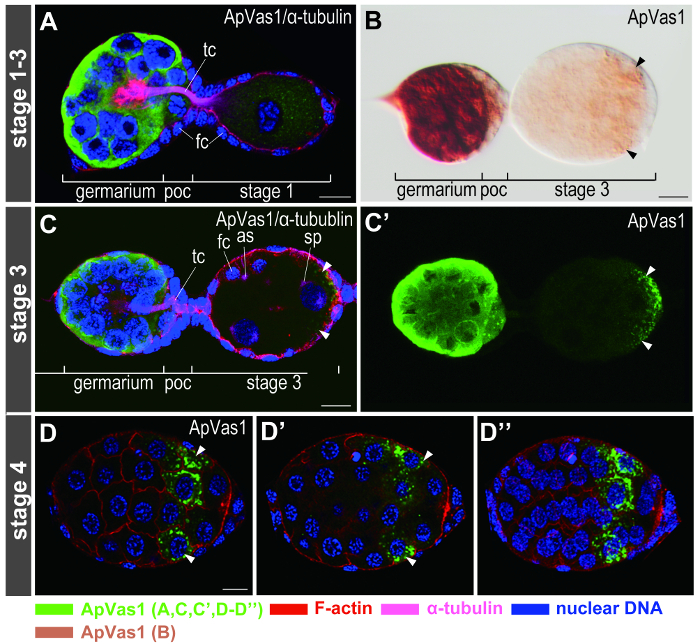 Figure 5. Immunofluorescence staining on early embryos. Anterior of egg chambers is to the left. Dilution ratios of ApVas1 antibody are 1:50 in (A, C-D) and 1:500 in (B). (A) Staining signals, which include ApVas1, α-tubulin, F-actin, and nuclear DNA, are detected using four channels with different wavelengths. Color keys of signals are shown at the bottom of the figure. (B) DIC image of a chromogenic result for comparison. ApVas1 is enriched within the germarium whereas the contrast intensity of posterior localization of ApVas1 (arrowheads) in the stage-3 embryo is not as clear as that shown in (C, C'). (C, C') Enrichment of ApVas1 signals in the egg posterior. Signals localized to the posterior region of the egg chamber (arrowheads) are enhanced by image stacking. Because signals of F-actin and α-tubulin partially mask those of ApVas1 in the posterior (C), an image produced by single-channeled scanning is shown in (C'). (D-D'') Confocal sectioning of ApVas1 localized in the posterior region of the stage-4 embryo. Images shown in (D) and (D') are ApVas1 detected in surface and central focal planes, respectively. (D") is the merged image of all sections from the same embryo as (D) and (D'). Abbreviations: as: aster; fc, follicle cells; poc, prospective oocyte; sp: spindle; tc, trophic cords. Scale bars: 10 µm. Please click here to view a larger version of this figure.
Figure 5. Immunofluorescence staining on early embryos. Anterior of egg chambers is to the left. Dilution ratios of ApVas1 antibody are 1:50 in (A, C-D) and 1:500 in (B). (A) Staining signals, which include ApVas1, α-tubulin, F-actin, and nuclear DNA, are detected using four channels with different wavelengths. Color keys of signals are shown at the bottom of the figure. (B) DIC image of a chromogenic result for comparison. ApVas1 is enriched within the germarium whereas the contrast intensity of posterior localization of ApVas1 (arrowheads) in the stage-3 embryo is not as clear as that shown in (C, C'). (C, C') Enrichment of ApVas1 signals in the egg posterior. Signals localized to the posterior region of the egg chamber (arrowheads) are enhanced by image stacking. Because signals of F-actin and α-tubulin partially mask those of ApVas1 in the posterior (C), an image produced by single-channeled scanning is shown in (C'). (D-D'') Confocal sectioning of ApVas1 localized in the posterior region of the stage-4 embryo. Images shown in (D) and (D') are ApVas1 detected in surface and central focal planes, respectively. (D") is the merged image of all sections from the same embryo as (D) and (D'). Abbreviations: as: aster; fc, follicle cells; poc, prospective oocyte; sp: spindle; tc, trophic cords. Scale bars: 10 µm. Please click here to view a larger version of this figure.
