

Cancer-associated cachexia (CAC) is a complex metabolic disorder with profound changes in energy balance. Here, Petruzzelli and Wagner review the molecules and pathways responsible for metabolic dysfunction in CAC and the ideas that led to the current understanding.
Keywords: cancer-associated cachexia (CAC), metabolic failure, white adipose tissue (WAT) browning, skeletal muscle atrophy
Abstract
Metabolic dysfunction contributes to the clinical deterioration observed in advanced cancer patients and is characterized by weight loss, skeletal muscle wasting, and atrophy of the adipose tissue. This systemic syndrome, termed cancer-associated cachexia (CAC), is a major cause of morbidity and mortality. While once attributed solely to decreased food intake, the present description of cancer cachexia is a disorder of multiorgan energy imbalance. Here we review the molecules and pathways responsible for metabolic dysfunction in CAC and the ideas that led to the current understanding.
Human cancers develop as a localized focus of uncontrolled cell growth and subsequently progress to a systemic disease (Fig. 1). Cancer research primarily focuses on the agents, events, and genetic alterations underlying tumor initiation, progression, and metastasis. However, the vast majority of end-stage cancer patients suffers a systemic illness defined as cachexia, a widespread but poorly understood condition (Lok 2015). The “most time honored symptom of cancer” (Editors 1929), cachexia is the prototype image that comes to mind when thinking of cancer. The loss of appetite, energy, and vigor; the languid and unsmiling face; the sallow and anemic aspect; and the skinny and wasted complexion are all too familiar to physicians treating cancer patients. Despite the obvious clinical picture, a formal definition of the diagnostic criteria has only recently been reached (Fearon et al. 2011). The current consensus for diagnosis is the unintentional loss of total body weight or skeletal muscle mass. Importantly, cancer-associated cachexia (CAC) is a complex metabolic disorder with profound changes in energy balance, which might be already irreversible at the time of obvious body weight loss. While cachexia itself is often rapidly progressive, marking the irreversible decline in health and survival, the time when cachexia appears in the clinical history of the cancer patients is, at present, mostly unpredictable. The severity of CAC is often unrelated to tumor size or stage, with small tumors commonly leading to severe wasting, as is the case, for example, for pancreatic and lung tumors. In contrast, widely disseminated cancers may cause death without any evidence of CAC. The reasons for this paradoxical lack of correlation between tumor burden and the degree of cancer cachexia are, at present, elusive. Furthermore, CAC often results in fewer completed cycles of chemotherapy with higher complication rates. Therefore, a better characterization of the metabolic changes in the organism affected by cancer is urgently needed in order to recognize the early events of CAC and improve its prognosis. In this review, we discuss the conceptual advances that shaped the current understanding of the systemic metabolic maladaptation to cancer.
Figure 1.
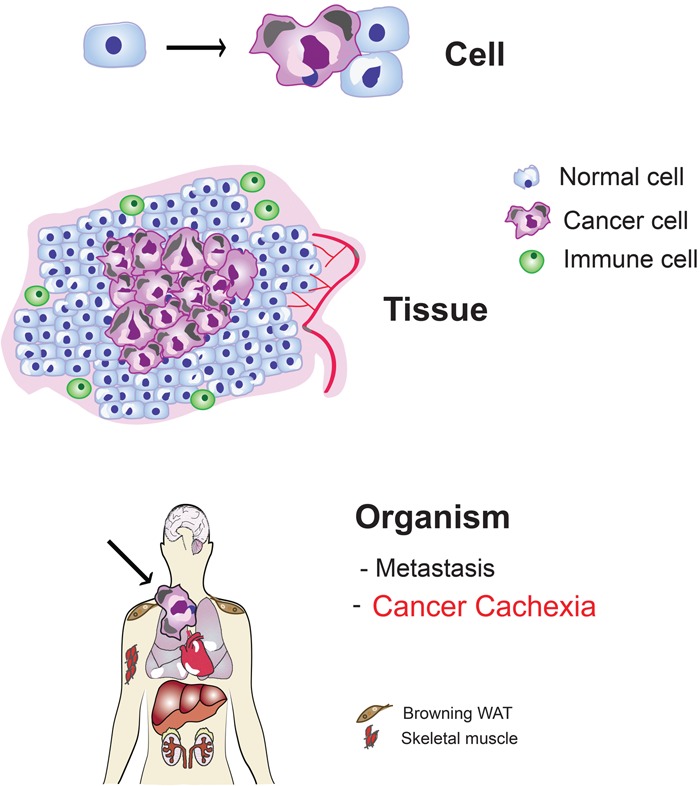
Multilevel cancer pathophysiology at a glance. Schematic representation of the evolution of cancer from a single transformed cell to a systemic disease. At the “cell” level, the transformed cell (purple) is characterized by the presence of genome instability and mutations, sustained proliferative signaling, avoidance of growth suppressors, replicative immortality, resistance to cell death, and deregulated cellular energetics. At the “tissue” level, the proliferation of the tumor mass is associated with induction of angiogenesis, activation of invasion, promotion of inflammation, and avoidance of immune destruction—all hallmarks of cancer as described in Hanahan and Weinberg (2011). At the “organism” level, the developing cancer induces changes in distant organs, including metastasis, metabolic failure, and cancer cachexia, which is not included in the hallmarks of cancer. Cancer cells are shown in purple, “normal” cells are indicated in blue, immune cells are shown in green, and blood vessels are indicated in red. The arrow at the “cell” level points to the conversion from a normal cell to a cancer cell, whereas the arrow pointing to the “organism” depicts a growing tumor (purple).
Research milestones in cancer cachexia
For centuries, the concept that a local malignant growth could be responsible for systemic effects has been under debate (E.F.B. 1909). Rather than a specific disease, the wasting associated with cancer was attributed to nonspecific pathological complications of the tumor, such as anorexia, hemorrhage, infection, or ulceration of the neoplastic tissue (Willis 1948). In contrast, those in favor of systemic alterations produced by the tumor on the host considered cachexia the result of either direct secretion by the tumor of some substances active in distant organs or uptake by the tumor of components from the blood that are essential for the correct functioning of distant organs (Greenstein 1947; Donovan 1954). Evidence supporting one hypothesis or the other was slim, and the contention was disputed on the basis of small clinical case series and anecdotal post-mortem findings (Donovan 1954).
During the past decades, a vast body of investigation has reshaped our understanding of CAC (Fig. 2). Experimental work with animal models of cancer rather than observations in the clinical setting led to the recognition of CAC as a legitimate entity independent of the effects of anorexia or mechanical interference of the tumor with the surrounding tissues. When tumor-bearing rats were force-fed a high-fat diet, weight loss was prevented. However, the development of anemia and the enlargement of the adrenal glands were not affected, thus showing the existence of systemic manifestations of cancer independent of nutritional intake (Begg and Dickinson 1951). Following the kinetics of tissue loss in tumor-bearing mice, it was noticed that adipose tissue wasting was an early event, occurring at a time when the tumor was barely palpable (Costa and Holland 1962). Surprisingly, fat loss could also be induced by nonviable tumor preparations, indicating that soluble components of tumor extracts can induce cancer cachexia (Costa and Holland 1962). The causes for the systemic effects associated with cancer were more sophisticated than just reduced food intake and needed to be sought in the complex relationship between the host and the tumor. Analogies between systemic responses to infectious agents and cancers were noted, including fever, leukocytosis, and increased serum levels of acute phase response proteins (Rosenthal and Franklin 1975). The first evidence that inflammatory mediators—namely, cytokines—were involved in the process of protein breakdown in isolated skeletal muscle and a potential role for interleukin-1 (IL-1) in muscle degradation during fever was published in 1983 (Baracos et al. 1983). Particular attention was received by the somewhat paradoxical increase in serum lipid levels despite the obvious loss of body weight in severely sick patients. It was found that such hypertriglyceridemia could be induced experimentally in animals by either injection of infective agents or transplantation of tumors (Rouzer and Cerami 1980; Kawakami and Cerami 1981). Hypertriglyceridemia was the result of lipoprotein lipase (LPL) inhibition and could be reproduced by injecting animals with conditioned medium from inflammatory cells incubated with endotoxin (Kawakami and Cerami 1981).
Figure 2.

Timeline of discoveries in cancer cachexia. In 1951, the first systemic manifestation of cancer was described in rats. In 1962, it was observed that injection of tumor preparations in mice was sufficient to induce fat atrophy. In 1983 and 1985, the first candidate molecules were identified. Seminal publications in 1993 and 2001 described a role for the ubiquitin pathway and myostatin in skeletal muscle atrophy. It was not until some years ago that an international consensus on the diagnostic criteria of CAC was reached. Promising results have been reported in late 2015 from the first phase III clinical trial targeting CAC with the ghrelin receptor agonist anamorelin (https://www.iaslc.org/news/results-phase-iii-trials-anamorelin-advanced-non-small-cell-lung-cancer-patients-cachexia).
In 1985, Bruce Beutler in Anthony Cerami's group (Cerami et al. 1985) provided definitive proof that circulating mediators could cause cachexia, showing that culture medium from endotoxin-activated macrophages caused body weight loss when injected into mice. The molecule in the conditioned medium causing cachexia was purified and termed “cachectin” (Beutler et al. 1985). Subsequent determination of the complete primary structure of cachectin revealed its identity with tumor necrosis factor-α (TNFα) (Fransen et al. 1985; Pennica et al. 1985). It should be noticed that these early preparations of conditioned medium contained multiple macrophage products, and it is therefore erroneous to attribute all of the cachexiogenic action to the effect of TNFα alone. Furthermore, TNFα causes systemic shock and the release of other cytokines, further confounding the attribution of the observed phenotype to a single identifiable factor. However, despite the technical limitations, these early studies contributed to a conceptual evolution in the field of cachexia research, and the wasting syndrome was finally regarded as the result of the host response to the tumor. Clinical evidence confirmed the conclusions of preclinical investigations showing that intravenous hyperalimentation could not alleviate cachexia in cancer patients (Evans et al. 1985). Remarkably, the focus of research had gradually shifted from the nature of the invasive agent (infection or cancer) to the quality of the response elicited in the organism. The immune system was the likely source of all mediators responsible for the systemic changes, and a variety of cytokines joined TNFα in the ability to cause systemic alterations (Beutler and Cerami 1986). However, despite the clear role played by cytokines in experimental cachexia, their involvement in human disease was less obvious, and clinical translation yielded ambiguous results (Balkwill et al. 1987; Socher et al. 1988). Perhaps as a consequence of the disappointment generated by the lack of clinical benefits from basic findings, the pace of research in the field of cancer cachexia progressively reduced. An additional explanation could be the rapid progress of molecular biology of cancer in the mid-1980s. That was the time when the first oncogenes were discovered, and the simplistic view of reducing cancer to a single base mutation was occupying the entire scene (Weinberg 2014). The war on cancer seemed to be close to a favorable ending, and the focus of research zoomed back to the tumor itself rather than the response that the tumor ignites in the organism.
