

Abstract
Objective:
To examine safety, tolerability, pharmacokinetics, and preliminary clinical efficacy of intrathecal nusinersen (previously ISIS-SMNRx), an antisense oligonucleotide designed to alter splicing of SMN2 mRNA, in patients with childhood spinal muscular atrophy (SMA).
Methods:
Nusinersen was delivered by intrathecal injection to medically stable patients with type 2 and type 3 SMA aged 2–14 years in an open-label phase 1 study and its long-term extension. Four ascending single-dose levels (1, 3, 6, and 9 mg) were examined in cohorts of 6–10 participants. Participants were monitored for safety and tolerability, and CSF and plasma pharmacokinetics were measured. Exploratory efficacy endpoints included the Hammersmith Functional Motor Scale Expanded (HFMSE) and Pediatric Quality of Life Inventory.
Results:
A total of 28 participants enrolled in the study (n = 6 in first 3 dose cohorts; n = 10 in the 9-mg cohort). Intrathecal nusinersen was well-tolerated with no safety/tolerability concerns identified. Plasma and CSF drug levels were dose-dependent, consistent with preclinical data. Extended pharmacokinetics indicated a prolonged CSF drug half-life of 4–6 months after initial clearance. A significant increase in HFMSE scores was observed at the 9-mg dose at 3 months postdose (3.1 points; p = 0.016), which was further increased 9–14 months postdose (5.8 points; p = 0.008) during the extension study.
Conclusions:
Results from this study support continued development of nusinersen for treatment of SMA.
Classification of evidence:
This study provides Class IV evidence that in children with SMA, intrathecal nusinersen is not associated with safety or tolerability concerns.
Spinal muscular atrophy (SMA) is an autosomal recessive disease characterized primarily by motor neuron degeneration, resulting in muscular atrophy and weakness involving limbs, and more variably, bulbar and respiratory muscles.1,2 The natural history of SMA includes several major phenotypes that are characterized by age at onset and achieved motor abilities.3 SMA is caused by deletions or loss-of-function mutations in the survival of motor neuron (SMN1) gene located on chromosome 5q13.4 Humans also have a paralogous SMN2 gene that has an identical coding sequence to SMN1 but differs by 11 nucleotides.5 One of the nucleotide changes is a C to T transition within exon 7, resulting in 80%–90% of the transcripts, excluding exon 7, producing a truncated protein that is rapidly degraded.5 As a result, the limited amount of full-length protein produced is insufficient to compensate for loss of the SMN1 gene, causing the SMA phenotype.5
Nusinersen (previously ISIS-SMNRx and also known as ISIS 396443) is an antisense oligonucleotide designed to bind to the SMN2 pre-mRNA and promote inclusion of exon 7 (figure 1).6,7 In mouse models of SMA, nusinersen enhanced exon 7 inclusion, increased SMN protein production, and improved function.6–9 Nusinersen is delivered by intrathecal injection as antisense oligonucleotide drugs do not cross an intact blood–brain barrier when delivered systemically.10 This first-in-human, open-label, single-ascending dose study was designed to assess safety, tolerability, pharmacokinetics, and clinical effects of intrathecal nusinersen in patients with childhood SMA.
Figure 1. Mechanism of action of nusinersen.

Nusinersen is a 2′-O-(2-methoxyethyl) modified ASO drug designed to target an hnRNP-A1/A2–dependent splicing silencer, ISS-N1, in intron 7 of the SMN pre-mRNA. Nusinersen displaces hnRNP proteins from the ISS-N1 site on the SMN2 pre-mRNA, facilitating accurate splicing of SMN2 transcripts (e.g., increasing the synthesis of transcripts containing exon 7) and resulting in increased production of full-length SMN protein. ASO = antisense oligonucleotide; hnRNP = heterogenous nuclear ribonucleoprotein; ISS = intronic splicing silencer; mRNA = messenger RNA; SMA = spinal muscular atrophy; SMN = survival of motor neuron.
METHODS
Standard protocol approvals, registrations, and participant consents.
Written informed parental consent and assent was obtained for all participants as required. Studies were initiated after institutional review board approvals of the participating study centers and carried out in accordance with Good Clinical Practice guidelines. A data and safety monitoring board (DSMB) monitored the studies. Studies were registered with ClinicalTrials.gov (NCT01494701; NCT01780246).
Study design.
