

In this study, Yan et al. used PCBP4-deficient mice to investigate the role of PCBP4 in tumor suppression. The results show that PCBP4-deficient mice are prone to lung adenocarcinoma, lymphoma, and kidney tumors and suggest that PCBP4 is a potential tissue-specific tumor suppressor.
Keywords: PCBP4, p53, ZFP871, ZNF709, lung cancer, lymphoma, kidney cancer
Abstract
Poly(C)-binding protein 4 (PCBP4), also called MCG10 and a target of p53, plays a role in the cell cycle and is implicated in lung tumor suppression. Here, we found that PCBP4-deficient mice are prone to lung adenocarcinoma, lymphoma, and kidney tumor and that PCBP4-deficient mouse embryo fibroblasts (MEFs) exhibit enhanced cell proliferation but decreased cellular senescence. We also found that p53 expression is markedly reduced in PCBP4-deficient MEFs and mouse tissues, suggesting that PCBP4 in turn regulates p53 expression. To determine how PCBP4 regulates p53 expression, PCBP4 targets were identified by RNA immunoprecipitation followed by RNA sequencing (RNA-seq). We found that the transcript encoding ZFP871 (zinc finger protein 871; also called ZNF709 in humans) interacts with and is regulated by PCBP4 via mRNA stability. Additionally, we found that ZFP871 physically interacts with p53 and MDM2 proteins. Consistently, ectopic expression of ZFP871 decreases—whereas knockdown of ZFP871 increases—p53 protein stability through a proteasome-dependent degradation pathway. Moreover, loss of ZFP871 reverses the reduction of p53 expression by lack of PCBP4, and thus increased expression of ZFP871 is responsible for decreased expression of p53 in the PCBP4-deficient MEFs and mouse tissues. Interestingly, we found that, like PCBP4, ZFP871 is also regulated by DNA damage and p53. Finally, we showed that knockdown of ZFP871 markedly enhances p53 expression, leading to growth suppression and apoptosis in a p53-dependent manner. Thus, the p53–PCBP4–ZFP871 axis represents a novel feedback loop in the p53 pathway. Together, we hypothesize that PCBP4 is a potential tissue-specific tumor suppressor and that ZFP871 is part of MDM2 and possibly other ubiquitin E3 ligases that target p53 for degradation.
PCBP4 is a member of the poly(C)-binding protein (PCBP) family (PCBP1–4) (Makeyev and Liebhaber 2000, 2002; Choi et al. 2009). PCBPs contain three hnRNP K homology (KH) domains, which are capable of binding to poly(C)-rich elements in DNA or RNA targets (Valverde et al. 2008). PCBP1 and PCBP2 physically interact and have been implicated in mRNA metabolism, including mRNA stability (Wang et al. 1995; Stefanovic et al. 1997; Paulding and Czyzyk-Krzeska 1999) and mRNA translation (Ostareck et al. 1997; Evans et al. 2003). PCBP4, also called MCG10, is identified as a target of p53 and suppresses cell proliferation by inducing apoptosis and cell cycle arrest in G2–M (Zhu and Chen 2000). In addition, ectopic expression of PCBP4 in lung cancer cells suppresses anchorage-independent cell proliferation, cell invasion, and in vivo growth of xenografts (Castano et al. 2008). Consistently, the ability of PCBP4 to recognize and bind to the poly(C) element via its KH domains is critical for its function in the cell cycle (Zhu and Chen 2000). Although a few RNA targets are found to be regulated by PCBP4 (Scoumanne et al. 2011), it remains unclear how PCBP4 is involved in tumor suppression. Thus, the identification of PCBP4 targets is necessary for elucidating the mechanisms by which PCBP4 deficiency promotes tumorigenesis.
Lung cancer is one of the most malignant and lethal tumors in the United States and throughout the world (Siegel et al. 2013). Many molecular alterations have been described in the multistep process of lung cancer development. Particularly, loss of chromosome 3p21 is one of the most prevalent genomic abnormalities in lung cancer, suggesting that chromosome 3p21 harbors one or more tumor suppressor genes whose inactivation is required for malignant transformation (Dammann et al. 2000; Tomizawa et al. 2001; Tai et al. 2006; Angeloni 2007; Hesson et al. 2007). Interestingly, the PCBP4 gene is located at chromosome 3p21. Loss of PCBP4 expression is common in poorly differentiated and highly proliferative lung adenocarcinoma and squamous cell carcinoma (Pio et al. 2004). These observations suggest that PCBP4 is a potential tissue-specific tumor suppressor in the lung. In this study, PCBP4-deficient mice were generated and used to test the role of PCBP4 in lung tumor suppression.
Results
PCBP4-deficient mice are prone to lung adenocarcinoma and lymphoma
Previously, we found that PCBP4 is a target of p53 in human cells (Zhu and Chen 2000). Thus, we examined whether PCBP4 is also regulated by p53 in mouse embryo fibroblasts (MEFs). Indeed, we showed that the level of PCBP4 was markedly decreased in p53-null MEFs compared with wild-type MEFs (Supplemental Fig. S1A). To analyze the biological function of PCBP4, a PCBP4-deficient mouse model was generated with an ES clone (D136D11) in which the PCBP4 gene was disrupted through insertion of a gene trap (Supplemental Fig. S1B). Upon injection to C57BL/6 blastocysts, multiple male chimeras were generated and then bred with C57BL/6 females for germline transmission. PCBP4-deficient mice and MEFs were genotyped by PCR (Supplemental Fig. S1C,D). PCBP4 protein was also measured by immunoblotting and found to be absent in PCBP4−/− MEFs and reduced in PCBP4+/− MEFs compared with that in wild-type MEFs (Supplemental Fig. S1E).
Mice heterozygous for PCBP4 were viable and appeared phenotypically normal and fertile. PCBP4−/− mice were observed at normal Mendelian ratios at birth, indicating that no embryonic lethality was associated with PCBP4 deletion. However, PCBP4−/− male and female mice were smaller than their wild-type counterparts at 15 d of age (Supplemental Fig. S1F). In addition, PCBP4+/− and PCBP4−/− female mice at 4–19 and 20–45 wk of age and male mice at 18–45 wk of age were smaller than their wild-type counterparts (Supplemental Fig. S1G–I; Supplemental Tables S1–S3).
To monitor long-term survival and potential propensity to tumor development, a cohort of wild-type, PCBP4+/−, and PCBP4−/− mice in the C57BL/6 background was generated (Supplemental Tables S4–S6). We found that the median survival time for PCBP4+/− (102.1 wk ± 3.2 wk) and PCBP4−/− (105.3 wk ± 9.8 wk) mice was ∼5–8 wk shorter than that for wild-type mice (110.4 wk ± 1.1 wk) (Fig. 1A). A log rank test showed that the difference was not statistically significant. In contrast, we found that 20% of PCBP4+/− mice and 26.7% of PCBP4−/− mice, but none of the wild-type mice, developed lung adenocarcinoma (Fig. 1B; Supplemental Fig. S2). A Fisher's exact test showed that the difference in lung adenocarcinomas between wild-type and PCBP4−/− mice was significant (P = 0.0498). Several representative images of lung tissues stained with hematoxylin and eosin (H&E) from wild-type, PCBP4+/−, and PCBP4−/− mice are shown in Figure 1C. Histology examination showed that all of the lung adenocarcinomas in PCBP4-deficient mice belonged to papillary predominant lung adenocarcinoma (Fig. 1C). These results suggest that mice deficient in PCBP4 are prone to lung cancer, which prompted us to examine the relationship of PCBP4 expression and the status of the p53 gene in human lung cancer with The Cancer Genome Atlas (TCGA) database. Bioinformatic analysis was performed with the program from the Galaxy Project (https://galaxyproject.org), an open, Web-based platform. From TCGA lung cancer database, RNA sequencing (RNA-seq) data for 1124 cases, microarray data for 188 cases, and gene mutation data for 408 cases were available and compared. We found that the level of PCBP4 transcript was measured by RNA-seq in 123 cases of lung cancer with wild-type p53 and 228 cases with mutant p53. Statistical analysis showed that PCBP4 was highly expressed in lung cancer with wild-type p53 compared with that in lung cancer with mutant p53 (Supplemental Table S7). The levels of p21 and Puma (both of which are p53 targets) were also higher in lung cancer with wild-type p53 than that in lung cancer with mutant p53 (Supplemental Table S7). Additionally, we found that the level of PCBP4 transcript was measured by RNA microarray assay in 30 cases of lung cancer with wild-type p53 and 114 cases with mutant p53. Statistical analysis showed that the level of PCBP4 transcript was much higher in lung cancer with wild-type p53 than that in lung cancer with mutant p53 (Supplemental Table S8). In contrast, the levels of p21 and Puma transcripts in lung cancer with wild-type p53 were similar to that in lung cancer with mutant p53 (Supplemental Table S8).