Cancer cachexia is an energy balance disorder linked to inflammation
Weight loss is a cardinal sign of cachexia and represents the main independent predictor of mortality in cancer patients (Fearon et al. 2012). The mechanisms for weight loss in cancer are multiple, including decreased nutrient intake, systemic metabolic dysfunction, and increased energy expenditure. Inflammation represents a common denominator in the pathophysiology of energy imbalance during cachexia. In mice, the peritoneal injection of cancer cells expressing TNFα has been shown to cause weight loss and cachexia. In contrast, mice injected with the same cells without TNFα do not lose body weight (Oliff et al. 1987). Similar results have been obtained with IL-6 in preclinical models (Strassmann et al. 1992). Both host- and tumor-derived cytokines cooperate in a complex way with the tumor microenvironment to sustain tumor growth and cachexia (Cahlin et al. 2000). In support of a role of cancer-derived cytokines, it has been shown recently that expression of the cytokine TNF-related weak inducer of apoptosis (TWEAK) by cancer cells causes cachexia, and the effect is similar in wild-type and TWEAK-deficient mice (Johnston et al. 2015).
As the cancer persists, it is assumed that ongoing local inflammation may reach a threshold when cytokines spill into the circulation, thus transforming the cancer disease from a localized tumor to a systemic impairment. Unfortunately, such a simplistic view does not stand present experimental validation. The levels of serum cytokines do not correlate with the appearance of cachexia in cancer patients (Fearon et al. 2012). Furthermore, treatments with antibodies targeting a single cytokine have failed so far to prevent or significantly ameliorate the wasting syndrome (Penna et al. 2010; Fearon et al. 2013). Very recent data emphasize the multifactorial etiology of CAC, showing now that a combination of cytokines and/or additional mediators is responsible for the cachectic phenotype (Schaefer et al. 2016).
Despite the absence of a simplistic threshold model linking cytokine levels to cachexia development, a rich body of evidence supports their causal role in the metabolic dysfunction observed in CAC. Mechanistically, cytokines were shown to increase the metabolic rate through activation of thermogenesis, inhibit adipocyte and skeletal myocyte differentiation, and reduce food intake (Guttridge et al. 2000; Li et al. 2002; Ruan et al. 2002; Arruda et al. 2010). However, weight loss in cancer patients cannot be attributed solely to decreased food intake, since dietary supplements fail to reverse cachexia (Bruera and Sweeney 2002). In contrast, a recent study in mice expressing high levels of the proinflammatory cytokine IL-18 suggests that high caloric feeding in the context of metabolic dysfunction may exacerbate weight loss and cause fatal cachexia (Murphy et al. 2016). In the context of cancer, metabolic dysfunction is caused by deregulated carbohydrate and lipid metabolism.
Altered carbohydrate metabolism in cancer cachexia
Carbohydrate intolerance in cancer patients has long been noted (Rohdenburg et al. 1919). While fasting blood sugar concentration between control and cancer groups did not differ significantly, intravenous glucose tolerance tests showed significantly decreased disappearance of glucose in cancer patients (Bishop and Marks 1959). In the first half of the last century, Cori and Cori (1925) compared glucose levels in the venous blood from the tumor-bearing arm and the unaffected arm of a patient with a sarcoma on the forearm. Glucose levels from the tumor-bearing arm were reduced, thus confirming in vivo the increased rate of tumor glycolysis (Cori and Cori 1925). Since tumor tissue takes up glucose, the decreased disappearance of glucose observed in the tolerance test must be sought in metabolic alterations in the host tissues associated with cancer development. Either increased hepatic glucose production or a decrease in peripheral utilization could account for the reduced glucose tolerance observed in cancer patients. Despite decreased hepatic glycogen stores, endogenous glucose production is increased in cachectic patients due to increased hepatic glucose recycling via lactate, a phenomenon termed the Cori cycle (Holroyde et al. 1984). Apart from these studies, clinical investigations on glucose metabolism in cachectic patients are noticeably thin. While one study suggests that glucose intolerance may worsen with the development of cachexia (Jasani et al. 1978), other studies found that glucose intolerance did not correlate with body weight loss (Yoshikawa et al. 2001; Agustsson et al. 2011). Very recently, elegant genetic studies in the fruit fly Drosophila have identified an important role of insulin signaling in inducing a cachexia-like systemic wasting following transplantation of Drosophila tumors (Figueroa-Clarevega and Bilder 2015; Kwon et al. 2015). Both studies have identified a tumor-secreted factor, ImpL2/IGFBP (an insulin-binding protein and antagonist of insulin/insulin-like growth factor [IGF] signaling), that is responsible for the wasting phenotypes in organs distant from the transplanted tumors (Wagner and Petruzzelli 2015).
Role of lipids, burning fat, and white adipose tissue (WAT) browning
Besides changes in carbohydrate metabolism, the handling of lipids between tissues is severely impaired in cancer patients. The deposition of triglycerides (TGs) in cytoplasmic lipid droplets represents the most efficient form to store lipids in WAT and many other cell types. Already in 1848, the French physiologist Claude Bernard discovered that TGs, commonly called fat, are digested in the gut before they can be absorbed. The hydrolysis of TGs, designated lipolysis, generates glycerol and fatty acids (FAs). The enzymes mediating intracellular lipolysis include adipose TG lipase (ATGL) and the hormone-sensitive lipase (HSL), while LPL is responsible for the hydrolysis of plasma TGs of lipoproteins in the vascular system (Young and Zechner 2013). FA uptake and TG synthesis decline in WAT in murine cancer models, whereas, in human CAC, it is associated with normal lipid synthesis but elevated lipolysis in WAT. This suggests that lipid catabolism is more relevant than inhibition of lipid synthesis for the loss of WAT in CAC (Dahlman et al. 2010). These findings were corroborated in an elegant study demonstrating that WAT lipolysis in cancer patients is increased due to elevated enzyme activities of ATGL and HSL (Das et al. 2011). Importantly, genetic deletion of Atgl in mice prevented increased lipolysis and the reduction of WAT and skeletal muscle mass in certain models of CAC. Similar results were also observed, although to a lesser extent, when HSL was inactivated (Das et al. 2011). Lipolysis in CAC is induced by many serum factors secreted by tumor or host cells, including hormones such as glucocorticoids and catecholamines; cytokines like TNFα, IL-1β, IL-6, prostaglandins; and a zinc–glycoprotein, ZAG, also called lipid-mobilizing factor (Tisdale 2010). How functional lipolysis impacts the development of cancer cachexia is the focus of ongoing investigations in several laboratories (for review, see Tsoli et al. 2015).
While quantitative changes in WAT content during cancer cachexia have long been recognized, only recently a qualitative change in the morphology and function of white adipocytes has been described. During the progression of cancer cachexia in preclinical models, WAT cells gradually convert to brown adipose tissue (BAT)-like cells, also called “beige” cells, in a process termed “browning” (Kir et al. 2014; Petruzzelli et al. 2014). Beige cells are characterized by high mitochondrial content and increased expression of uncoupling protein 1 (UCP1), which is responsible for uncoupling the use of mitochondrial electron transport from ATP synthesis to thermogenesis (Nedergaard and Cannon 2014). The phenomenon of browning was initially described as an adaptive response to prolonged exposure to cold environments (Cousin et al. 1992). When exposed to cold temperatures, mice deficient in the ability to activate thermogenesis rapidly lose core body temperature and are more susceptible to cold-induced damage (Nguyen et al. 2011). The induction of browning in humans was initially hypothesized on the basis of increased fluorodeoxyglucose (FDG) uptake in WAT depots using positron emission tomography (PET) (Nedergaard et al. 2007) and later confirmed at the histological level (Cypess et al. 2009; van Marken Lichtenbelt et al. 2009; Virtanen et al. 2009). Recent investigations have shown that the role of browning is not limited to cold acclimatization. In preclinical models of diet-induced obesity, browning promotes systemic energy expenditure, which results in body weight loss and improved insulin sensitivity. The protection conferred by browning against high-fat diet-induced obesity suggests pharmacological enhancement of browning as a promising therapeutic strategy for metabolic disorders due to excess of nutrients (Feldmann et al. 2009; Yoneshiro et al. 2013). While the effect of browning is identical in both obesity and cancer, the metabolic result is the opposite. Increased lipid mobilization and energy expenditure are favorable in obesity while being deleterious in cancer (Fig. 3). In fact, different from obesity and the metabolic syndrome, browning in the context of cancer exacerbates the metabolic dysfunction, enhancing energy dissipation and contributing to the progression of CAC (Kir et al. 2014; Petruzzelli et al. 2014). Browning in cancer-bearing mice is a systemic event manifested in multiple WAT depots. It precedes the onset of skeletal muscle atrophy and determines a hypermetabolic state characterized by high resting energy expenditure. Notably, browning is not restricted to one experimental model and is not associated with one specific cancer type, since it was documented in complementary model systems, including genetically engineered mouse models (GEMMs), carcinogen-induced cancers, syngeneic transplants of murine cancer cells, and xenogeneic transplants of human cancer tissue (Petruzzelli et al. 2014). Recently, browning of WAT has been shown to take part in the pathogenesis of hypermetabolism commonly observed in other morbid conditions, like post-burn injury, severe adrenergic stress, and kidney failure (Kir et al. 2015; Patsouris et al. 2015; Sidossis et al. 2015). Treatment of mice with a synthetic thyroid hormone receptor agonist induces adaptive thermogenesis in subcutaneous WAT, thus suggesting a role for WAT browning also in hyperthyroidism (Lin et al. 2015).