In this open-label, escalating-dose, phase 1 study, participants received a single intrathecal injection of 5 mL nusinersen following standard lumbar puncture (LP) techniques (over 1–3-minutes). Four dose levels (1, 3, 6, 9 mg) were evaluated in sequential groups: dose escalation was dependent on DSMB review of safety data through study day 8 for the preceding dose group. Participants were eligible to re-enroll in an open-label extension study at 9–14 months after their initial nusinersen dose to receive additional study drug; data included in this report include baseline evaluations for the follow-up study only.
Participants.
Male and female patients aged 2–14 years with symptomatic SMA and documented SMN1 homozygous gene deletion were eligible to participate. Participants were enrolled at 4 study centers: Columbia University, University of Utah, Boston Children's Hospital, and University of Texas Southwestern Medical School. Inclusion/exclusion criteria were designed to enroll patients who were medically stable and in whom safety concerns could be clearly assessed. Briefly, participants needed to be able to complete all study procedures, meet age-appropriate institutional guidelines for LP procedures, and have a life expectancy of >2 years per investigator judgement. Participants were excluded for respiratory insufficiency, hospitalization for surgery or pulmonary event within the past 2 months, active infection at screening, history of brain or spinal cord disease or bacterial meningitis, presence of implanted CSF drainage shunt, clinically significant laboratory abnormalities, any ongoing medical condition that would interfere with the conduct and assessments of the study, or treatment with another investigational drug ≤1 month of screening.
Study procedures.
LP was performed under anesthesia/sedation per institutional guidelines. Before intrathecal injection of study drug, 5–6 mL of CSF was collected for analysis. Following injection, participants were observed for 24 hours. Follow-up visits were performed on days 8 and 29 for all participants and on day 85 for participants in the 6- and 9-mg dose groups. A second LP to collect CSF for safety and pharmacokinetics was performed on day 8 in the 1-, 3-, and 6-mg dose groups and on days 8 or 29 in the 9-mg dose group (n = 5 at each time point). Participants were assessed 9–14 months postdose at enrollment into the long-term extension study, using assessments identical to those employed in the single-dose study.
Safety assessments.
Safety assessments included collection of adverse events (AEs), physical/neurologic examinations, vital signs, clinical laboratory tests (serum chemistry, hematology, urinalysis; analyzed centrally at PPD, Wilmington, NC), and ECGs. CSF safety laboratory tests (cell counts, protein, glucose) were assessed at 7 or 28 days and 9–14 months postdose (analyzed in local laboratories), and CSF cytokines (interleukin-6, tumor necrosis factor–α, monocyte chemotactic protein 1; Aushon BioSystems, Billerica, MA) at 7 or 28 days postdose. Plasma samples for participants in the 6- and 9-mg cohorts were collected on days 1, 8 or 29, and 85 and 9–14 months postdose for the analysis of anti–nusinersen antibodies using an immunogenicity assay validated to be specific for the detection of anti–nusinersen antibodies (Charles River Laboratories, Wilmington, MA).
Pharmacokinetic and SMN protein assessments.
Plasma pharmacokinetic specimens were collected on day 1 predose, at 1, 2, 4, 6, 8, 12, and 20 hours postdose, and on day 8 in the 6- and 9-mg cohorts. CSF pharmacokinetic specimens were collected on day 1 predose, days 8 and 29, and 9–14 months. Nusinersen concentrations were determined using a variation of the hybridization ELISA method11 or an electrochemiluminescence method; both methods were validated (PPD). SMN protein concentration was determined using an Erenna Immunoassay System validated for human CSF (Singulex, Inc., Alameda, CA). CSF elimination half-life was estimated for each individual using concentrations determined at 7 or 29 days and 9–14 months by using the following equation:
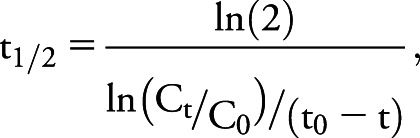 |
where t1/2 is half-life in days; Ct and C0 are CSF concentrations at 9–14 months and 7 or 29 days, respectively; and (t0 − t) is time in days between measurements.
Clinical outcome measures.