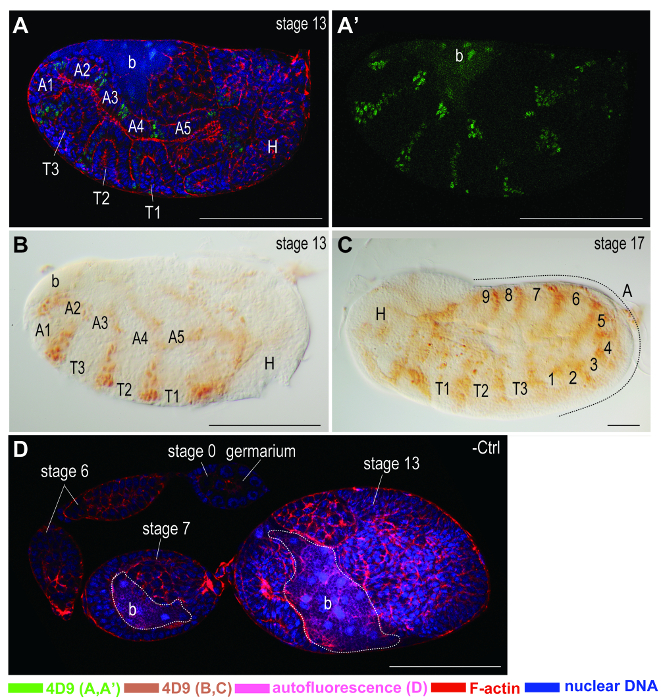 Figure 6. Immunostaining on embryonic segments. Anterior of egg chambers is to the left. PK-treated embryos are stained with the monoclonal antibody 4D9 against Engrailed/Invected, which are expressed in embryonic segments. Color keys of signals are shown at the bottom of the figure.(A, A') Immunofluorescence staining on a stage-13 embryo. (A), a confocal image stacked from images of Engrailed/Invected, α-tubulin, F-actin, and nuclear DNA stainings. (A') a confocal image showing the staining of Engrailed/Invected only. Without the interference of signals from other channels, signals are better displayed. (B, C) Chromogenic staining on embryos at stages 13 and 17 of development, respectively. (B) Stage-13 embryo. (C) Stage-17 embryo. From stages 13 to 17, the growing numbers of segments in the abdomen are labeled by 4D9. (D) Negative control (-Ctrl) staining only with secondary antibody conjugated with Alexa Fluor 633. Almost no immunostaining signals are detected on ovariole without primary antibody. However, autofluorescence of bacteria (dash line) within stage 7 and 13 of egg chambers are detected at the excitation wavelength 488 nm. Abbreviations: A: abdomen; b: bacteria; H: head; T: thorax. Scale bars: 100 µm. Please click here to view a larger version of this figure.
Figure 6. Immunostaining on embryonic segments. Anterior of egg chambers is to the left. PK-treated embryos are stained with the monoclonal antibody 4D9 against Engrailed/Invected, which are expressed in embryonic segments. Color keys of signals are shown at the bottom of the figure.(A, A') Immunofluorescence staining on a stage-13 embryo. (A), a confocal image stacked from images of Engrailed/Invected, α-tubulin, F-actin, and nuclear DNA stainings. (A') a confocal image showing the staining of Engrailed/Invected only. Without the interference of signals from other channels, signals are better displayed. (B, C) Chromogenic staining on embryos at stages 13 and 17 of development, respectively. (B) Stage-13 embryo. (C) Stage-17 embryo. From stages 13 to 17, the growing numbers of segments in the abdomen are labeled by 4D9. (D) Negative control (-Ctrl) staining only with secondary antibody conjugated with Alexa Fluor 633. Almost no immunostaining signals are detected on ovariole without primary antibody. However, autofluorescence of bacteria (dash line) within stage 7 and 13 of egg chambers are detected at the excitation wavelength 488 nm. Abbreviations: A: abdomen; b: bacteria; H: head; T: thorax. Scale bars: 100 µm. Please click here to view a larger version of this figure.