Figure 3.
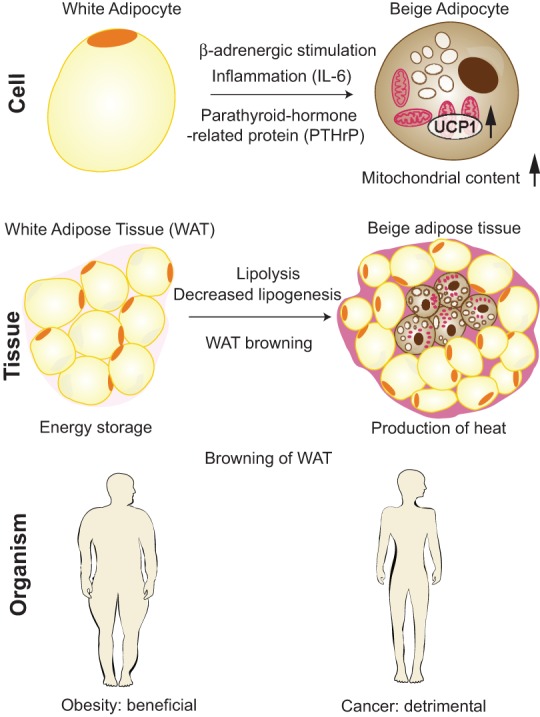
Mechanisms and consequences of WAT browning in cancer cachexia. At the “cell” level, beige adipocytes are induced in WAT by a combination of signaling pathways, including β-adrenergic stimulation, inflammation mediated by IL-6, and the presence of parathyroid-related peptide (PTHrP); as a result, UCP1 levels and mitochondrial content are increased. At the “tissue” level, CAC is associated with the appearance of islets of beige adipocytes in WAT, surrounded by white adipocytes of reduced size due to ongoing lipolysis. WAT browning and lipolysis result in decreased energy storage and increased production of heat. In the context of obesity, WAT browning is beneficial, while in cancer patients, it is detrimental.
Tumors can directly activate thermogenesis in beige cells through the secretion of parathyroid-related peptide (PTHrP), which has been identified in the supernatants from a murine lung carcinoma cell line and shown to drastically induce the expression of UCP1 (Kir et al. 2014). At the molecular level, transformation of white adipocytes into beige cells requires the function of the transcriptional coregulator PRDM16. Interestingly, fat-specific Prdm16-deficient mice challenged in a model of cancer cachexia showed a significant reduction of browning, thermogenic activity, and WAT atrophy. Importantly, injection of cachectic xenotransplant mice with a neutralizing antibody specific for PTHrP was beneficial, reducing the intensity of cancer cachexia and skeletal muscle atrophy. In lung and colorectal cancer patients, higher plasma PTHrP concentrations are associated with increased energy expenditure and enhanced lean tissue wasting, thus confirming the therapeutic potential of inhibiting PTHrP in human cancer (Kir et al. 2014). While treatment of cachectic mice with a PTHrP antibody ameliorated the severity of cachexia, it did not inhibit it completely, thus suggesting that other tumor-derived or host-derived molecules collaborate with PTHrP in the induction of browning and systemic wasting.
Next to direct activation of browning through tumor-derived PTHrP, systemic inflammation and activation of the β-adrenergic pathway represent complementary mechanisms involved in the pathogenesis of browning during CAC (Petruzzelli et al. 2014). Plasma levels of IL-6 are increased in cachectic mice, and genetic blockade of IL-6 by stable incorporation of a shRNA led to a drastic reduction of the severity of cancer cachexia in a xenogeneic cancer model. In addition, IL-6 receptor (IL-6-R) knockout mice implanted with melanoma cells displayed reduced browning when compared with control mice, further corroborating the role of IL-6 in the activation of the thermogenic program in white adipocytes (Petruzzelli et al. 2014). While the direct induction of UCP1 expression by incubation of adipocytes in the presence of recombinant IL-6 is modest, indirect mechanisms are likely to enhance IL-6-induced browning, such as alternative activation of macrophages (Mauer et al. 2014). These cells have been shown to sustain adaptive thermogenesis by means of enhanced recruitment of β-adrenergic fibers (Nguyen et al. 2011). Indeed, macrophages infiltrate the WAT of cachectic mice and express markers of alternative activation. The link between the immune system and adipose tissue biology is further supported by recent investigations showing WAT browning following microbiota depletion (Suarez-Zamorano et al. 2015; Yeoh and Vijay-Kumar 2015). Interestingly, colonization of the intestine by different strains of bacteria has been shown to modulate disease severity and cachexia development in mouse models (Schieber et al. 2015). A role for therapeutic agents targeting intestinal function in CAC remains largely speculative at present (Klein et al. 2013). Whether inhibition of browning may indirectly affect tumor metabolism is not known. Systemic alterations of the metabolism in the host are predicted to affect local metabolic pathways of cancer cells, although more experimental data are needed.
In addition to browning, many studies using murine cancer models have demonstrated that lipolysis induces the activation of interscapular BAT during cancer cachexia, further contributing to energy uncoupling in mitochondria with the subsequent worsening of the negative energy balance (Tsoli and Robertson 2012). BAT has a key role in thermogenesis and energy balance and therefore may well participate in energy expenditure in cancer patients. BAT has been shown to be present in adult humans, and a role for BAT in CAC is possible but is by no means definitively proven (Bauwens et al. 2014).
Muscle wasting in cancer cachexia
CAC is characterized by muscle atrophy, which severely impairs the patient's mobility because of fatigue and weakness (Cohen et al. 2015). Early labeling experiments have shown that different mechanisms cause skeletal muscle atrophy in different conditions. Increased myofibrillar degradation was observed in skeletal muscle atrophy caused by denervation, while a combination of decreased synthesis and increased degradation was responsible for cortisone-induced muscle atrophy (Goldberg and Goodman 1969). In animal models, glucocorticoids did not cause skeletal muscle atrophy at physiological concentrations but only at increased concentrations under pathological conditions (Tomas et al. 1979). Therefore, increased adrenal activity and glucocorticoid levels in cancer patients were hypothesized as likely to be responsible for skeletal muscle-wasting during cancer cachexia. However, adrenalectomy did not prevent skeletal muscle atrophy in tumor-bearing animals, thus arguing against a role for adrenal hyperfunction in muscle atrophy during experimental CAC (Svaninger et al. 1987). Conversely, microscopic examination of skeletal muscle from cancer cachexia patients did not show evidence of degeneration of muscular or intramuscular nerve bundles, thus excluding also a role for denervation (Marin and Denny-Brown 1962). The factors responsible for skeletal muscle atrophy in CAC remained elusive until the important role of cytokines was finally identified.
Administration of TNFα or IL-1 in mice was found to cause loss of skeletal muscle mass similar to what was observed in cachectic cancer patients (Fong et al. 1989). However, while treatment of tumor-bearing rats with anti-cytokine immunoglobulins reduced skeletal muscle atrophy, the protection against systemic wasting was only partial (Costelli et al. 1993). Evidence accumulated pointing to the idea that, in cachexia, the synergistic action of multiple cytokines and other mediators was responsible for skeletal muscle atrophy and likely most of the other components of the wasting syndrome (Argiles and Lopez-Soriano 1999). At the molecular level, the ubiquitin-dependent proteasome pathway (UPP) was identified as one important mechanism underlying muscle breakdown in pathologic states, such as prolonged fasting and metabolic acidosis (Wing and Goldberg 1993; Mitch et al. 1994). Similarly, activation of the UPP was observed in preclinical models of cancer cachexia (Temparis et al. 1994; Baracos et al. 1995). At the genetic level, deletion of muscle-specific E3 ligases Atrogin-1/MAFbx or Murf1 (muscle RING finger protein 1) protected skeletal muscle against experimental atrophy (Bodine et al. 2001). In contrast, muscle-specific activation of NF-kB caused skeletal muscle wasting (Cai et al. 2004). In vitro studies have shown a role for TNFα in the activation of NF-kB, which results in inhibition of myocyte differentiation (Guttridge et al. 2000). In addition, cytokines cause a reduction in myofibrillar protein by decreasing the expression of nuclear transcription factor MyoD and through activation of UPP (Acharyya et al. 2004). A large body of evidence implicates the FOXO family of transcription factors as key mediators of skeletal muscle atrophy during CAC as well as during fasting and other pathological conditions (Egerman and Glass 2014; Cohen et al. 2015). The catabolic effects of FOXO transcription factors are mediated by induction of the atrophy-related ubiquitin ligase Atrogin-1/MAFbx (Sandri et al. 2004) and Murf1 (Zhao et al. 2007; Cohen et al. 2009). A third E3 ligase, Mul1, has been shown to be involved in the reduction of oxidative capacity in cachectic muscles by controlling mitochondrial protein degradation (Lokireddy et al. 2012).
Compelling evidence shows that the atrophy-related genes, also called atrogenes, are directly responsible for skeletal muscle atrophy due to conditions different from CAC, such as denervation, diabetes, or renal failure. Therefore, this points to a concept that a common transcriptional program underlies the loss of skeletal muscle mass independently of the triggering factor (Lecker et al. 2004; Sandri et al. 2006). Skeletal muscle activation of atrogenes in experimental cachexia may also be the result of cross-talk mechanisms between distant organs. As previously noticed, genetic inhibition of lipolysis ameliorates skeletal muscle atrophy in mouse models of CAC (Das et al. 2011). Lipolysis determines an elevated flux of FAs from adipose tissue, and increased FA uptake in the skeletal muscle leads to ceramide synthesis, reduced mTOR activity, and Atrogin and Murf expression (Corcoran et al. 2007; De Larichaudy et al. 2012). In this regard, intramyocellar lipid droplets have been described in skeletal muscle of cancer patients, and its overall content was associated with the extent of body weight loss (Stephens et al. 2011).