Exploratory clinical assessments included Hammersmith Functional Motor Scale Expanded (HFMSE)12,13 performed at baseline, day 29, day 85 (6- and 9-mg cohorts), and at 9–14 months; and Pediatric Quality of Life Inventory (PedsQL) Measurement 4.0 Generic Core Scales and 3.0 Neuromuscular Module14,15 at baseline, day 29, and day 85 (6- and 9-mg cohorts). For HFMSE, physical therapist evaluators were trained annually with a standardized procedure manual. Intraclass correlation coefficients for interrater reliability among all evaluators were very high (>0.951; computed using 1-way random effects analysis of variance models of 3 videotaped assessments).
Sample size and statistical analysis.
Sample size was selected based upon prior experience with phase 1 single-dose studies of antisense oligonucleotide. There was no statistically based rationale for the number of participants chosen. All participants who received the single dose were included in safety, tolerability, pharmacokinetic, and efficacy analyses. Exploratory efficacy endpoints were assessed using Wilcoxon signed-rank test.
Classification of evidence.
This study provides Class IV evidence that in children with SMA, a single dose of nusinersen up to 9 mg is not associated with safety or tolerability concerns on a follow-up of 9–14 months. A single dose of 9 mg nusinersen resulted in an increase in HFMSE scores in children with SMA; however, the study was open-label and results would need to be confirmed in a controlled trial.
RESULTS
Participants.
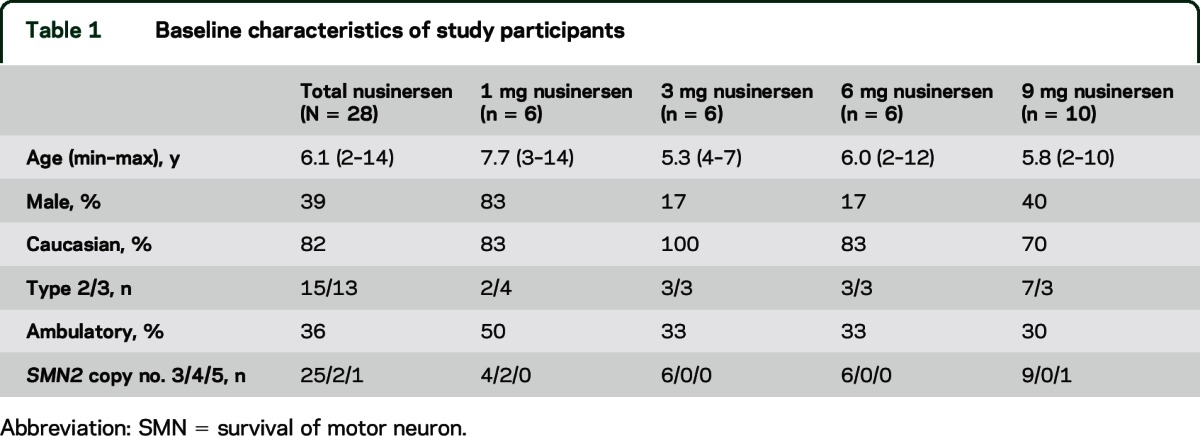
Six participants were enrolled in the 1-, 3-, and 6-mg dose groups, and 10 participants in the 9-mg group. All participants completed treatment and follow-up evaluations. Demographics/background disease characteristics are presented in table 1. Twenty-four of 28 participants in the phase 1 study were enrolled in the long-term extension study 9–14 months after initial dose and thus, their demographics were similar to those in the single-dose study.
Table 1.
Baseline characteristics of study participants
Safety and tolerability.