Figure 1.

PCBP4-deficient mice are prone to lung cancer and lymphoma. (A) Kaplan-Meier survival curve for wild-type (WT) (median survival time 110.4 wk ± 1.1 wk), PCBP4+/− (median survival time 102.1 wk ± 3.2 wk), and PCBP4−/− (median survival time 105.3 wk ± 9.8 wk) mice. (B) PCBP4+/− and PCBP4−/− mice are prone to lung cancer and lymphoma. (*) P < 0.05. (C) Representative images of H&E-stained normal lungs in wild-type mice and lung adenocarcinomas (ADCs) in PCBP4+/− and PCBP4−/− mice.
We also found that PCBP4+/− and PCBP4−/− mice were highly prone to lymphomas compared with wild-type mice (Fig. 1B; Supplemental Tables S4–S6). A Fisher's exact test showed that the penetrance of lymphoma between wild-type and PCBP4+/− mice (P = 0.038) or PCBP4−/− mice (P = 0.002) was statistically significant. Additionally, we found that, unlike the thymic lymphomas that frequently developed in p53-deficient mice (Donehower et al. 1992; Jacks et al. 1994), the majority of lymphomas in PCBP4-deficient mice was associated with visceral organs such as the spleen, liver, kidney, and lungs (Supplemental Fig. S3). To determine the origin of lymphomas, immunofluorescence staining was performed for kidney sections of PCBP4−/− mice with antibodies against B-cell marker B220 and T-cell marker CD3, respectively. We found that the lymphoma in the kidney of the PCBP4−/− mouse was positive for B220 with scattered CD3-positive T cells (Supplemental Fig. S4), suggesting that lymphomas in visceral organs of PCBP4-deficient mice originated from B cells. Furthermore, we found that one PCBP4+/− and one PCBP4−/− mouse developed a kidney tumor (Supplemental Fig. S5), consistent with the TCGA database data that 11.8% of renal clear cell carcinomas have deletion of the PCBP4 gene. These data suggest that PCBP4 plays a role in kidney tumorigenesis.
To test whether PCBP4 is expressed in a tissue-specific manner, the levels of PCBP4 transcript were measured in eight mouse tissues. We found that PCBP4 was highly expressed in the brain, lungs, and lymph nodes compared with in the liver, spleen, kidneys, heart, and thymus (Supplemental Fig. S6A,B). Due to the fact that PCBP4-deficient mice are prone to lung cancer and B-cell lymphoma, the expression pattern of PCBP4 in various organs suggests that PCBP4 is a potential tissue-specific tumor suppressor.
PCBP4-deficient MEFs exhibit enhanced cell proliferation but decreased cellular senescence along with decreased expression of p53
Since loss of PCBP4 predisposes mice to spontaneous tumors, we assessed whether PCBP4 deficiency modulates cell proliferation and cellular senescence with wild-type and PCBP4−/− MEFs at passage 5. We found that, over a 5-d period, the rate of cell proliferation for PCBP4−/− MEFs was much higher than that in the wild type (Fig. 2A,B). We also found that the number of senescence-associated β-galactosidase (SA-β-gal)-positive cells was significantly decreased in PCBP4−/− MEFs as compared with that in the wild type (Fig. 2C,D). Consistently, we found that the levels of promyelocytic leukemia protein (PML) and p130, both of which are associated with cellular senescence, were much lower in PCBP4−/− MEFs than that in the wild type (Fig. 2E). However, PCBP4 deficiency had little if any effect on the expression levels of PAI-1 and phosphorylated AKT (Fig. 2E).
Figure 2.

PCBP4 deficiency promotes cell proliferation but inhibits cellular senescence. (A) Representative images of wild-type and PCBP4−/− MEFs grown on a plate for 5 d. (B) The number of wild-type and PCBP4−/− MEFs over a 5-d period was counted and is presented as mean ± SD from three separate experiments. (C) Representative images of SA-β-Gal-stained wild-type and PCBP4−/− MEFs. (D) The percentage of SA-β-Gal-positive cells shown in C. (*) P < 0.05. (E) The protein levels of PCBP4, PML, p130, PAI-1, total AKT, and phosphorylated AKT were measured in wild-type and PCBP4−/− littermate MEFs at passage 5. (F) The levels of p53 and PCBP4 proteins were measured in two sets of wild-type and PCBP4−/− littermate MEFs at passage 3. (G) The levels of p53 and PCBP4 proteins were measured in thymus tissues from two pairs of wild-type and PCBP4−/− littermate mice. (H,I) The levels of p53 and PCBP4 proteins were measured in lung (H) and kidney (I) tissues from one pair of wild-type and PCBP4−/− littermate mice.
Since the above phenotypes observed in PCBP4-deficient mice and MEFs have many similarities to that in p53-deficient mice and MEFs, we hypothesized that, as a target of p53, PCBP4 may in turn regulate p53 expression. To test this, we measured p53 expression and found that the level of p53 protein was much lower in PCBP4−/− MEFs than that in wild-type littermates at passage 3 (Fig. 2F). It is well known that wild-type p53 is one of the master regulators in cellular senescence (Odell et al. 2010). The decreased expression of wild-type p53 in PCBP4-deficient low-passage primary MEFs might lead to partial senescence bypass in MEFs, as observed in Figure 2, C and D. Consistently, we also found that the level of p53 protein was lower in the PCBP4−/− mouse thymus (Fig. 2G), lungs (Fig. 2H) and kidneys (Fig. 2I) than that in wild-type tissues.
PCBP4 interacts with and modulates the stability of the transcript encoding ZFP871 (zinc finger protein 871)
As a PCBP, it is likely that PCBP4 regulates a set of poly(C)-containing transcripts whose products would then mediate PCBP4 to regulate p53 expression and tumor suppression. Although a few targets are found to be regulated by PCBP4 (Scoumanne et al. 2011), it remains unclear what mediates the activity of PCBP4 in tumor suppression. Thus, cell extracts from wild-type and PCBP4−/− MEFs were immunoprecipitated with anti-PCBP4 and then subjected to RNA-seq. A number of potential PCBP4-associated transcripts were identified and are listed in Supplemental Table S9. From these potential targets, ∼20 transcripts with a poly(C) motif in their 3′ untranslated regions (UTRs) were chosen for further validation by RT–PCR. Eight transcripts (ZFP871, CCNI, ARF3, Rhoq, Rab1, Plor3gl, BMP4, and BMPr1a) were confirmed to be enriched in anti-PCBP4 immunoprecipitates from wild-type MEFs compared with PCBP4−/− MEFs (Fig. 3A). Additionally, these transcripts were absent in Ig control immunoprecipitates (Fig. 3A). Next, quantitative RT–PCR (qRT–PCR) was performed to measure the relative levels of PCBP4-binding transcripts in wild-type and PCBP4−/− MEFs. We found that PCBP4 deficiency led to increased expression for ZFP871, CCNI, BMP4, and BMPr1a and decreased expression for Rhoq and Rab1 but no change for ARF3 and Plor3gl (Fig. 3B). To confirm this, we measured the effect of PCBP4 on ZFP871, CCNI, and BMPr1a in one wild-type and two PCBP4−/− MEFs derived from the same litter (Fig. 3C). Consistently, PCBP4 deficiency led to increased expression for ZFP871, CCNI, and BMPr1a (Fig. 3D). Additionally, we found that the effect of PCBP4 on its target mRNAs is independent of p53, as the levels of transcripts for ZFP871, CCNI, and BMPr1a were increased by PCBP4 deficiency in the absence of p53 (p53−/−; PCBP4−/− vs. p53−/−) (Fig. 3E).
Figure 3.