While there is considerable experimental evidence for the contribution of atrophy-related UPP in preclinical models, its direct role in human disease and human CAC in particular is, at present, controversial. Conflicting evidence comes from studies that have measured the expression levels of the different UPP components in cancer cachexia patients. Arguing against a direct role, individual components of the UPP were actually found to be unchanged or even down-regulated in cancer patients with suppression of both anabolic and catabolic processes, indicative of reduced muscle turnover that was restored to normal levels following tumor resection (Stephens et al. 2010; Gallagher et al. 2012). On the contrary, different studies reported an increase in the expression levels of proteasome subunits in skeletal muscle of cancer patients with weight loss (Williams et al. 1999; Khal et al. 2005). Besides overexpression of the ubiquitin gene, direct measurement of the proteasome proteolytic activity showed enhancement in skeletal muscles of patients with gastric cancer when compared with noncancer surgical controls and was associated with advanced tumor stage and poor nutritional status (Bossola et al. 2003). These conflicting data comparing animal models and patients with CAC may be due to differences in timing of examination of the skeletal muscle. The analysis in rodents was performed during or at the end of rapid skeletal muscle wasting, while, in cachectic humans, it was performed in the final stage that follows the period of dramatic wasting. It has been shown that changes in expression levels of atrogenes are maximal during the periods of rapid changes in skeletal muscle mass, while further weight loss is associated with reduced gene expression (Khal et al. 2005). Multiple time points during skeletal muscle atrophy in human cachexia must be measured before conclusions can be drawn. Additional mechanisms of muscle atrophy in cachexia have been suggested, including activation of the JAK/STAT3 pathway (Bonetto et al. 2012; Shum and Polly 2012), induction of apoptosis (He et al. 2014), mitochondrial dysfunction (White et al. 2011), and the direct effect of cancer chemotherapy (Le Bricon et al. 1995).
Besides the factors responsible for skeletal muscle atrophy, studies on factors relevant for muscle hypertrophy have also provided important insights into the mechanisms underlying muscle wasting in CAC. Insulin is the main anabolic factor opposing the catabolic effects of glucocorticoids, and the absence of insulin in rats contributes to skeletal muscle atrophy (Price et al. 1996). At the molecular level, IGF-1 activates insulin receptor substrate 1, which signals through PI3K–AKT to induce protein synthesis by activating mTOR (Rommel et al. 2001). Skeletal muscle hypertrophy is also observed in the presence of inactivating mutations in Myostatin (Schuelke et al. 2004), while forced expression of Myostatin causes muscle atrophy in adult mice (Zimmers et al. 2002). The muscle hypertrophy observed in myostatin-deficient mice is abolished after inhibition of bone morphogenetic protein (BMP) signaling, which results in up-regulation of the muscle ubiquitin ligase of the SCF complex in atrophy-1 (MUSA1) (Sartori et al. 2013). Myostatin and Activin are members of the transforming growth factor β (TGFβ) family that were shown to be involved in skeletal muscle atrophy by binding to the Myostatin/Activin type II receptor B (ActRIIB) (Benny Klimek et al. 2010). Interestingly, expression of a dominant-negative ActRIIB in transgenic mice results in skeletal muscle hypertrophy (Lee and McPherron 2001). Furthermore, expression of Myostatin is increased upon inflammatory signaling, whereas it inhibits myoblast differentiation and increases Foxo activation and the expression of ubiquitin ligases (Sartori et al. 2009; Trendelenburg et al. 2009). A recently identified PGC1α isoform, Pgc1α4, has been shown to be highly expressed in exercised muscle and was able to prevent skeletal muscle atrophy by repressing Myostatin activity. Notably, mice with skeletal muscle expression of Pgc1α4 were protected from CAC (Ruas et al. 2012). As an additional mechanism for skeletal muscle dysfunction in cancer, TGFβ release from bone metastasis has been demonstrated to lower intracellular calcium signaling and reduce the force of muscle contraction (Waning et al. 2015).
From a therapeutic perspective, recent clinical trials have provided proof of principle that it is possible to promote skeletal muscle anabolism in cancer patients. A high-protein diet supplemented with leucine has been shown to increase muscle fractional synthetic rate in a small randomized trial in cancer patients (Deutz et al. 2011). However, it has been reported that leucine supplementation increases pancreatic cancer growth in mice, a mechanism mediated by activation of mTOR (Liu et al. 2014). The landmark study by Zhou et al. (2010) has shown that pharmacological blockade of ActRIIB in mouse models of CAC ameliorates skeletal muscle atrophy and prevents atrophy of cardiac muscle. Importantly, ActRIIB blockade significantly prolonged survival even in the absence of direct effects on tumor growth and cytokine secretion. At present, it is not clear whether Myostatin inhibition may also ameliorate skeletal muscle atrophy by direct stimulation of stem cell proliferation. Protection against skeletal muscle atrophy and regrowth of skeletal muscle myocytes are observed after ActRIIB blockade, although a causative role of Myostatin inhibition has yet to be proven. Impaired regenerative capacity of myogenic cells has been recently described in CAC, a process mediated by NF-kB-dependent expression of the self-renewing factor Pax7 (He et al. 2013). Furthermore, inhibition of ActRIIB by a humanized monoclonal antibody has been shown to increase skeletal muscle mass and prevent glucocorticoid-induced atrophy in mice (Lach-Trifilieff et al. 2014). The beneficial effects of inhibiting the Myostatin/Activin pathway is not limited to ameliorating skeletal muscle atrophy but was also shown to improve other pathological conditions in preclinical models, such as insulin resistance and systemic inflammation. The translational potential of Myostatin/Activin antagonism is currently being evaluated in multiple clinical settings (Han et al. 2013; Cohen et al. 2015). Implementation of these findings in clinical practice is anticipated to potentially ameliorate the prognosis of cancer patients.
The role of the liver in cancer cachexia
Next to WAT and skeletal muscle, the liver is of primary importance in the control of systemic metabolism. However, the nature and extent of liver damage during CAC has received little attention. Similarly, the contribution of the liver to the metabolic dysfunction observed in cachexia is currently poorly characterized. Enhanced liver inflammation during CAC is suggested by increased infiltration of macrophages in the livers of pancreatic cancer patients with cachexia when compared with pancreatic cancer patients without cachexia (Martignoni et al. 2009). Activated macrophages in the liver parenchyma may provide a local source of IL-6 production, which stimulates the synthesis of hepatic acute-phase protein (Castell et al. 1989). Preclinical investigations have shown that hepatic oxidative phosphorylation is reduced in a rat model of peritoneal carcinosis, concomitant with increased energy wasting and production of reactive oxygen species (Dumas et al. 2010). Furthermore, clinical investigations have shown that hepatic gluconeogenesis is increased in cancer patients (Yoshikawa et al. 1999). Last, hepatic steatosis has been documented in CAC patients (Teli et al. 1995). At the molecular level, hepatic gene expression of the transcription factor TGFβ1-stimulated clone 22 D4 (TSC22D4) is increased in experimental cachexia and correlates with the degree of systemic wasting (Jones et al. 2013). Gene expression levels of the nuclear receptor cofactor receptor-interacting protein 140 (RIP140) are also increased in CAC and may contribute to liver steatosis by preventing the release of TG stores (Berriel Diaz et al. 2008).
Endocrine routes to cancer cachexia
Activation of neuroendocrine responses plays a major role in CAC (Fearon et al. 2012). The role of the hypothalamus in cachexia has been the focus of intense investigations due to the critical function that this gland has in the central control of food intake and appetite. Cytokine expression in the brain is very low under physiological conditions but is strongly induced in response to peripheral inflammation and is prominent in the hypothalamus (Grossberg et al. 2009). Feed-forward mechanisms amplify and maintain the inflammatory response in the brain through local production of cytokines and neurotransmitters. Neurons in the arcuate nucleus of the hypothalamus and in the nucleus tractus solitarus of the brainstem compose the central melanocortin system (Fan et al. 1997), and inhibition of this neural network has been demonstrated to alleviate the severity of CAC in preclinical models (Wisse et al. 2001; Cheung et al. 2005). For instance, treatment with the gastrointestinal hormone ghrelin inhibits the central melanocortin system and reduces hypothalamic inflammation, resulting in weight gain and increased lean body mass in tumor-implanted rats (DeBoer et al. 2007). The central mechanisms responsible for cytokine-induced body weight loss include reduction of food intake and increased metabolic rate through activation of thermogenesis (Li et al. 2002; Arruda et al. 2010). Less is known on the role of the hypothalamus in the pathogenesis of endocrine dysregulation observed in an organism affected by cancer, although a potential contribution of the hypothalamic–pituitary–adrenal axis is suspected. Chronically elevated IL-6 levels in the brain have been shown to activate the hypothalamic–pituitary–adrenal axis and cause adrenal gland hyperplasia (Raber et al. 1997). Furthermore, increased serum levels of hormones from the cortical adrenal gland (cortisol) and medulla (norepinephrine and epinephrine) have been reported in patients with cachexia associated with chronic heart failure, a condition termed cardiac cachexia (Anker and Coats 1999).
Conclusions and perspectives
Our understanding of CAC has changed dramatically over the past three decades (Fig. 4). It suffices to look at the differences between reviews published a few decades ago to realize the conceptual leap forward. In 1977, “cancer aggression” had a minor metabolic component for cachexia development, which was caused solely by the cancer tissue (Costa 1977). The present description envisions CAC as a complex and multifactorial syndrome resulting from the interaction and mutual effects of the tumor and host tissues (Fearon et al. 2012). Experiments in animal models proved instrumental in revealing the mechanisms by which the tumor perturbs host homeostasis—mechanisms that reach far beyond reduction of food intake or local damage at the site of tumor growth. Studies in preclinical GEMMs helped to define the molecular mechanisms involved in key manifestations of systemic wasting. During the past years, many scientists thought that the basic pathological events had been characterized and that the responsible factors had been enumerated. The wealth of knowledge generated on the molecular mechanisms underlying CAC pathophysiology has paved the way to novel therapeutic approaches, and new candidate molecules hold promise for clinical use (Table 1). However, to date, the therapeutic application of basic discoveries has proven elusive (Lok 2015), and current therapeutic management of cachectic patients is palliative, based on appetite improvement and best supportive care (Ma et al. 2014).