Discussion
We identified optimal conditions critical to successful immunostaining in the pea aphid A. pisum, an emerging model organism for genomic and developmental studies3,15. Optimized conditions for increasing tissue permeability and reducing background staining enhanced the intensity and specificity of signals. They differ from standard protocols for immunostaining in other animal models in the steps for creating pores in cell membranes and blocking non-specific antibody binding.
For immunostaining and in situ hybridization, proteinase K (PK) digestion is one of the common approaches for increasing tissue permeability16-19. In antibody staining of aphid tissues, our results showed that: (1) PK digestion significantly enhanced immunostaining signals in the parthenogenetic viviparous embryos during germ band extension (stages 11-14) (Figure 3B'', C''), although weak signals were still visible in embryos devoid of PK treatment (Figure 3B, C); and (2) PK digestion was obligatory for embryos from katatrepsis onward (stages 15-20) (Figure 4C, F and Figure 6C). For asexual viviparous embryos prior to katatrepsis, the volume of the egg chambers is unlikely the cause of the penetration problem because successful immunostaining on early sexual oviparous embryos residing in the laid eggs-which are almost equivalent in size to the most mature asexual egg chamber-does not require PK digestion20. Therefore, the elongating and folding of the germ band (stages 11 to 14) in the limited space of the asexual egg chambers may hinder the entrance of antibody, and it is PK that allows the penetration of antibody by creating pores in cell membranes between the jammed embryonic structures. By contrast, the straight line extension of the germ band in the sexual eggs may explain why it is stained even in the absence of PK. Removal of the vitelline envelope/chorion can also make sexual oviparous embryos more accessible to antibody. For embryos after katatrepsis (stages 16 to 20), PK treatment is essential for both asexual and sexual morphs because embryos are thick cuticled19 (Figure 4C, F and Figure 6C).
Nonetheless, the suggested condition for PK digestion (1 µg/ml for 10 min) requires further adjustment if the integrity of embryonic structures or the intensity of signals is not satisfactory. This is mainly caused by the PK activity that may vary from batch to batch. For example, if embryonic tissues become damaged after PK digestion, concentration of PK can be reduced to 0.5 µg/ml. Likewise, if signal intensity is weak, extension of incubation to 15-20 min is thus recommended. The most mature embryos (stage 20) subjected to the prolonged PK incubation up to 20 min at 1.0 µg/ml concentration, however, usually display weak signals, suggesting that PK digestion is yet incomplete. Prolonged digestion can enhance staining signals but is optional, especially when expression patterns detected in the stage 18 embryos are similar to those in the stage 20 embryos. In comparison with embryos at stage 20 of development, the stage 18 embryos with analogous morphology yet with lesser amount of body cuticle generally can yield stronger signal intensity.
During signal development, background staining can be elevated by the activity of endogenous enzymes that can cross-react with the substrates. In the pea aphid embryos, activity of the endogenous alkaline phosphatase (AP) and peroxidase (POD), both of which are common antibody conjugates for digesting signal substrates, was detected. Hydrolase activity of the endogenous AP could be effectively suppressed throughout embryogenesis by levamisole (data not shown). Nevertheless, treatment of embryos with 0.3-0.6% hydrogen peroxide (H2O2) could only eliminate the endogenous POD activity prior to katatrepsis but was not sufficient for older embryos (Figure 4A, D). Methanol incubation, an alternative approach for denaturing endogenous proteins, was demonstrated to be effective in the aphids21 (Figure 4B, E). Unlike H2O2, which damages aphid tissues during prolonged incubation of 30 min or longer, methanol does not digest or deform the embryos during any stage of development (Figure 4C, F). Consequently, methanol is a better alternative for suppressing endogenous POD activity in aphid embryos. If residual background staining is detected, which usually occurs to the mature embryos with a thickened layer of cuticle, O/N incubation in methanol at 4 °C-according to our experience-can further reduce the endogenous POD activity.