Identification of ZFP871 as a target of PCBP4. (A) Interaction of PCBP4 protein with its target transcripts. Rabbit anti-PCBP4 antibody was used to immunoprecipitate PCBP4–RNA complexes from wild-type and PCBP4−/− MEFs along with IgG as a control. The binding of PCBP4 to the transcripts was measured by RT–PCR. Actin was used as a negative control. (B) qRT–PCR was performed to measure the level of PCBP4 target transcripts in wild-type and PCBP4−/− MEFs. Data are presented as mean ± SD normalized to actin mRNA from three separate experiments. (*) P < 0.05. (C) Generation of one wild-type and two PCBP4−/− MEFs from the same litter. (D) The level of ZFP871, CCNI, and BMPR1a transcripts in one wild-type and two PCBP4−/− MEFs shown in C was measured by qRT–PCR. (E) The level of ZFP871, CCNI, and BMPR1a transcripts in p53−/− and p53−/−; PCBP4−/− MEFs was measured by qRT–PCR. (F) Schematic presentation of the ZFP871 transcript, the predicted zinc finger domain in the ZFP871 protein, and the location of fragments A–F along with a CU-rich element in the ZFP871 3′ UTR. (G) The PCBP4 protein binds to probe D (nucleotides 6521–8070) but not probe A (nucleotides 1995–3480), B (nucleotides 3481–5000), C (nucleotides 5001–6520), E (nucleotides 8071–9582), or F (nucleotides 9583–10,857). RNA electrophoretic mobility shift assay (REMSA) was performed by mixing a 32P-labeled RNA probe with recombinant glutathione S-transferase (GST) or GST-fused PCBP4 protein. The binding of PCBP4 to the p21 3′ UTR was used as a positive control. The bracket indicates RNA–protein complexes (RPC). (H) REMSA competition assay was performed by adding an excess amount of unlabeled p21 cold probe to a reaction mixture containing the PCBP4 protein and 32P-labeled probe D. (I) REMSA was performed with probe DD, which lacks the CU-rich element (nucleotides 7324–7365). (J) The half-life of the ZFP871 transcript was measured by qRT–PCR in p53−/− and p53−/−; PCBP4−/− MEFs treated with 5 µg/mL actinomycin (Act D), an inhibitor of transcription. Data are presented as mean ± SD normalized to actin mRNA from three separate experiments. (K, left panel) The PCBP4 protein was measured by Western blot analysis in p53−/−; PCBP4−/− MEFs transiently transfected with pcDNA3 or pcDNA3-PCBP4. (Right panel) The level of ZFP871 transcript was measured by qRT–PCR in p53−/−; PCBP4−/− MEFs treated as in the left panel. Data are presented as mean ± SD after being normalized to actin mRNA from three separate experiments. P < 0.05. (L) The experiments were performed as in K except that C2C12 cells were used.
To test whether ZFP871 is a direct target of PCBP4, RNA electrophoretic mobility shift assay (REMSA) was performed to identify potential PCBP4-binding sites in the mouse ZFP871 transcript. The ZFP871 3′ UTR has ∼9000 nucleotides (nt) and thus was amplified in six fragments (A–F) as probes for REMSA (Fig. 3F). We showed that only the D fragment was recognized by glutathione S-transferase (GST)-fused PCBP4 protein but not GST protein (Fig. 3G). The p21 3′ UTR, which is known to contain a PCBP4-binding site (Scoumanne et al. 2011), was used as a positive control (Fig. 3G). The specificity of PCBP4 binding to the ZFP871 3′ UTR was confirmed by a competition assay in which an excess amount of an unlabeled fragment from the p21 3′ UTR decreased the interaction of PCBP4 with the 32P-labeled probe D (Fig. 3H). Upon analyzing the nucleotide sequence in the D fragment, we found a region rich in the CU dinucleotide (nucleotides 7324–7365). To determine whether the CU-rich element is required for the interaction of PCBP4 with the D fragment, we generated a mutated D fragment, called DD, in which the CU-rich element was deleted (Fig. 3F). We found that the DD fragment was incapable of forming a complex with the PCBP4 protein (Fig. 3I).
To explore how PCBP4 regulates ZFP871 expression, the half-life of ZFP871 mRNA was examined in p53−/− and p53−/−; PCBP4−/− MEFs treated with 5 µg/mL actinomycin D (Act D). p53-deficient MEFs were used to avoid the potential interference of p53 on ZFP871 mRNA stability, and Act D is known to inhibit nascent RNA synthesis (Bensaude 2011). We showed that the half-life of ZFP871 mRNA was increased from 1.6 h in p53−/− MEFs to 2.5 h in p53−/−; PCBP4−/− MEFs (Fig. 3J), suggesting that the PCBP4 protein modulates ZFP871 mRNA stability. To further test this, we examined the effect of ectopic PCBP4 on ZFP871 expression and found that ZFP871 expression was inhibited by PCBP4 in p53−/−; PCBP4−/− MEFs (Fig. 3K) as well as in C2C12 immortalized but untransformed murine myoblasts (Fig. 3L).
The above observations indicate that ZFP871 expression is directly regulated by PCBP4. Since PCBP4 is a target of p53, we examined whether activation of the p53 pathway by DNA damage has any effect on ZFP871 expression. To test this, FL83B and C2C12 cells were treated with camptothecin or doxorubicin to activate the p53 pathway. We found that the levels of the p53 protein and ZFP871 mRNA were markedly increased by DNA damage in both FL83B and C2C12 cells (Supplemental Fig. S7A,B). Consistently, we found that knockdown of p53 significantly decreased the level of the p53 protein and ZFP871 mRNA in C2C12 cells (Supplemental Fig. S7C,D).
ZFP871 modulates cell growth by targeting p53 for degradation
Zinc finger proteins are known to exert diverse activities through interaction with a variety of molecules (Laity et al. 2001; Krishna et al. 2003; Loughlin and Mackay 2006), but the activity for ZFP871 has not been explored. Since PCBP4 regulates both p53 and ZFP871 expression, we explored whether ZFP871 plays a role in the PCBP4-mediated regulation of p53 expression. To test this, HA-tagged ZFP871 was transiently expressed in C2C12 cells. Since the p53 gene is prone to mutation in immortalized PCBP4-deficient MEFs (Supplemental Fig. S8), mouse C2C12, FL83B, and TS20 cell lines were used in the following experiments. Upon treatment with doxorubicin, a topoisomerase II inhibitor that is known to induce DNA double-strand breaks, p53 was accumulated along with increased expression of p21 (Fig. 4A, cf. lanes 1 and 3), suggesting that the endogenous p53 gene in C2C12 cells is wild type and functional. Interestingly, we found that ectopic expression of ZFP871 suppressed p53 expression under basal and DNA damage conditions (Fig. 4A, cf. lanes 1,3 and 2,4, respectively). Conversely, upon knockdown of ZFP871 by two unique ZFP871 targeting siRNAs in C2C12 cells, the levels of p53 and p21 proteins were increased (Fig. 4B). Similarly, knockdown of ZFP871 led to increased expression of p53 and p21 in FL83B (Fig. 4C) and TS20 (Fig. 4D) cells, both of which also carry wild-type p53. Since ZFP871 expression was increased by PCBP4 deficiency (Fig. 3) and since p53 expression is regulated by ZFP871 (Fig. 4A–D), we tested whether the decreased expression of p53 by PCBP4 deficiency can be reversed by ZFP871 knockdown. Indeed, we found that the decreased expression of p53 induced by PCBP4 deficiency was substantially mitigated by ZFP871 knockdown (Fig. 4E, cf. lanes 4 and 1–3, respectively). Consistent with the above observations (Figs. 2F–I, 4B–D), we found that p53 expression was decreased by PCBP4 knockdown but increased by ZFP871 knockdown (Fig. 4E, cf. lanes 1 and 2,3, respectively). The efficacy of ZFP871 knockdown by siRNA was measured and is shown in Figure 4F. Additionally, ZFP871 expression was increased upon PCBP4 knockdown (Fig. 4F, cf. the Scr column and siPCBP4 column).
Figure 4.

p53 expression was suppressed by PCBP4 deficiency via ZFP871. (A) The levels of p53 and p21 proteins were measured in C2C12 cells transfected with pcDNA3 or pcDNA3-HA-ZFP871 in the absence or presence of 0.35 µg/mL doxorubicin for 24 h. Actin was used as a loading control. (B) The levels of p53 and p21 proteins were measured in C2C12 cells transfected with two unique scrambled siRNAs or siRNAs against ZFP871. (C,D) The levels of p53 and p21 proteins were measured in murine FL83B (C) and TS20 (D) cells transfected with a scrambled siRNA (Scr-1) or a siRNA against ZFP871 (siZFP871-1) in the absence or presence of 0.35 µg/mL doxorubicin for 24 h. (E) The levels of p53 and PCBP4 proteins were measured in C2C12 cells transiently transfected with a scrambled siRNA (Scr-1), siRNA against PCBP4 (siPCBP4), siRNA against ZFP871 (siZFP871-1), or both siPCBP4 and siZFP871 for 3 d. (F) The level of ZFP871 transcript was measured by qRT–PCR in C2C12 cells treated as in E.