Figure 4.
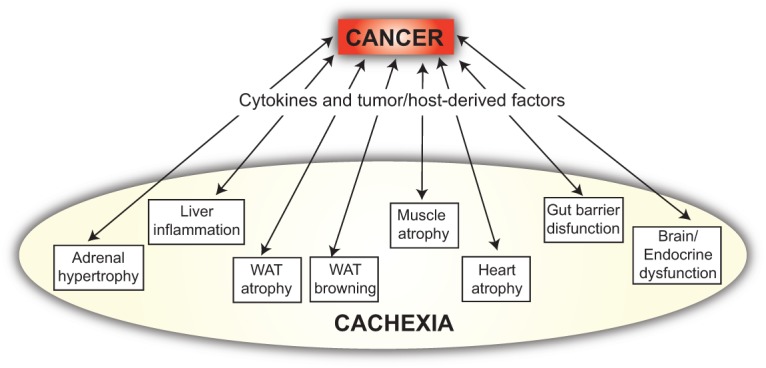
Conceptual evolution of the understanding of cancer cachexia. The scheme depicts the way we envision multifactorial cancer cachexia in 2015, involving reciprocal compounding interactions between the tumor and the organism, which result in inflammatory and metabolic changes distant from the pathological sites of tumor growth. This way is very different from the unidirectional way that “cancer aggression” was viewed decades ago (Costa 1977).
Table 1.
Novel pharmacological agents and potential future treatment strategies for CAC

We now know that prevention and treatment of CAC needs to be multifactorial, as targeting single mediators has repeatedly failed. To increase the chances of success, treatment has to start early in the clinical history of cancer patients, before obvious evidence of metabolic dysfunction. A phase of adapting and adjusting must occur between the tumor and the host in the “unaffected” weight-stable cancer patient. Characterization of these early events at multiple organ levels is essential for understanding the pathophysiology of the host–tumor interaction, including the neuroendocrine axis (Lainscak et al. 2008). Similarly limited is our knowledge of the metabolic cross-talk between the tumor and the host, which is the starting point for understanding the progression from a local malignant growth to a systemic disease. The characterization of these events requires a new level of “systemic approaches” to design the right experiments for a scientific field that has historically been studying one phenotype or organ and one tumor at the time. Future investigations focusing more on the “whole” and less on the “parts” will go beyond the dichotomy tumor–organism and provide the conceptual framework to devise new therapeutic strategies for treating the organism in addition to just killing the tumor. Such holistic approaches will likely lead to a better understanding of the metabolic dysfunction in cancer cachexia for the benefit of cancer patients.
Acknowledgments
We are very grateful to Dr. Doug Hanahan, Dr. Anna Hupalowska, Dr. Graham Robertson, Dr. Martina Schweiger, Dr. Marta Shahbazi, Dr. Rudolf Zechner, and our laboratory colleagues Dr. Latifa Bakiri, Dr. Oezge Uluckan, and Dr. Sebastian Hasenfuss for critical reading and comments on our manuscript. E.F.W. is supported by a grant from the Spanish Ministry of Economy (BFU2012-40230) and a European Research Council Advanced Grant (ERC FCK/2008/37).
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.276733.115.
References
- Acharyya S, Ladner KJ, Nelsen LL, Damrauer J, Reiser PJ, Swoap S, Guttridge DC. 2004. Cancer cachexia is regulated by selective targeting of skeletal muscle gene products. J Clin Invest 114: 370–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agustsson T, D'Souza MA, Nowak G, Isaksson B. 2011. Mechanisms for skeletal muscle insulin resistance in patients with pancreatic ductal adenocarcinoma. Nutrition 27: 796–801. [DOI] [PubMed] [Google Scholar]
- Anker SD, Coats AJ. 1999. Cardiac cachexia: a syndrome with impaired survival and immune and neuroendocrine activation. Chest 115: 836–847. [DOI] [PubMed] [Google Scholar]
- Argiles JM, Lopez-Soriano FJ. 1999. The role of cytokines in cancer cachexia. Med Res Rev 19: 223–248. [DOI] [PubMed] [Google Scholar]
- Arruda AP, Milanski M, Romanatto T, Solon C, Coope A, Alberici LC, Festuccia WT, Hirabara SM, Ropelle E, Curi R, et al. 2010. Hypothalamic actions of tumor necrosis factor α provide the thermogenic core for the wastage syndrome in cachexia. Endocrinology 151: 683–694. [DOI] [PubMed] [Google Scholar]
- Balkwill F, Osborne R, Burke F, Naylor S, Talbot D, Durbin H, Tavernier J, Fiers W. 1987. Evidence for tumour necrosis factor/cachectin production in cancer. Lancet 2: 1229–1232. [DOI] [PubMed] [Google Scholar]
- Baracos V, Rodemann HP, Dinarello CA, Goldberg AL. 1983. Stimulation of muscle protein degradation and prostaglandin E2 release by leukocytic pyrogen (interleukin-1). A mechanism for the increased degradation of muscle proteins during fever. N Engl J Med 308: 553–558. [DOI] [PubMed] [Google Scholar]
- Baracos VE, DeVivo C, Hoyle DH, Goldberg AL. 1995. Activation of the ATP-ubiquitin-proteasome pathway in skeletal muscle of cachectic rats bearing a hepatoma. Am J Physiol 268: E996–E1006. [DOI] [PubMed] [Google Scholar]
- Bauwens M, Wierts R, van Royen B, Bucerius J, Backes W, Mottaghy F, Brans B. 2014. Molecular imaging of brown adipose tissue in health and disease. Eur J Nucl Med Mol Imaging 41: 776–791. [DOI] [PubMed] [Google Scholar]
- Bayliss TJ, Smith JT, Schuster M, Dragnev KH, Rigas JR. 2011. A humanized anti-IL-6 antibody (ALD518) in non-small cell lung cancer. Expert Opin Biol Ther 11: 1663–1668. [DOI] [PubMed] [Google Scholar]
- Begg RW, Dickinson TE. 1951. Systemic effects of tumors in force-fed rats. Cancer Res 11: 409–412. [PubMed] [Google Scholar]
- Benny Klimek ME, Aydogdu T, Link MJ, Pons M, Koniaris LG, Zimmers TA. 2010. Acute inhibition of myostatin-family proteins preserves skeletal muscle in mouse models of cancer cachexia. Biochem Biophys Res Commun 391: 1548–1554. [DOI] [PubMed] [Google Scholar]
- Berriel Diaz M, Krones-Herzig A, Metzger D, Ziegler A, Vegiopoulos A, Klingenspor M, Muller-Decker K, Herzig S. 2008. Nuclear receptor cofactor receptor interacting protein 140 controls hepatic triglyceride metabolism during wasting in mice. Hepatology 48: 782–791. [DOI] [PubMed] [Google Scholar]
- Beutler B, Cerami A. 1986. Cachectin and tumour necrosis factor as two sides of the same biological coin. Nature 320: 584–588. [DOI] [PubMed] [Google Scholar]
- Beutler B, Mahoney J, Le Trang N, Pekala P, Cerami A. 1985. Purification of cachectin, a lipoprotein lipase-suppressing hormone secreted by endotoxin-induced RAW 264.7 cells. J Exp Med 161: 984–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop JS, Marks PA. 1959. Studies on carbohydrate metabolism in patients with neoplastic disease. II. Response to insulin administration. J Clin Invest 38: 668–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, et al. 2001. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294: 1704–1708. [DOI] [PubMed] [Google Scholar]
- Bonetto A, Aydogdu T, Jin X, Zhang Z, Zhan R, Puzis L, Koniaris LG, Zimmers TA. 2012. JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL-6 and in experimental cancer cachexia. Am J Physiol Endocrinol Metab 303: E410–E421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossola M, Muscaritoli M, Costelli P, Grieco G, Bonelli G, Pacelli F, Rossi Fanelli F, Doglietto GB, Baccino FM. 2003. Increased muscle proteasome activity correlates with disease severity in gastric cancer patients. Ann Surg 237: 384–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruera E, Sweeney C. 2002. Palliative care models: international perspective. J Palliat Med 5: 319–327. [DOI] [PubMed] [Google Scholar]
- Cahlin C, Korner A, Axelsson H, Wang W, Lundholm K, Svanberg E. 2000. Experimental cancer cachexia: the role of host-derived cytokines interleukin (IL)-6, IL-12, interferon-γ, and tumor necrosis factor α evaluated in gene knockout, tumor-bearing mice on C57 Bl background and eicosanoid-dependent cachexia. Cancer Res 60: 5488–5493. [PubMed] [Google Scholar]
- Cai D, Frantz JD, Tawa NE Jr, Melendez PA, Oh BC, Lidov HG, Hasselgren PO, Frontera WR, Lee J, Glass DJ, et al. 2004. IKKβ/NF-κB activation causes severe muscle wasting in mice. Cell 119: 285–298. [DOI] [PubMed] [Google Scholar]
- Castell JV, Gomez-Lechon MJ, David M, Andus T, Geiger T, Trullenque R, Fabra R, Heinrich PC. 1989. Interleukin-6 is the major regulator of acute phase protein synthesis in adult human hepatocytes. FEBS Lett 242: 237–239. [DOI] [PubMed] [Google Scholar]
- Cerami A, Ikeda Y, Le Trang N, Hotez PJ, Beutler B. 1985. Weight loss associated with an endotoxin-induced mediator from peritoneal macrophages: the role of cachectin (tumor necrosis factor). Immunol Lett 11: 173–177. [DOI] [PubMed] [Google Scholar]
- Cheung W, Yu PX, Little BM, Cone RD, Marks DL, Mak RH. 2005. Role of leptin and melanocortin signaling in uremia-associated cachexia. J Clin Invest 115: 1659–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Brault JJ, Gygi SP, Glass DJ, Valenzuela DM, Gartner C, Latres E, Goldberg AL. 2009. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. J Cell Biol 185: 1083–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Nathan JA, Goldberg AL. 2015. Muscle wasting in disease: molecular mechanisms and promising therapies. Nat Rev Drug Discov 14: 58–74. [DOI] [PubMed] [Google Scholar]
- Corcoran MP, Lamon-Fava S, Fielding RA. 2007. Skeletal muscle lipid deposition and insulin resistance: effect of dietary fatty acids and exercise. Am J Clin Nutr 85: 662–677. [DOI] [PubMed] [Google Scholar]