Residual background staining was detected in embryos blocked with serum proteins such as normal donkey serum (NDS), normal goat serum (NGS), bovine serum albumin (BSA), or their mixtures13 (Figure 3A'-C'). Adopting a commercially supplied blocking reagent used for in situ hybridization, by contrast, the remaining background during immunostaining could be significantly eliminated5 (Figure 3A''-C''). Although the ingredients of the blocking reagent are proprietary, it likely contains non-serum proteins that can block non-specific epitopes of the pea aphid proteins. The blocking reagent also worked much better than animal sera in the green peach aphid Myzus persicae. Likewise, in M. persicae PK digestion and methanol incubation resulted in satisfactory antibody penetration and background reduction - similar to the improved results achieved in A. pisum (data not shown). We anticipate that the conditions optimized for increasing tissue permeability and reducing background, whether one uses chromogenic staining or fluorescence, will apply to many other aphid species.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We are grateful to Chau-Ti Ting (Fly Core in Taiwan) for providing the monoclonal 4D9 antibody, Technology Commons (TechComm) of the College of Life Science NTU for confocal microscopy, Hsiao-Ling Lu for proofreading the manuscript, and Chen-yo Chung for helping filming. CC particularly thanks Charles E. Cook for providing strategic suggestions and for critical editing of the manuscript. This work was supported by the Ministry of Science and Technology (101-2313-B-002-059-MY3 and 104-2313-B-002-022-MY3 for GWL and CC), and the National Taiwan University (NTU-CESRP 101R4602D3 for CC; 103R4000 for GWL).
References
- Blackman RL. Reproduction, cytogenetics and development. In: Minks AK, Harrewijn P, editors. Aphids: their biology, natural enemies. Amsterdam: Elsevier Press; 1987. pp. 163–195. [Google Scholar]
- Davis GK. Cyclical parthenogenesis and viviparity in aphids as evolutionary novelties. J. Exp. Zool. 2012;318:448–459. doi: 10.1002/jez.b.22441. [DOI] [PubMed] [Google Scholar]
- The International Aphid Genomics Consortium. Genome sequence of the pea aphid Acyrthosiphon pisum. PLoS Biol. 2010;8(2):e1000313. doi: 10.1371/journal.pbio.1000313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abzhanov A, et al. Are we there yet? Tracking the development of new model systems. Trends Genet. 2008;24(7):353–360. doi: 10.1016/j.tig.2008.04.002. [DOI] [PubMed] [Google Scholar]
- Chang C, et al. Apvasa marks germ-cell migration in the parthenogenetic pea aphid Acyrthosiphon pisum (Hemiptera: Aphidoidea)) Dev. Genes Evol. 2007;217(4):275–287. doi: 10.1007/s00427-007-0142-7. [DOI] [PubMed] [Google Scholar]
- Chang C, et al. Whole-mount identification of gene transcripts in aphids: Protocols and evaluation of probe accessibility. Arch. Insect Biochem. 2008;68(4):186–196. doi: 10.1002/arch.20243. [DOI] [PubMed] [Google Scholar]
- Chung CY, Cook CE, Lin GW, Huang TY, Chang C. Reliable protocols for whole-mount fluorescent in situ hybridization (FISH) in the pea aphid Acyrthosiphon pisum: a comprehensive survey and analysis. Insect Sci. 2014;21(3):265–277. doi: 10.1111/1744-7917.12086. [DOI] [PubMed] [Google Scholar]