To investigate how ZFP871 regulates p53 expression, we examined the effect of ZFP871 knockdown on the level of the p53 transcript and the stability of the p53 protein in C2C12 cells. We found that knockdown of ZFP871 had little if any effect on the level of p53 mRNA (Supplemental Fig. S9). However, the half-life of the p53 protein was markedly increased by ZFP871 knockdown—from ∼22 to ∼105 min (Fig. 5A,B). To investigate whether ZFP871 promotes p53 degradation via ubiquitination-mediated proteolysis, p53 and Flag-tagged ubiquitin were coexpressed in C2C12 cells with or without HA-tagged ZFP871 followed by treatment with 5 µM MG132 for 6 h. Consistent with the observation above (Fig. 4A), the level of exogenous p53 protein in C2C12 cells was decreased by ectopic expression of ZFP871 (Fig. 5C). In contrast, the level of polyubiquitinated p53 was markedly increased by ectopic expression of ZFP871 in C2C12 cells (Fig. 5D,E).
Figure 5.

ZFP871 targets p53 for degradation via a ubiquitin-dependent proteolysis. (A) The half-life of the p53 protein was measured in C2C12 cells that were transfected with a scrambled siRNA (Scr-1) or a siRNA against ZFP871 (siZFP871-1) for 3 d and then treated with 50 µg/mL cycloheximide (CHX) for 0–120 min. (B) The relative levels of the p53 protein measured in A were normalized by levels of actin protein and then plotted versus time. (C) C2C12 cells were transfected with pcDNA3-p53 and pcDNA3-Flag-ubiquitin with or without pcDNA3-HA-ZFP871 followed by treatment with 5 µM MG132 for 6 h. The levels of ectopically expressed p53 and HA-tagged ZFP871 proteins in whole-cell lysates were measured by anti-p53 and anti-HA (to detect ZFP871), respectively. (D,E) C2C12 cells were treated as in C. The level of polyubiquitinated p53 in whole-cell lysates (WCLs) was immunoprecipitated with rabbit anti-p53 antibody followed by Western blotting with mouse anti-ubiquitin (D) or mouse anti-p53 (E). (F) TS20 cells were transiently transfected with HA-ZFP871 for 24 h at 35°C and then incubated for an additional 8 h at 35°C or 39°C in the presence or absence of 5 µM MG132. The levels of HA-ZFP871 and p53 were measured by mouse anti-HA and mouse anti-p53 antibodies, respectively. (G) C2C12 (left panel) or FL83B (right panel) cells were transfected with pcDNA3-HA-ZFP871. The interaction between endogenous p53 and ectopic HA-tagged ZFP871 in C2C12 and FL83B cells was measured by coimmunoprecipitation with rabbit anti-p53 antibody followed by Western blotting with mouse anti-p53 to detect p53 protein or anti-HA to detect HA-tagged ZFP871. Rabbit IgG was used as a control for immunoprecipitation. (H) GST pull-down assay was performed with nuclear extracts from C2C12 cells and GST or GST-tagged ZFP871. The GST and GST-ZFP871 beads were then washed, followed by Western blot analysis with antibody against p53, MDM2, or GST.
To further determine whether ZFP871 inhibits p53 expression via a ubiquitination-dependent proteasomal degradation, ectopic ZFP871 was expressed in temperature-sensitive murine TS20 cells in which the E1 ubiquitin-activating enzyme is not active at the restrictive temperature of 39°C (Chowdary et al. 1994). We found that the level of p53 was increased at the restrictive temperature (39°C) (Fig. 5F, cf. lanes 1 and 3), consistent with a previous report that p53 is accumulated in TS20 cells (Chowdary et al. 1994). Importantly, we found that ZFP871 inhibited p53 expression at the permissive temperature (35°C) (Fig. 5F, cf. lanes 1 and 2) but much less at 39°C (Fig. 5F, cf. lanes 3 and 4). In addition, we found that the stability of the p53 protein was restored upon MG132 treatment (Fig. 5F, lanes 5–8).
Next, we examined whether ZFP871 physically interacts with p53 and then regulates p53 expression. We found that endogenous p53 and ectopic HA-tagged ZFP871 were coimmunoprecipitated with anti-p53 antibody in C2C12 and FL83B cells (Fig. 5G). To confirm this, GST pull-down assay was performed and showed that endogenous p53 in C2C12 cells interacted with the GST-ZFP871 fusion protein but not the GST protein (Fig. 5H). In addition, we found that MDM2 was pulled down by the GST-ZFP871 fusion protein but not the GST protein (Fig. 5H), suggesting that ZFP871 may regulate p53 expression as a modulator of MDM2 E3 ligase.
Knockdown of ZFP871 leads to growth suppression and apoptosis in a p53-dependent manner
Since PCBP4 deficiency leads to increased ZFP871 expression along with enhanced cell proliferation, we examined whether knockdown of ZFP871 is capable of suppressing cell proliferation in C2C12 cells. Indeed, upon knockdown of ZFP871 with three unique siRNAs (Fig. 6A), cell proliferation was markedly inhibited (Fig. 6B). To test how knockdown of ZFP871 leads to growth suppression, TUNEL assay was performed in C2C12 and TS20 cells. We found that knockdown of ZFP871 significantly increased the percentage of apoptotic cells (Supplemental Fig. S10A–C). As a positive control, we found that DNA damage induced by treatment with doxorubicin also significantly increased the percentage of apoptotic cells (Supplemental Fig. S10D–F). Since knockdown of ZFP871 decreases cell proliferation potentially via increased expression of p53 (Fig. 4B–D), we examined whether the decreased cell proliferation by knockdown of ZFP871 can be reversed by knockdown of p53. Consistent with the above observations (Fig. 4B–D), we showed that knockdown of ZFP871 led to increased expression of p53 (Fig. 6C, cf. lanes 1,2), which was decreased by p53 siRNA (Fig. 6C, cf. lanes 2 and 4). While cell proliferation was inhibited by ZFP871 knockdown (Fig. 6D, siZFP871 column), knockdown of p53 alone had little effect on cell proliferation (Fig. 6D, sip53 column). Interestingly, the decreased cell proliferation induced by knockdown of ZFP871 was substantially suppressed by knockdown of p53 in C2C12 cells (Fig. 6D, siZFP871 and sip53 columns). To confirm this, we examined whether knockdown of ZFP871 leads to apoptosis in a p53-dependent manner. To test this, the percentage of cells in sub-G1 was measured and used to represent the extent of apoptosis. We showed that knockdown of p53 alone had no effect on apoptosis in C2C12 cells (Fig. 6E, sip53 column), consistent with the above observation that knockdown of p53 alone had no significant effect on cell proliferation (Fig. 6D, sip53 column). In contrast, we found that knockdown of ZFP871 promoted apoptosis (13.4%), which was inhibited by knockdown of p53 (5.2%) (Fig. 6E).
Figure 6.