- Cori CF, Cori GT. 1925. The carbohydrate metabolism of tumors: II. Changes in the sugar, lactic acid, and CO2-combining power of blood passing through a tumor. J Biol Chem 65: 397–405. [Google Scholar]
- Costa G. 1977. Cachexia, the metabolic component of neoplastic diseases. Cancer Res 37: 2327–2335. [PubMed] [Google Scholar]
- Costa G, Holland JF. 1962. Effects of Krebs-2 carcinoma on the lipide metabolism of male Swiss mice. Cancer Res 22: 1081–1083. [PubMed] [Google Scholar]
- Costelli P, Carbo N, Tessitore L, Bagby GJ, Lopez-Soriano FJ, Argiles JM, Baccino FM. 1993. Tumor necrosis factor-α mediates changes in tissue protein turnover in a rat cancer cachexia model. J Clin Invest 92: 2783–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousin B, Cinti S, Morroni M, Raimbault S, Ricquier D, Penicaud L, Casteilla L. 1992. Occurrence of brown adipocytes in rat white adipose tissue: molecular and morphological characterization. J Cell Sci 103: 931–942. [DOI] [PubMed] [Google Scholar]
- Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, Kuo FC, Palmer EL, Tseng YH, Doria A, et al. 2009. Identification and importance of brown adipose tissue in adult humans. N Engl J Med 360: 1509–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlman I, Mejhert N, Linder K, Agustsson T, Mutch DM, Kulyte A, Isaksson B, Permert J, Petrovic N, Nedergaard J, et al. 2010. Adipose tissue pathways involved in weight loss of cancer cachexia. Br J Cancer 102: 1541–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das SK, Eder S, Schauer S, Diwoky C, Temmel H, Guertl B, Gorkiewicz G, Tamilarasan KP, Kumari P, Trauner M, et al. 2011. Adipose triglyceride lipase contributes to cancer-associated cachexia. Science 333: 233–238. [DOI] [PubMed] [Google Scholar]
- DeBoer MD, Zhu XX, Levasseur P, Meguid MM, Suzuki S, Inui A, Taylor JE, Halem HA, Dong JZ, Datta R, et al. 2007. Ghrelin treatment causes increased food intake and retention of lean body mass in a rat model of cancer cachexia. Endocrinology 148: 3004–3012. [DOI] [PubMed] [Google Scholar]
- De Larichaudy J, Zufferli A, Serra F, Isidori AM, Naro F, Dessalle K, Desgeorges M, Piraud M, Cheillan D, Vidal H, et al. 2012. TNF-α- and tumor-induced skeletal muscle atrophy involves sphingolipid metabolism. Skelet Muscle 2: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutz NE, Safar A, Schutzler S, Memelink R, Ferrando A, Spencer H, van Helvoort A, Wolfe RR. 2011. Muscle protein synthesis in cancer patients can be stimulated with a specially formulated medical food. Clin Nutr 30: 759–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobs AS, Boccia RV, Croot CC, Gabrail NY, Dalton JT, Hancock ML, Johnston MA, Steiner MS. 2013. Effects of enobosarm on muscle wasting and physical function in patients with cancer: a double-blind, randomised controlled phase 2 trial. Lancet Oncol 14: 335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan H. 1954. Malignant cachexia. Proc R Soc Med 47: 27–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumas JF, Goupille C, Julienne CM, Pinault M, Chevalier S, Bougnoux P, Servais S, Couet C. 2010. Efficiency of oxidative phosphorylation in liver mitochondria is decreased in a rat model of peritoneal carcinosis. J Hepatol 54: 320–327. [DOI] [PubMed] [Google Scholar]
- Ebner N, Steinbeck L, Doehner W, Anker SD, von Haehling S. 2014. Highlights from the 7th cachexia conference: muscle wasting pathophysiological detection and novel treatment strategies. J Cachexia Sarcopenia Muscle 5: 27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Editors. 1929. Cachexia in cancer. N Engl J Med 201: 956–957. [Google Scholar]
- E.F.B. 1909. The natural history of cancer, with special reference to its causation and prevention lectures on the pathology of cancer. Nature 79: 391–392. [Google Scholar]
- Egerman MA, Glass DJ. 2014. Signaling pathways controlling skeletal muscle mass. Crit Rev Biochem Mol Biol 49: 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans WK, Makuch R, Clamon GH, Feld R, Weiner RS, Moran E, Blum R, Shepherd FA, Jeejeebhoy KN, DeWys WD. 1985. Limited impact of total parenteral nutrition on nutritional status during treatment for small cell lung cancer. Cancer Res 45: 3347–3353. [PubMed] [Google Scholar]
- Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. 1997. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature 385: 165–168. [DOI] [PubMed] [Google Scholar]
- Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, Jatoi A, Loprinzi C, MacDonald N, Mantovani G, et al. 2011. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol 12: 489–495. [DOI] [PubMed] [Google Scholar]
- Fearon KC, Glass DJ, Guttridge DC. 2012. Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab 16: 153–166. [DOI] [PubMed] [Google Scholar]
- Fearon K, Arends J, Baracos V. 2013. Understanding the mechanisms and treatment options in cancer cachexia. Nat Rev Clin Oncol 10: 90–99. [DOI] [PubMed] [Google Scholar]
- Feldmann HM, Golozoubova V, Cannon B, Nedergaard J. 2009. UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab 9: 203–209. [DOI] [PubMed] [Google Scholar]
- Figueroa-Clarevega A, Bilder D. 2015. Malignant Drosophila tumors interrupt insulin signaling to induce cachexia-like wasting. Dev Cell 33: 47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong Y, Moldawer LL, Marano M, Wei H, Barber A, Manogue K, Tracey KJ, Kuo G, Fischman DA, Cerami A, et al. 1989. Cachectin/TNF or IL-1 α induces cachexia with redistribution of body proteins. Am J Physiol 256: R659–R665. [DOI] [PubMed] [Google Scholar]
- Fransen L, Muller R, Marmenout A, Tavernier J, Van der Heyden J, Kawashima E, Chollet A, Tizard R, Van Heuverswyn H, Van Vliet A, et al. 1985. Molecular cloning of mouse tumour necrosis factor cDNA and its eukaryotic expression. Nucleic Acids Res 13: 4417–4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher IJ, Stephens NA, MacDonald AJ, Skipworth RJ, Husi H, Greig CA, Ross JA, Timmons JA, Fearon KC. 2012. Suppression of skeletal muscle turnover in cancer cachexia: evidence from the transcriptome in sequential human muscle biopsies. Clin Cancer Res 18: 2817–2827. [DOI] [PubMed] [Google Scholar]
- Garcia JM, Boccia RV, Graham CD, Yan Y, Duus EM, Allen S, Friend J. 2015. Anamorelin for patients with cancer cachexia: an integrated analysis of two phase 2, randomised, placebo-controlled, double-blind trials. Lancet Oncol 16: 108–116. [DOI] [PubMed] [Google Scholar]
- Goldberg AL, Goodman HM. 1969. Relationship between cortisone and muscle work in determining muscle size. J Physiol 200: 667–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenstein JP. 1947. Biochemistry of Cancer. Academic Press, Inc, New York. [Google Scholar]
- Grossberg AJ, Scarlett JM, Zhu X, Bowe DD, Batra AK, Braun TP, Marks DL. 2009. Arcuate nucleus proopiomelanocortin neurons mediate the acute anorectic actions of leukemia inhibitory factor via gp130. Endocrinology 151: 606–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttridge DC, Mayo MW, Madrid LV, Wang CY, Baldwin AS Jr. 2000. NF-κB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science 289: 2363–2366. [DOI] [PubMed] [Google Scholar]
- Han HQ, Zhou X, Mitch WE, Goldberg AL. 2013. Myostatin/activin pathway antagonism: molecular basis and therapeutic potential. Int J Biochem Cell Biol 45: 2333–2347. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144: 646–674. [DOI] [PubMed] [Google Scholar]
- He WA, Berardi E, Cardillo VM, Acharyya S, Aulino P, Thomas-Ahner J, Wang J, Bloomston M, Muscarella P, Nau P, et al. 2013. NF-κB-mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J Clin Invest 123: 4821–4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He WA, Calore F, Londhe P, Canella A, Guttridge DC, Croce CM. 2014. Microvesicles containing miRNAs promote muscle cell death in cancer cachexia via TLR7. Proc Natl Acad Sci 111: 4525–4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holroyde CP, Skutches CL, Boden G, Reichard GA. 1984. Glucose metabolism in cachectic patients with colorectal cancer. Cancer Res 44: 5910–5913. [PubMed] [Google Scholar]
- Hong DS, Hui D, Bruera E, Janku F, Naing A, Falchook GS, Piha-Paul S, Wheler JJ, Fu S, Tsimberidou AM, et al. 2014. MABp1, a first-in-class true human antibody targeting interleukin-1α in refractory cancers: an open-label, phase 1 dose-escalation and expansion study. Lancet Oncol 15: 656–666. [DOI] [PubMed] [Google Scholar]
- Jasani B, Donaldson LJ, Ratcliffe JG, Sokhi GS. 1978. Mechanism of impaired glucose tolerance in patients with neoplasia. Br J Cancer 38: 287–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston AJ, Murphy KT, Jenkinson L, Laine D, Emmrich K, Faou P, Weston R, Jayatilleke KM, Schloegel J, Talbo G, et al. 2015. Targeting of Fn14 prevents cancer-induced cachexia and prolongs survival. Cell 162: 1365–1378. [DOI] [PubMed] [Google Scholar]