- Mutti NS, Park Y, Reese JC, Reeck GR. RNAi knockdown of a salivary transcript leading to lethality in the pea aphid, Acyrthosiphon pisum. J. Insect Sci. 2006;6:38. doi: 10.1673/031.006.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaubert-Possamai S, et al. Gene knockdown by RNAi in the pea aphid Acyrthosiphon pisum. BMC Biotechnol. 2007;7:63. doi: 10.1186/1472-6750-7-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapountzis P, et al. New insight into the RNA interference response against cathepsin-L gene in the pea aphid, Acyrthosiphon pisum: Molting or gut phenotypes specifically induced by injection or feeding treatments. Insect Biochem. Mol. Biol. 2014;51:20–32. doi: 10.1016/j.ibmb.2014.05.005. [DOI] [PubMed] [Google Scholar]
- Braendle C, et al. Developmental origin and evolution of bacteriocytes in the aphid-Buchnera symbiosis. PLoS Biol. 2003;1(1):e21. doi: 10.1371/journal.pbio.0000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura T, et al. A comparison of parthenogenetic and sexual embryogenesis of the pea aphid Acyrthosiphon pisum (Hemiptera: Aphidoidea) J. Exp. Zool. 2003;295(1):59–81. doi: 10.1002/jez.b.3. [DOI] [PubMed] [Google Scholar]
- Chang C, Lee WC, Cook CE, Lin GW, Chang T. Germ-plasm specification and germline development in the parthenogenetic pea aphid Acyrthosiphon pisum: Vasa and Nanos as markers. Int. J. Dev. Biol. 2006;50(4):413–421. doi: 10.1387/ijdb.052100cc. [DOI] [PubMed] [Google Scholar]
- Koshy AA, Cabral CM. 3-D imaging and analysis of neurons infected in vivo with Toxoplasma gondii. J. Vis. Exp. 2014. p. e52237. [DOI] [PMC free article] [PubMed]
- Brisson JA, Stern DL. The pea aphid, Acyrthosiphon pisum: an emerging genomic model system for ecological, developmental and evolutionary studies. Bioessays. 2006;28(7):747–755. doi: 10.1002/bies.20436. [DOI] [PubMed] [Google Scholar]
- Tautz D, Pfeifle C. A non-radioactive in situ hybridization method for the localization of specific RNAs in Drosophila embryos reveals translational control of the segmentation gene hunchback. Chromosoma. 1989;98(2):81–85. doi: 10.1007/BF00291041. [DOI] [PubMed] [Google Scholar]
- Harland RM. In-situ hybridization: an improved whole-mount method for Xenopus embryos. Method. Cell Biol. 1991;36:685–695. doi: 10.1016/s0091-679x(08)60307-6. [DOI] [PubMed] [Google Scholar]
- Wolff C, Sommer R, Schroder R, Glaser G, Tautz D. Conserved and divergent expression aspects of the Drosophila segmentation gene hunchback in the short germ band embryo of the flour beetle Tribolium. Development. 1995;121(12):4227–4236. doi: 10.1242/dev.121.12.4227. [DOI] [PubMed] [Google Scholar]
- Jowett T. Double in situ hybridization techniques in zebrafish. Methods. 2001;23(4):345–358. doi: 10.1006/meth.2000.1147. [DOI] [PubMed] [Google Scholar]
- Lin GW, Cook CE, Miura T, Chang C. Posterior localization of ApVas1 positions the preformed germ plasm in the sexual oviparous pea aphid Acyrthosiphon pisum. Evodevo. 2014;5:18. doi: 10.1186/2041-9139-5-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straus W. Inhibition of peroxidase by methanol and by methanol-nitroferricyanide for use in immunoperoxidase procedures. J. Histochem. Cytochem. 1971;19(11):682–688. doi: 10.1177/19.11.682. [DOI] [PubMed] [Google Scholar]