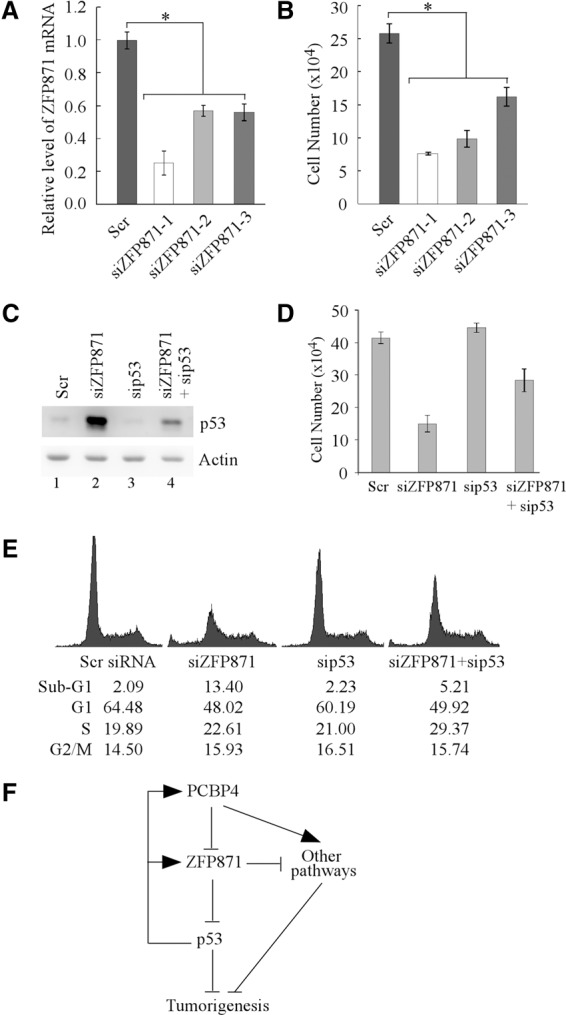
ZFP871 modulates cell growth by targeting p53 for degradation. (A) The level of the ZFP871 transcript was measured by qRT–PCR in C2C12 cells transiently transfected with a scrambled siRNA (Scr) or three unique siRNAs against ZFP871 (siZFP871) for 3 d. Data were presented as mean ± SD after being normalized to actin mRNA from three separate experiments. (*) P < 0.05. (B) C2C12 cells were treated as in A. The number of cells was counted at day 3. Data are presented as mean ± SD of three separate wells. (*) P < 0.05. (C) The level of p53 protein was measured in C2C12 cells transfected with a scrambled siRNA (Scr), a siRNA against ZFP871 (siZFP871), a siRNA against p53 (sip53), or both siZFP871 and siZFP871. (D) C2C12 cells were treated as in C, and the surviving cells were counted at day 3. Data are presented as mean ± SD from three separate wells. (E) C2C12 cells were treated as in C. Both floating dead cells in the medium and live cells on the plate were stained with propidium iodide. The stained cells were analyzed in a fluorescence-activated cell sorter. The percentage of cells in the sub-G1, G1, S, and G2–M phases was determined using the CELLQuest program. (F) A model for the p53–PCBP4–ZFP871 axis in tumor suppression.
Discussion
The p53–PCBP4–ZFP871 axis in tumor suppression
Previous studies suggest that loss of the PCBP4 gene is correlated with lung cancer (Pio et al. 2004). Additionally, loss of heterozygosity (LOH) in chromosome 3p21 is present in 60% of non-small-cell lung cancer, and the p53 mutation is associated with 3p21 LOH (Marsit et al. 2004). Interestingly, allelic losses on 17p13 (p53 region) and 3p21 (PCBP4 region) in early lung squamous cell carcinoma were found to be associated with the depth of invasion and longitudinal extension of the carcinoma (Endo et al. 2000). We and others also found that overexpression of PCBP4 in lung cancer cell lines leads to apoptosis and cell cycle arrest in G2–M (Zhu and Chen 2000; Castano et al. 2008). These data suggest that loss of both PCBP4 and p53 may cooperate to promote lung cancer progression. Here, we found that mice deficient in PCBP4 are prone to lung adenocarcinomas, lymphomas, and kidney tumors. Consistently, PCBP4-deficient MEFs exhibit enhanced cell proliferation but decreased cellular senescence. Furthermore, we provided evidence: (1) PCBP4 directly interacts with ZFP871 transcript and modulates ZFP871 expression via mRNA stability. (2) ZFP871 interacts with p53 and modulates p53 expression via protein stability. (3) ZFP871 modulates cell proliferation and apoptosis in a p53-dependent manner. (4) p53 expression is modulated by PCBP4 in a ZFP871-dependent manner. Due to the fact that PCBP4 and ZFP871 are regulated by DNA damage and p53 (Supplemental Fig. S7; Zhu and Chen 2000), both of which in turn regulate p53 expression, we postulate that the p53–PCBP4–ZFP871 axis represents a novel feedback loop in the p53 pathway (Fig. 6F). We also hypothesize that, in the early stage of carcinogenesis, genomic abnormality in chromosome 3p21 may lead to loss of the PCBP4 gene, which promotes ZFP871 expression via mRNA stability, leading to decreased expression of p53 and subsequently tumor formation in the lungs, lymphoid tissues, and kidneys. Since PCBP4 is expressed at relatively higher levels in the lungs and lymph nodes, PCBP4 may function as a tissue-specific tumor suppressor. In the current study, wild-type and PCBP4-deficient low-passage primary MEFs were used to analyze the role of PCBP4 in senescence. However, due to the limitation of MEFs for senescence assay and the frequent mutation of the p53 gene in MEFs, further studies need to be performed with PCBP4 knockout primary human fibroblasts to address how PCBP4 deficiency overcomes the cellular senescence.
ZFP871 targets p53 for degradation
p53 is a short half-life protein, and protein stability plays a key role in p53 regulation and function (Vousden and Prives 2009; Manfredi 2010). Over the years, several ubiquitin E3 ligases have been identified to regulate p53 protein stability, such as MDM2–MDMX (Haupt et al. 1997; Kubbutat et al. 1997; Brooks and Gu 2006), Pirh2 (Leng et al. 2003), ARF-BP1 (Chen et al. 2005), COP1 (Dornan et al. 2004), and WWP1 (Laine and Ronai 2007). Zinc finger proteins represent a group of protein families with an array of functions in transcription and protein degradation (Laity et al. 2001; Krishna et al. 2003; Loughlin and Mackay 2006). Interestingly, MDM2, MDMX, and Pirh2 are proteins containing a zinc finger domain (Yu et al. 2006; Lee and Gu 2010; Shloush et al. 2011). Mutations in the MDM2 zinc finger domain abrogate the activity of MDM2 to target p53 for degradation (Lindstrom et al. 2007). However, the structure and function of ZFP871 have not been explored. Here, our results indicate that ZFP871 physically interacts with the p53 protein, and disruption of ZFP871 increases p53 protein stability. Consistently, overexpression of ZFP871 decreases the level of the p53 protein but increases the level of the polyubiquitinated p53 protein. Thus, we postulate that ZFP871 is part of a ubiquitin E3 ligase, such as MDM2, that targets p53 for proteosomal degradation. Indeed, zinc finger proteins often serve as a scaffold for protein–protein interactions (Laity et al. 2001; Loughlin and Mackay 2006; Tian et al. 2009). We also found that ZFP871 physically interacts with MDM2 and p53, and thus ZFP871 may be a part of the MDM2 E3 ligase that targets p53 for degradation. Nevertheless, it remains possible that ZFP871 may recruit other p53 regulators and indirectly modulate p53 protein stability. It should be mentioned that ZFP871 has three homologous genes in humans: ZNF709 (48% identity in amino acid sequence), ZFP14 (44%), and gonadotropin inducible transcriptional repressor 4 (44%). Our preliminary studies showed that knockdown of human ZNF709 led to increased expression of p53, but whether p53 expression is regulated by ZFP14 and gonadotropin inducible transcriptional repressor 4 has not been explored. Also, ZFP871 may regulate other proteins critical for tumor suppression in addition to p53. Due to the sequence and structural similarities among the p53 family proteins, it is likely that ZFP871 may regulate other p53 family members, such as TAp63 and TAp73. Thus, further studies are needed to address these issues in order to fully understand the mechanisms by which PCBP4 exerts tumor suppression in both p53-dependent and p53-independent pathways.