- Jones A, Friedrich K, Rohm M, Schafer M, Algire C, Kulozik P, Seibert O, Muller-Decker K, Sijmonsma T, Strzoda D, et al. 2013. TSC22D4 is a molecular output of hepatic wasting metabolism. EMBO Mol Med 5: 294–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami M, Cerami A. 1981. Studies of endotoxin-induced decrease in lipoprotein lipase activity. J Exp Med 154: 631–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khal J, Hine AV, Fearon KC, Dejong CH, Tisdale MJ. 2005. Increased expression of proteasome subunits in skeletal muscle of cancer patients with weight loss. Int J Biochem Cell Biol 37: 2196–2206. [DOI] [PubMed] [Google Scholar]
- Kir S, White JP, Kleiner S, Kazak L, Cohen P, Baracos VE, Spiegelman BM. 2014. Tumour-derived PTH-related protein triggers adipose tissue browning and cancer cachexia. Nature 513: 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kir S, Komaba H, Garcia AP, Economopoulos KP, Liu W, Lanske B, Hodin RA, Spiegelman BM. 2015. PTH/PTHrP receptor mediates cachexia in models of kidney failure and cancer. Cell Metab 10.1016/j.cmet.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein GL, Petschow BW, Shaw AL, Weaver E. 2013. Gut barrier dysfunction and microbial translocation in cancer cachexia: a new therapeutic target. Curr Opin Support Palliat Care 7: 361–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon Y, Song W, Droujinine IA, Hu Y, Asara JM, Perrimon N. 2015. Systemic organ wasting induced by localized expression of the secreted insulin/IGF antagonist ImpL2. Dev Cell 33: 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lach-Trifilieff E, Minetti GC, Sheppard K, Ibebunjo C, Feige JN, Hartmann S, Brachat S, Rivet H, Koelbing C, Morvan F, et al. 2014. An antibody blocking activin type II receptors induces strong skeletal muscle hypertrophy and protects from atrophy. Mol Cell Biol 34: 606–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lainscak M, Filippatos GS, Gheorghiade M, Fonarow GC, Anker SD. 2008. Cachexia: common, deadly, with an urgent need for precise definition and new therapies. Am J Cardiol 101: 8E–10E. [DOI] [PubMed] [Google Scholar]
- Le Bricon T, Gugins S, Cynober L, Baracos VE. 1995. Negative impact of cancer chemotherapy on protein metabolism in healthy and tumor-bearing rats. Metabolism 44: 1340–1348. [DOI] [PubMed] [Google Scholar]
- Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, Price SR, Mitch WE, Goldberg AL. 2004. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J 18: 39–51. [DOI] [PubMed] [Google Scholar]
- Lee SJ, McPherron AC. 2001. Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci 98: 9306–9311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Klein RL, Matheny M, King MA, Meyer EM, Scarpace PJ. 2002. Induction of uncoupling protein 1 by central interleukin-6 gene delivery is dependent on sympathetic innervation of brown adipose tissue and underlies one mechanism of body weight reduction in rats. Neuroscience 115: 879–889. [DOI] [PubMed] [Google Scholar]
- Lin JZ, Martagon AJ, Cimini SL, Gonzalez DD, Tinkey DW, Biter A, Baxter JD, Webb P, Gustafsson JA, Hartig SM, et al. 2015. pharmacological activation of thyroid hormone receptors elicits a functional conversion of white to brown fat. Cell Rep 13: 1528–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu KA, Lashinger LM, Rasmussen AJ, Hursting SD. 2014. Leucine supplementation differentially enhances pancreatic cancer growth in lean and overweight mice. Cancer Metab 2: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lok C. 2015. Cachexia: the last illness. Nature 528: 182–183. [DOI] [PubMed] [Google Scholar]
- Lokireddy S, Wijesoma IW, Teng S, Bonala S, Gluckman PD, McFarlane C, Sharma M, Kambadur R. 2012. The ubiquitin ligase Mul1 induces mitophagy in skeletal muscle in response to muscle-wasting stimuli. Cell Metab 16: 613–624. [DOI] [PubMed] [Google Scholar]
- Ma JD, Heavey SF, Revta C, Roeland EJ. 2014. Novel investigational biologics for the treatment of cancer cachexia. Expert Opin Biol Ther 14: 1113–1120. [DOI] [PubMed] [Google Scholar]
- Marin OS, Denny-Brown D. 1962. Changes in skeletal muscle associated with cachexia. Am J Pathol 41: 23–39. [PMC free article] [PubMed] [Google Scholar]
- Martignoni ME, Dimitriu C, Bachmann J, Krakowski-Rosen H, Ketterer K, Kinscherf R, Friess H. 2009. Liver macrophages contribute to pancreatic cancer-related cachexia. Oncol Rep 21: 363–369. [PubMed] [Google Scholar]
- Mauer J, Chaurasia B, Goldau J, Vogt MC, Ruud J, Nguyen KD, Theurich S, Hausen AC, Schmitz J, Bronneke HS, et al. 2014. Signaling by IL-6 promotes alternative activation of macrophages to limit endotoxemia and obesity-associated resistance to insulin. Nat Immunol 15: 423–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitch WE, Medina R, Grieber S, May RC, England BK, Price SR, Bailey JL, Goldberg AL. 1994. Metabolic acidosis stimulates muscle protein degradation by activating the adenosine triphosphate-dependent pathway involving ubiquitin and proteasomes. J Clin Invest 93: 2127–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy AJ, Kraakman MJ, Kammoun HL, Dragoljevic D, Lee MK, Lawlor KE, Wentworth JM, Vasanthakumar A, Gerlic M, Whitehead LW, et al. 2016. IL-18 production from the NLRP1 inflammasome prevents obesity and metabolic syndrome. Cell Metab 23: 155–164. [DOI] [PubMed] [Google Scholar]
- Nedergaard J, Cannon B. 2014. The browning of white adipose tissue: some burning issues. Cell Metab 20: 396–407. [DOI] [PubMed] [Google Scholar]
- Nedergaard J, Bengtsson T, Cannon B. 2007. Unexpected evidence for active brown adipose tissue in adult humans. Am J Physiol Endocrinol Metab 293: E444–E452. [DOI] [PubMed] [Google Scholar]
- Nguyen KD, Qiu Y, Cui X, Goh YP, Mwangi J, David T, Mukundan L, Brombacher F, Locksley RM, Chawla A. 2011. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature 480: 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliff A, Defeo-Jones D, Boyer M, Martinez D, Kiefer D, Vuocolo G, Wolfe A, Socher SH. 1987. Tumors secreting human TNF/cachectin induce cachexia in mice. Cell 50: 555–563. [DOI] [PubMed] [Google Scholar]
- Paspaliaris V, Langan B, DeAndrea R, Wood J, Tsouvelekas A, Demosthenes B. 2011. Phase I/II study of IP-1510 a novel interleukin-1 receptor antagonist in the management of cancer-related cachexia. J Cachexia Sarcopenia Muscle 2: 209–261.22476871 [Google Scholar]
- Patsouris D, Qi P, Abdullahi A, Stanojcic M, Chen P, Parousis A, Amini-Nik S, Jeschke MG. 2015. Burn induces browning of the subcutaneous white adipose tissue in mice and humans. Cell Rep 13: 1538–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penna F, Minero VG, Costamagna D, Bonelli G, Baccino FM, Costelli P. 2010. Anti-cytokine strategies for the treatment of cancer-related anorexia and cachexia. Expert Opin Biol Ther 10: 1241–1250. [DOI] [PubMed] [Google Scholar]
- Pennica D, Hayflick JS, Bringman TS, Palladino MA, Goeddel DV. 1985. Cloning and expression in Escherichia coli of the cDNA for murine tumor necrosis factor. Proc Natl Acad Sci 82: 6060–6064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petruzzelli M, Schweiger M, Schreiber R, Campos-Olivas R, Tsoli M, Allen J, Swarbrick M, Rose-John S, Rincon M, Robertson G, et al. 2014. A switch from white to brown fat increases energy expenditure in cancer-associated cachexia. Cell Metab 20: 433–447. [DOI] [PubMed] [Google Scholar]
- Price SR, Bailey JL, Wang X, Jurkovitz C, England BK, Ding X, Phillips LS, Mitch WE. 1996. Muscle wasting in insulinopenic rats results from activation of the ATP-dependent, ubiquitin-proteasome proteolytic pathway by a mechanism including gene transcription. J Clin Invest 98: 1703–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raber J, O'Shea RD, Bloom FE, Campbell IL. 1997. Modulation of hypothalamic-pituitary-adrenal function by transgenic expression of interleukin-6 in the CNS of mice. J Neurosci 17: 9473–9480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohdenburg GL, Bernhard A, Krehbiel O. 1919. Sugar tolerance in cancer. J Am Med Assoc 72: 1528–1530. [Google Scholar]
- Rommel C, Bodine SC, Clarke BA, Rossman R, Nunez L, Stitt TN, Yancopoulos GD, Glass DJ. 2001. Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat Cell Biol 3: 1009–1013. [DOI] [PubMed] [Google Scholar]
- Rosenthal CJ, Franklin EC. 1975. Variation with age and disease of an amyloid A protein-related serum component. J Clin Invest 55: 746–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouzer CA, Cerami A. 1980. Hypertriglyceridemia associated with Trypanosoma brucei brucei infection in rabbits: role of defective triglyceride removal. Mol Biochem Parasitol 2: 31–38. [DOI] [PubMed] [Google Scholar]
- Ruan H, Hacohen N, Golub TR, Van Parijs L, Lodish HF. 2002. Tumor necrosis factor-α suppresses adipocyte-specific genes and activates expression of preadipocyte genes in 3T3-L1 adipocytes: nuclear factor-κB activation by TNF-α is obligatory. Diabetes 51: 1319–1336. [DOI] [PubMed] [Google Scholar]