Materials and methods
PCBP4- and p53-deficient mice and primary MEFs
To generate a PCBP4-deficient mouse model, the PCBP4 gene was disrupted through insertion of a gene trap (rFRosaβgeo + 1s) into the first intron (Supplemental Fig. S1B; Schnutgen et al. 2005). Embryonic stem cells (no. D136D11; TBV-2 [129/SvPas]) were obtained from the German Gene Trap Consortium (Helmholtz Zentrum München) and microinjected into C57BL/6 blastocysts at the University of California at Davis Murine Targeted Genomics Laboratory. Male chimeras were bred with C57BL/6 females (Jackson Laboratory), and pups were tested for germline transmission. Genotyping of progeny was performed by PCR analysis of toe clipping samples using three primers. For wild-type mice, a 718-base-pair (bp) fragment was detected using forward primer GM103F (5′-CAGGCTGCAGAGAGGATGGCTT-3′) and reverse primer GM103R (5′-AAAGCCTCCTCCTCCACGGA-3′). For PCBP4 knockout mice, a 160-bp fragment was detected using forward primer GM103F and reverse primer GM104F (5′-GCTAGCTTGCCAAACCTACAGGT). Primer GM104F is located within the targeting cassette. β-Hemoglobin was measured as an internal control with primers 5′-CAGAGGTCTGCTTTCCAGCA-3′ and 5′-TGTCACCCTGGCATAAAAGC-3′. Mice heterozygous for PCBP4 were bred, and MEFs were isolated from 13.5-d-old embryos as described previously (Scoumanne et al. 2011). Primary MEFs were cultured in DMEM supplemented with 10% fetal bovine serum (FBS). All animals were housed at the University of California at Davis Campus Veterinary Services vivarium facility. All animal care and use protocols were approved by the University of California at Davis Institutional Animal Care and Use Committee.
p53 knockout mice (C57BL/6J strain), developed in the laboratory of Dr. Tyler Jacks (Jacks et al. 1994), were purchased from the Jackson Laboratory. PCBP4+/− mice were bred with p53+/− mice to generate PCBP4+/−; p53+/− mice. To generate p53−/− and p53−/−; PCBP4−/− MEFs, PCBP4+/−; p53+/− mice were bred, and MEFs were isolated from 13.5-d-old embryos.
Histology and immunofluorescence staining
Mouse tissues were dissected, fixed in buffered formalin (pH 6.8–7.0) for 24 h, and then stored in 70% ethanol. Tissues were paraffin-embedded and sectioned. The 5-μm-thick sections were deparaffinized, rehydrated, and then stained with H&E.
To determine the origin of the lymphomas, immunofluorescence staining was performed with rat anti-B220 (B-cell marker) (BD Pharmingen) and rabbit anti-CD3 (T-cell marker) (Dako), respectively. Briefly, tissue sections were deparaffinized and antigen was retrieved with sodium citrate buffer (10 mM sodium citrate, 0.05% Tween 20 at pH 6.0) followed by blockage of endogenous peroxidase with 0.3% H2O2 in TBS (50 mM Tris HCl, 0.9% NaCl at pH 7.6). After being blocked in TBS with 10% FBS for 2 h at room temperature, the sections were incubated with primary antibodies overnight at 4°C. Next, the sections were incubated with fluorophore-conjugated secondary antibodies for 1 h at room temperature in a dark environment and mounted with mounting solution containing DAPI (4′, 6-diamidino-2-phenylindole).
Plasmids, siRNA oligos, and RT–PCR
To generate the GST-tagged mouse PCBP4-expressing vector, the coding region of PCBP4 was amplified by PCR using cDNA as a template with the forward primer 5′-GAATTCATGAGCAGTTCAGATGCGG-3′ and reverse primer 5′-CTCGAGTCAGTAGGGGGAGAATTTCTG-3′. To generate the pcDNA3-PCBP4-expressing vector, the same strategy as above was used except that the forward prime was replaced with 5′-AAGCTTACCATGAGCAGTTCAGATGCGG-3′. To generate the GST-tagged mouse ZFP871-expressing vector, the coding region of ZFP871 (accession no. NM_172458) was amplified by PCR using cDNA as a template with the forward primer 5′-GGATCCATGGAGTCAGTGGCCTTTG-3′ and reverse primer 5′-GCGGCCGCTAAAAAACACTGGAATCCAGG-3′. To generate HA-tagged mouse ZFP871 expressing vector, the same strategy as above was used except that the forward primer was replaced with 5′-GGATCCACCATGTACCCATACGACGTACCAGATTACGCTATGGAGTCAGTGGCCTTTG-3′. The PCR products were subcloned into pGEM-T easy vector. After confirmation by sequencing, the coding regions were then cloned into pcDNA3 or pGEX4T-1.
All siRNAs were purchased from Dharmacon RNA Technologies and transfected into cells at a final concentration of 100 nM for 3 d. The two scrambled siRNAs used were 5′-GCAGUGUCUCCACGUACUAdTdT-3′ (Scr-siRNA-1) and 5′-UUCUCCGAACGUGUCACGUdTdT-3′ (Scr-siRNA-2). To transiently knock down ZFP871 (accession no. NM_172458), three siRNAs against ZFP871 were used to rule out potential off-target effects. These were siZFP871-1 (5′-GGGCCAAGAAAUGCUAUAUdTdT-3′; nucleotides 784–802), siZFP871-2 (5′-GUGCGAAGAUCAGGAAUAdTdT-3′; nucleotides 537–555), and siZFP871-3 (5′-CACC CUUAUUCCUGUAAGUdTdT-3′; nucleotides 10,778–10,796). The siRNA against mouse p53 was 5′-ACCACUUGAUGGAGAGUAUdTdT-3′.
Total RNA was isolated from cells using TRIzol reagent (Invitrogen). cDNA was synthesized using the Iscript cDNA synthesis kit (Bio-Rad). To measure the levels of PCBP4 target mRNAs, RT–PCR was performed with the primers listed in Supplemental Table S10. Actin was measured as an internal control with the forward primer 5′-TCCATCATGAAGTGTGACGT-3′ and reverse primer 5′-TGATCCACATCTGCTGGAAG-3′.
Cell proliferation and cellular senescence assays
To determine whether knockdown of ZFP871 inhibits cell proliferation, mouse C2C12 myoblasts were transfected with scrambled siRNAs or siRNAs against ZFP871 for 3 d. Surviving cells were collected and counted. Cellular senescence assay was performed as previously described (Qian et al. 2008). The rate of senescent cells was calculated based on the number of positive cells among 2000 cells.
Immunoprecipitation and Western blot analysis
Immunoprecipitation was carried out as previously described (Jung et al. 2013). Briefly, C2C12 or FL83B cells were washed with phosphate-buffered saline (PBS), lysed in lysis buffer (50 mM Tris-Cl at pH 8.0, 150 mM NaCl, 1 mM EDTA, 1% Nonidet P-40, 1× protease inhibitor cocktail [PIC] [Sigma], 1 mM PMSF), sonicated, and clarified by centrifugation. Cell lysates (500 µg of total proteins) were incubated with rabbit anti-p53 (FL-393, Santa Cruz Biotechnology) for 4 h at 4°C and then with protein G-agarose beads (Sigma) for 2 h. After being washed with lysis buffer, immunoprecipitated protein complexes were subjected to SDS-PAGE along with whole-cell lysates as a control. For each set, 5% of the whole-cell lysates used for immunoprecipitation were used as an input control. IgG antibody was used as a negative control. Immunoblots were visualized by SuperSignal West femto chemiluminiscent detection reagents (Pierce). The antibodies used for Western blot assays included rabbit anti-p130 (C-20, Santa Cruz Biotechnology), rabbit anti-PML (H238, Santa Cruz Biotechnology), mouse anti-p21 (SX118, Santa Cruz Biotechnology), rabbit anti-KAI-1 (H135, Santa Cruz Biotechnology), rabbit anti-AKT or p-AKT (Cell Signaling), mouse anti-HA (Covance), rabbit anti-actin (Sigma), and mouse anti-GST (B14, Santa Cruz Biotechnology),
RNA–protein immunoprecipitation assay and high-throughput RNA-seq
RNA–protein immunoprecipitation assay was carried out as previously described (Peritz et al. 2006). Briefly, wild-type or PCBP4−/− MEFs at passage 3 were collected and lysed at 4°C with a lysis buffer (50 mM Tris-HCl at pH 7.4, 1% NP-40, 150 mM NaCl, 1× PIC, 1 mM PMSF, 0.5 U/μL RNasin). Five percent of the cell extracts were used for total RNA isolation, and the remaining extracts were incubated with protein A/G beads conjugated with rabbit anti-PCBP4 (ProSci) or a control IgG overnight at 4°C. Following four washes with a buffer containing RNase-free DNase, RNAs on the beads were purified with TRIzol reagent. The quality of RNA samples was analyzed with a bioanalyzer. The residual rRNA in the RNA samples was depleted with a Ribo-Zero rRNA removal kit (Epicentre Technologies Corp.). High-throughput RNA-seq was performed with HiSeq 2000 in the Functional Genomics Laboratory at the University of California at Berkeley.
REMSA
ZFP871 and p21 3′ UTRs were PCR-amplified using forward primers containing the T7 promoter sequence (5′-GGATCCTAA TACGACTCACTATAGGGAG-3′) and reverse primers listed in Supplemental Table S11. RNA probes were made from in vitro transcription by T7 RNA polymerase in the presence of α-32P-UTP. GST-tagged PCBP4 protein was purified as described previously (Scoumanne et al. 2011). REMSA was performed with 200 nM recombinant protein, 1 mg/mL yeast tRNA, and 50,000 cpm 32P-labeled RNA probe in a reaction buffer (10 mM HEPES at pH 8.0, 10 mM KCl, 10 mM MgCl2, 1 mM DTT) for 20 min at 25°C. RNA–protein complexes were digested with 100 U RNaseT1 for 15 min at 37°C and then separated in 6% native PAGE. RNA–protein complexes were visualized by autoradiography. To test the specificity of PCBP4 binding to the ZFP871 3′ UTR, the competition assay was performed by adding an excess amount of unlabeled p21 3′ UTR cold probe into the reaction mixture with a 32P-labeled probe as described previously (Cho et al. 2010).
Protein half-life assay
C2C12 cells were transfected with a scrambled siRNA or a siRNA against ZFP871 for 3 d and then treated with 50 µg/mL cycloheximide to inhibit de novo protein synthesis for various times. The relative levels of p53 protein were quantified by Western blotting and normalized by levels of actin protein, which were then plotted versus time (in minutes) to calculate the half-life of the p53 protein.
Nuclear extraction, GST-ZFP871 fusion protein purification, and GST pull-down assay
C2C12 cells cultured in a 10-cm plate were washed twice with cold PBS and then lysed with 1 mL of cytoplasmic lysis buffer (10 mM HEPES at pH 7.9, 10 mM KCl, 0.1 mM EDTA,1 mM DTT, 1×PIC, 0.4% NP-40) for 10 min on ice. After centrifugation at 12,000g for 1 min, the pellet (nucleus) was resuspended with 100 µL of GST-binding buffer (25 mM Tris·HCl at pH 7.5, 150 mM NaCl, 1×PIC, 1% Triton-X100) and stored at −80°C for pull-down assay.
Recombinant GST-tagged ZFP871 protein was expressed in Escherichia coli BL21(DE3) and purified according to a standard protocol (Rebay and Fehon 2009). After washing, GST and GST-ZFP871 beads were resuspended in GST-binding buffer to make 50% slurry and stored at 4°C for pull-down assay.
For GST pull-down assay, 0.4 mL of GST-binding buffer containing 400 µg of nuclear extracts was added into 20 µL of sepharose 4B beads for preclearing and then added into 20 µL of GST beads or GST-ZFP871 beads. After incubation overnight at 4°C, the mixture was centrifuged to precipitate the beads at 500g for 5 min. The beads were washed four times with GST-binding buffer and then resuspended in 40 µL of 2× SDS loading buffer for SDS-PAGE and Western blot analysis.
Ubiquitination assay
The in vivo ubiquitination assay was performed as described previously (Leng et al. 2003). C2C12 cells were transfected with pcDNA3-p53 and pcDNA3-Flag-ubiquitin with or without pcDNA3-HA-ZFP871 followed by treatment with 5 µM MG132 for 6 h. Cell lysates were immunoprecipitated with rabbit anti-p53 antibody followed by Western blot analysis with mouse anti-ubiquitin or anti-p53 antibody to detect polyubiquitinated p53.
DNA histogram analysis and TUNEL assay
C2C12 cells were seeded at 2 × 105 per 6-cm plate and then transfected with scrambled siRNA, siRNA against ZFP871, siRNA against p53, or both siRNA against ZFP871 and siRNA against p53 for 3 d. Both floating dead cells in the medium and live cells on the plate were collected and fixed with 70% ethanol for 24 h at 4°C. The fixed cells were centrifuged and resuspended in 0.3 mL of PBS containing 50 μg/mL RNase A and 50 μg/mL propidium iodide (PI). The stained cells were analyzed using a fluorescence-activated cell sorter. The percentages of cells in the sub-G1, G1, S, and G2–M phases were determined using the CellQuest program (BD Biosciences).
TUNEL assay was performed with an in situ cell death detection kit (Roche) according to the manual. Briefly, cells were fixed with 4% paraformaldehyde for 1 h and then permeabilized with 0.1% Triton X-100 in 0.1% sodium citrate for 2 min on ice. After being washed twice with PBS, cells were incubated with 50 µL of TUNEL reaction mixture for 60 min at 37°C in a humidified atmosphere in the dark. After being washed three times with PBS, the cells were counterstained with DAPI and then analyzed under a fluorescence microscope.
Statistics
Fisher's exact test was used for comparison between tumor incidences in wild-type and PCBP4-deficient mice. The log rank test was used to determine the differences in median survival of different genotypes. Two-group comparisons were analyzed by two-sided Student's t-test. P-values were calculated, and P < 0.05 was considered significant.
Supplementary Material
Acknowledgments
We thank Dr. Huaijun Zhou and Dr. Ying Wang for analyzing RNA-seq data, Dr. Kent Lloyd and the Mouse Biology Program at University of California at Davis for generating the PCBP4-deficient mouse model, and Dr. Mingyi Chen for interpreting the histopathological data of the PCBP4-deficient mice. This study is supported in part by National Institutes of Health grants CA076069, CA081237, CA121137, and CA123227. W.Y., A.S., Y.-S.J., E.X., J.Z., Y.Z., C.R., and P.S. performed the experiments. W.Y., A.S., and X.C. designed the experiments and analyzed the data. W.Y. and X.C. wrote the manuscript. A.S. and J.Z. proofread and edited the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and pubication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.271890.115.
References
- Angeloni D. 2007. Molecular analysis of deletions in human chromosome 3p21 and the role of resident cancer genes in disease. Brief Funct Genomic Proteomic 6: 19–39. [DOI] [PubMed] [Google Scholar]
- Bensaude O. 2011. Inhibiting eukaryotic transcription: which compound to choose? How to evaluate its activity? Transcription 2: 103–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks CL, Gu W. 2006. p53 ubiquitination: Mdm2 and beyond. Mol Cell 21: 307–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castano Z, Vergara-Irigaray N, Pajares MJ, Montuenga LM, Pio R. 2008. Expression of αCP-4 inhibits cell cycle progression and suppresses tumorigenicity of lung cancer cells. Int J Cancer 122: 1512–1520. [DOI] [PubMed] [Google Scholar]
- Chen D, Kon N, Li M, Zhang W, Qin J, Gu W. 2005. ARF-BP1/Mule is a critical mediator of the ARF tumor suppressor. Cell 121: 1071–1083. [DOI] [PubMed] [Google Scholar]
- Cho SJ, Zhang J, Chen X. 2010. RNPC1 modulates the RNA-binding activity of, and cooperates with, HuR to regulate p21 mRNA stability. Nucleic Acids Res 38: 2256–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HS, Hwang CK, Song KY, Law PY, Wei LN, Loh HH. 2009. Poly(C)-binding proteins as transcriptional regulators of gene expression. Biochem Biophys Res Commun 380: 431–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdary DR, Dermody JJ, Jha KK, Ozer HL. 1994. Accumulation of p53 in a mutant cell line defective in the ubiquitin pathway. Mol Cell Biol 14: 1997–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dammann R, Li C, Yoon JH, Chin PL, Bates S, Pfeifer GP. 2000. Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat Genet 25: 315–319. [DOI] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr, Butel JS, Bradley A. 1992. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356: 215–221. [DOI] [PubMed] [Google Scholar]
- Dornan D, Wertz I, Shimizu H, Arnott D, Frantz GD, Dowd P, O'Rourke K, Koeppen H, Dixit VM. 2004. The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature 429: 86–92. [DOI] [PubMed] [Google Scholar]
- Endo C, Sato M, Fujimura S, Sakurada A, Aikawa H, Takahashi S, Usuda K, Saito Y, Sagawa M. 2000. Allelic loss on 17p13 (TP53) and allelic loss on 3p21 in early squamous cell carcinoma of the lung. Surg Today 30: 695–699. [DOI] [PubMed] [Google Scholar]