- Ruas JL, White JP, Rao RR, Kleiner S, Brannan KT, Harrison BC, Greene NP, Wu J, Estall JL, Irving BA, et al. 2012. A PGC-1α isoform induced by resistance training regulates skeletal muscle hypertrophy. Cell 151: 1319–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL. 2004. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 117: 399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandri M, Lin J, Handschin C, Yang W, Arany ZP, Lecker SH, Goldberg AL, Spiegelman BM. 2006. PGC-1α protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc Natl Acad Sci 103: 16260–16265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartori R, Milan G, Patron M, Mammucari C, Blaauw B, Abraham R, Sandri M. 2009. Smad2 and 3 transcription factors control muscle mass in adulthood. Am J Physiol Cell Physiol 296: C1248–C1257. [DOI] [PubMed] [Google Scholar]
- Sartori R, Schirwis E, Blaauw B, Bortolanza S, Zhao J, Enzo E, Stantzou A, Mouisel E, Toniolo L, Ferry A, et al. 2013. BMP signaling controls muscle mass. Nat Genet 45: 1309–1318. [DOI] [PubMed] [Google Scholar]
- Schaefer M, Oeing CU, Rohm M, Baysal-Temel E, Lehmann LH, Bauer R, Volz HC, Boutros M, Sohn D, Sticht C, et al. 2016. Ataxin-10 is part of a cachexokine cocktail triggering cardiac metabolic dysfunction in cancer cachexia. Mol Metab 10.1016/j.molmet.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schieber AM, Lee YM, Chang MW, Leblanc M, Collins B, Downes M, Evans RM, Ayres JS. 2015. Disease tolerance mediated by microbiome E. coli involves inflammasome and IGF-1 signaling. Science 350: 558–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuelke M, Wagner KR, Stolz LE, Hubner C, Riebel T, Komen W, Braun T, Tobin JF, Lee SJ. 2004. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med 350: 2682–2688. [DOI] [PubMed] [Google Scholar]
- Shum AM, Polly P. 2012. Cancer cachexia: molecular targets and pathways for diagnosis and drug intervention. Endocr Metab Immune Disord Drug Targets 12: 247–259. [DOI] [PubMed] [Google Scholar]
- Sidossis LS, Porter C, Saraf MK, Borsheim E, Radhakrishnan RS, Chao T, Ali A, Chondronikola M, Mlcak R, Finnerty CC, et al. 2015. Browning of subcutaneous white adipose tissue in humans after severe adrenergic stress. Cell Metab 22: 219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Socher SH, Martinez D, Craig JB, Kuhn JG, Oliff A. 1988. Tumor necrosis factor not detectable in patients with clinical cancer cachexia. J Natl Cancer Inst 80: 595–598. [DOI] [PubMed] [Google Scholar]
- Stephens NA, Gallagher IJ, Rooyackers O, Skipworth RJ, Tan BH, Marstrand T, Ross JA, Guttridge DC, Lundell L, Fearon KC, et al. 2010. Using transcriptomics to identify and validate novel biomarkers of human skeletal muscle cancer cachexia. Genome Med 2: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens NA, Skipworth RJ, Macdonald AJ, Greig CA, Ross JA, Fearon KC. 2011. Intramyocellular lipid droplets increase with progression of cachexia in cancer patients. J Cachexia Sarcopenia Muscle 2: 111–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strassmann G, Fong M, Kenney JS, Jacob CO. 1992. Evidence for the involvement of interleukin 6 in experimental cancer cachexia. J Clin Invest 89: 1681–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez-Zamorano N, Fabbiano S, Chevalier C, Stojanovic O, Colin DJ, Stevanovic A, Veyrat-Durebex C, Tarallo V, Rigo D, Germain S, et al. 2015. Microbiota depletion promotes browning of white adipose tissue and reduces obesity. Nat Med 21: 1497–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svaninger G, Gelin J, Lundholm K. 1987. Tumor–host wasting not explained by adrenal hyperfunction in tumor-bearing animals. J Natl Cancer Inst 79: 1135–1141. [PubMed] [Google Scholar]
- Teli MR, James OF, Burt AD, Bennett MK, Day CP. 1995. The natural history of nonalcoholic fatty liver: a follow-up study. Hepatology 22: 1714–1719. [PubMed] [Google Scholar]
- Temparis S, Asensi M, Taillandier D, Aurousseau E, Larbaud D, Obled A, Bechet D, Ferrara M, Estrela JM, Attaix D. 1994. Increased ATP-ubiquitin-dependent proteolysis in skeletal muscles of tumor-bearing rats. Cancer Res 54: 5568–5573. [PubMed] [Google Scholar]
- Tisdale MJ. 2010. Are tumoral factors responsible for host tissue wasting in cancer cachexia? Future Oncol 6: 503–513. [DOI] [PubMed] [Google Scholar]
- Tomas FM, Munro HN, Young VR. 1979. Effect of glucocorticoid administration on the rate of muscle protein breakdown in vivo in rats, as measured by urinary excretion of N τ-methylhistidine. Biochem J 178: 139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trendelenburg AU, Meyer A, Rohner D, Boyle J, Hatakeyama S, Glass DJ. 2009. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am J Physiol Cell Physiol 296: C1258–C1270. [DOI] [PubMed] [Google Scholar]
- Tsoli M, Robertson G. 2012. Cancer cachexia: malignant inflammation, tumorkines, and metabolic mayhem. Trends Endocrinol Metab 24: 174–183. [DOI] [PubMed] [Google Scholar]
- Tsoli M, Swarbrick MM, Robertson GR. 2015. Lipolytic and thermogenic depletion of adipose tissue in cancer cachexia. Semin Cell Dev Biol 10.1016/j.semcdb.2015.10.039. [DOI] [PubMed] [Google Scholar]
- van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JM, Kemerink GJ, Bouvy ND, Schrauwen P, Teule GJ. 2009. Cold-activated brown adipose tissue in healthy men. N Engl J Med 360: 1500–1508. [DOI] [PubMed] [Google Scholar]
- Virtanen KA, Lidell ME, Orava J, Heglind M, Westergren R, Niemi T, Taittonen M, Laine J, Savisto NJ, Enerback S, et al. 2009. Functional brown adipose tissue in healthy adults. N Engl J Med 360: 1518–1525. [DOI] [PubMed] [Google Scholar]
- Wagner EF, Petruzzelli M. 2015. Cancer metabolism: a waste of insulin interference. Nature 521: 430–431. [DOI] [PubMed] [Google Scholar]
- Waning DL, Mohammad KS, Reiken S, Xie W, Andersson DC, John S, Chiechi A, Wright LE, Umanskaya A, Niewolna M, et al. 2015. Excess TGF-β mediates muscle weakness associated with bone metastases in mice. Nat Med 21: 1262–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg RA. 2014. Coming full circle-from endless complexity to simplicity and back again. Cell 157: 267–271. [DOI] [PubMed] [Google Scholar]
- White JP, Baltgalvis KA, Puppa MJ, Sato S, Baynes JW, Carson JA. 2011. Muscle oxidative capacity during IL-6-dependent cancer cachexia. Am J Physiol Regul Integr Comp Physiol 300: R201–R211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams A, Sun X, Fischer JE, Hasselgren PO. 1999. The expression of genes in the ubiquitin-proteasome proteolytic pathway is increased in skeletal muscle from patients with cancer. Surgery 126: 744–749. [PubMed] [Google Scholar]
- Willis RA. 1948. Pathology of Tumors. C.V. Mosby Co, St. Louis, MO. [Google Scholar]
- Wing SS, Goldberg AL. 1993. Glucocorticoids activate the ATP-ubiquitin-dependent proteolytic system in skeletal muscle during fasting. Am J Physiol 264: E668–E676. [DOI] [PubMed] [Google Scholar]
- Wisse BE, Frayo RS, Schwartz MW, Cummings DE. 2001. Reversal of cancer anorexia by blockade of central melanocortin receptors in rats. Endocrinology 142: 3292–3301. [DOI] [PubMed] [Google Scholar]
- Yeoh BS, Vijay-Kumar M. 2015. Debugging the host browns the fat. Nat Med 21: 1390–1391. [DOI] [PubMed] [Google Scholar]
- Yoneshiro T, Aita S, Matsushita M, Kayahara T, Kameya T, Kawai Y, Iwanaga T, Saito M. 2013. Recruited brown adipose tissue as an antiobesity agent in humans. J Clin Invest 123: 3404–3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa T, Noguchi Y, Doi C, Makino T, Okamoto T, Matsumoto A. 1999. Insulin resistance was connected with the alterations of substrate utilization in patients with cancer. Cancer Lett 141: 93–98. [DOI] [PubMed] [Google Scholar]
- Yoshikawa T, Noguchi Y, Doi C, Makino T, Nomura K. 2001. Insulin resistance in patients with cancer: relationships with tumor site, tumor stage, body-weight loss, acute-phase response, and energy expenditure. Nutrition 17: 590–593. [DOI] [PubMed] [Google Scholar]
- Young SG, Zechner R. 2013. Biochemistry and pathophysiology of intravascular and intracellular lipolysis. Genes Dev 27: 459–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, Lecker SH, Goldberg AL. 2007. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab 6: 472–483. [DOI] [PubMed] [Google Scholar]
- Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q, Rosenfeld R, Chen Q, Boone T, Simonet WS, et al. 2010. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 142: 531–543. [DOI] [PubMed] [Google Scholar]
- Zimmers TA, Davies MV, Koniaris LG, Haynes P, Esquela AF, Tomkinson KN, McPherron AC, Wolfman NM, Lee SJ. 2002. Induction of cachexia in mice by systemically administered myostatin. Science 296: 1486–1488. [DOI] [PubMed] [Google Scholar]