- Evans JR, Mitchell SA, Spriggs KA, Ostrowski J, Bomsztyk K, Ostarek D, Willis AE. 2003. Members of the poly (rC) binding protein family stimulate the activity of the c-myc internal ribosome entry segment in vitro and in vivo. Oncogene 22: 8012–8020. [DOI] [PubMed] [Google Scholar]
- Haupt Y, Maya R, Kazaz A, Oren M. 1997. Mdm2 promotes the rapid degradation of p53. Nature 387: 296–299. [DOI] [PubMed] [Google Scholar]
- Hesson LB, Cooper WN, Latif F. 2007. Evaluation of the 3p21.3 tumour-suppressor gene cluster. Oncogene 26: 7283–7301. [DOI] [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. 1994. Tumor spectrum analysis in p53-mutant mice. Curr Biol 4: 1–7. [DOI] [PubMed] [Google Scholar]
- Jung YS, Qian Y, Yan W, Chen X. 2013. Pirh2 E3 ubiquitin ligase modulates keratinocyte differentiation through p63. J Invest Dermatol 133: 1178–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna SS, Majumdar I, Grishin NV. 2003. Structural classification of zinc fingers: survey and summary. Nucleic Acids Res 31: 532–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubbutat MH, Jones SN, Vousden KH. 1997. Regulation of p53 stability by Mdm2. Nature 387: 299–303. [DOI] [PubMed] [Google Scholar]
- Laine A, Ronai Z. 2007. Regulation of p53 localization and transcription by the HECT domain E3 ligase WWP1. Oncogene 26: 1477–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laity JH, Lee BM, Wright PE. 2001. Zinc finger proteins: new insights into structural and functional diversity. Curr Opin Struct Biol 11: 39–46. [DOI] [PubMed] [Google Scholar]
- Lee JT, Gu W. 2010. The multiple levels of regulation by p53 ubiquitination. Cell Death Differ 17: 86–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng RP, Lin Y, Ma W, Wu H, Lemmers B, Chung S, Parant JM, Lozano G, Hakem R, Benchimol S. 2003. Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell 112: 779–791. [DOI] [PubMed] [Google Scholar]
- Lindstrom MS, Jin A, Deisenroth C, White Wolf G, Zhang Y. 2007. Cancer-associated mutations in the MDM2 zinc finger domain disrupt ribosomal protein interaction and attenuate MDM2-induced p53 degradation. Mol Cell Biol 27: 1056–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loughlin FE, Mackay JP. 2006. Zinc fingers are known as domains for binding DNA and RNA. Do they also mediate protein–protein interactions? IUBMB Life 58: 731–733. [DOI] [PubMed] [Google Scholar]
- Makeyev AV, Liebhaber SA. 2000. Identification of two novel mammalian genes establishes a subfamily of KH-domain RNA-binding proteins. Genomics 67: 301–316. [DOI] [PubMed] [Google Scholar]
- Makeyev AV, Liebhaber SA. 2002. The poly(C)-binding proteins: a multiplicity of functions and a search for mechanisms. RNA 8: 265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfredi JJ. 2010. The Mdm2–p53 relationship evolves: Mdm2 swings both ways as an oncogene and a tumor suppressor. Genes Dev 24: 1580–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsit CJ, Hasegawa M, Hirao T, Kim DH, Aldape K, Hinds PW, Wiencke JK, Nelson HH, Kelsey KT. 2004. Loss of heterozygosity of chromosome 3p21 is associated with mutant TP53 and better patient survival in non-small-cell lung cancer. Cancer Res 64: 8702–8707. [DOI] [PubMed] [Google Scholar]
- Odell A, Askham J, Whibley C, Hollstein M. 2010. How to become immortal: let MEFs count the ways. Aging 2: 160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostareck DH, Ostareck-Lederer A, Wilm M, Thiele BJ, Mann M, Hentze MW. 1997. mRNA silencing in erythroid differentiation: hnRNP K and hnRNP E1 regulate 15-lipoxygenase translation from the 3′ end. Cell 89: 597–606. [DOI] [PubMed] [Google Scholar]
- Paulding WR, Czyzyk-Krzeska MF. 1999. Regulation of tyrosine hydroxylase mRNA stability by protein-binding, pyrimidine-rich sequence in the 3′-untranslated region. J Biol Chem 274: 2532–2538. [DOI] [PubMed] [Google Scholar]
- Peritz T, Zeng F, Kannanayakal TJ, Kilk K, Eiriksdottir E, Langel U, Eberwine J. 2006. Immunoprecipitation of mRNA–protein complexes. Nat Protoc 1: 577–580. [DOI] [PubMed] [Google Scholar]
- Pio R, Zudaire I, Pino I, Castano Z, Zabalegui N, Vicent S, Garcia-Amigot F, Odero MD, Lozano MD, Garcia-Foncillas J, et al. 2004. αCP-4, encoded by a putative tumor suppressor gene at 3p21, but not its alternative splice variant αCP-4a, is underexpressed in lung cancer. Cancer Res 64: 4171–4179. [DOI] [PubMed] [Google Scholar]
- Qian Y, Zhang J, Yan B, Chen X. 2008. DEC1, a basic helix-loop-helix transcription factor and a novel target gene of the p53 family, mediates p53-dependent premature senescence. J Biol Chem 283: 2896–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebay I, Fehon RG. 2009. Preparation of soluble GST fusion proteins. Cold Spring Harb Protoc 10.1101/pdb.prot4996. [DOI] [PubMed] [Google Scholar]
- Schnutgen F, De-Zolt S, Van Sloun P, Hollatz M, Floss T, Hansen J, Altschmied J, Seisenberger C, Ghyselinck NB, Ruiz P, et al. 2005. Genomewide production of multipurpose alleles for the functional analysis of the mouse genome. Proc Natl Acad Sci 102: 7221–7226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scoumanne A, Cho SJ, Zhang J, Chen X. 2011. The cyclin-dependent kinase inhibitor p21 is regulated by RNA-binding protein PCBP4 via mRNA stability. Nucleic Acids Res 39: 213–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shloush J, Vlassov JE, Engson I, Duan S, Saridakis V, Dhe-Paganon S, Raught B, Sheng Y, Arrowsmith CH. 2011. Structural and functional comparison of the RING domains of two p53 E3 ligases, Mdm2 and Pirh2. J Biol Chem 286: 4796–4808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R, Naishadham D, Jemal A. 2013. Cancer statistics, 2013. CA Cancer J Clin 63: 11–30. [DOI] [PubMed] [Google Scholar]
- Stefanovic B, Hellerbrand C, Holcik M, Briendl M, Aliebhaber S, Brenner DA. 1997. Posttranscriptional regulation of collagen α1(I) mRNA in hepatic stellate cells. Mol Cell Biol 17: 5201–5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai AL, Mak W, Ng PK, Chua DT, Ng MY, Fu L, Chu KK, Fang Y, Qiang Song Y, Chen M, et al. 2006. High-throughput loss-of-heterozygosity study of chromosome 3p in lung cancer using single-nucleotide polymorphism markers. Cancer Res 66: 4133–4138. [DOI] [PubMed] [Google Scholar]
- Tian C, Xing G, Xie P, Lu K, Nie J, Wang J, Li L, Gao M, Zhang L, He F. 2009. KRAB-type zinc-finger protein Apak specifically regulates p53-dependent apoptosis. Nat Cell Biol 11: 580–591. [DOI] [PubMed] [Google Scholar]
- Tomizawa Y, Sekido Y, Kondo M, Gao B, Yokota J, Roche J, Drabkin H, Lerman MI, Gazdar AF, Minna JD. 2001. Inhibition of lung cancer cell growth and induction of apoptosis after reexpression of 3p21.3 candidate tumor suppressor gene SEMA3B. Proc Natl Acad Sci 98: 13954–13959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valverde R, Edwards L, Regan L. 2008. Structure and function of KH domains. FEBS J 275: 2712–2726. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Prives C. 2009. Blinded by the light: the growing complexity of p53. Cell 137: 413–431. [DOI] [PubMed] [Google Scholar]
- Wang X, Kiledjian M, Weiss IM, Liebhaber SA. 1995. Detection and characterization of a 3′ untranslated region ribonucleoprotein complex associated with human α-globin mRNA stability. Mol Cell Biol 15: 1769–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu GW, Allen MD, Andreeva A, Fersht AR, Bycroft M. 2006. Solution structure of the C4 zinc finger domain of HDM2. Protein Sci 15: 384–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Chen X. 2000. MCG10, a novel p53 target gene that encodes a KH domain RNA-binding protein, is capable of inducing apoptosis and cell cycle arrest in G2-M. Mol Cell Biol 20: 5602–5618. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.