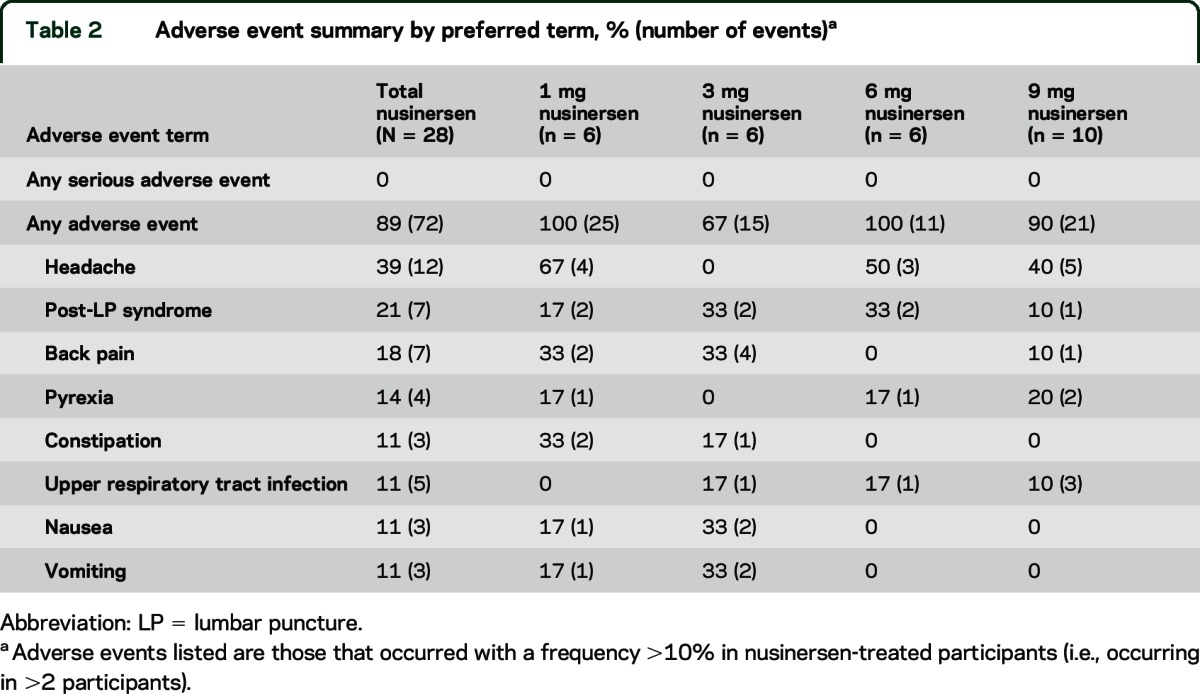
There were no serious AEs during the course of the study and no participant experienced an AE leading to discontinuation. Overall, 89% of participants reported AEs (table 2); most AEs were mild in severity, with only 5 participants reporting an event that was moderate in severity and none that was severe. The most frequently observed AEs by prevalence were headache (39.3% of participants), post-LP headache (21.4%), and back pain (17.9%). There were 2 AEs considered potentially related to the study drug: a mild paresthesia in the 1-mg group and a report of palpitations in the 3-mg group, both of which resolved spontaneously without sequelae. The incidence of post-LP headache was 10.9% (6 events reported for 55 LPs performed). No clinically significant changes in vital signs, neurologic or physical examinations, clinical laboratory tests, or ECGs were reported. There were no clinically significant changes in CSF safety laboratory tests or CSF cytokines. All samples were negative for the presence of anti–nusinersen antibodies, indicating that an immunogenic response to nusinersen was not observed at 9–14 months after a single intrathecal dose of nusinersen.
Table 2.
Adverse event summary by preferred term, % (number of events)a
Pharmacokinetic profile.
Nusinersen concentrations were measurable over 24 hours postdose in plasma and 7 days postdose in CSF (figure 2), as well as 29 days postdose in CSF in the 9-mg group (2.33 ± 0.928 ng/mL). Nusinersen concentrations 7 days after dosing increased dose dependently but less than dose proportionally (3.7-fold) over the evaluated dose range. A substantial portion of intrathecally administered nusinersen rapidly transferred from the site of administration (CSF) into systemic circulation, with dose-dependent mean peak plasma levels observed within a few hours after dosing. After peaking, nusinersen plasma concentrations declined slowly ≤20 hours after intrathecal dosing, followed by a slower period of decline over the next 7 days. By 7 days postdose, concentrations in the 6-mg cohort had decreased to below the limit of quantification (1 ng/mL), and to <1% of the 20-hour postdose concentration in the 9-mg cohort. Dose-normalized total plasma exposure measures decreased ∼3-fold over a ∼3-fold increase in total body weight. A similar relationship between dose-normalized total plasma exposure and age was observed. No differences were observed between males and females for untransformed total plasma exposure (area under the concentration time curve from 0 to 20 hours [AUC0–20h]) measures stratified by cohort or for dose-normalized total plasma exposure (AUC0–20h) measures. At the 9- to 14-month evaluation, CSF nusinersen concentrations were still measurable. Apparent terminal half-life in CSF was estimated to be 132–166 days (mean ± SD days: 132 ± 42, 1-mg group; 135 ± 14.8, 3-mg group; 163 ± 26.5, 6-mg group; 177 ± 41.3, 9-mg group).
Figure 2. CSF and plasma concentrations of nusinersen.
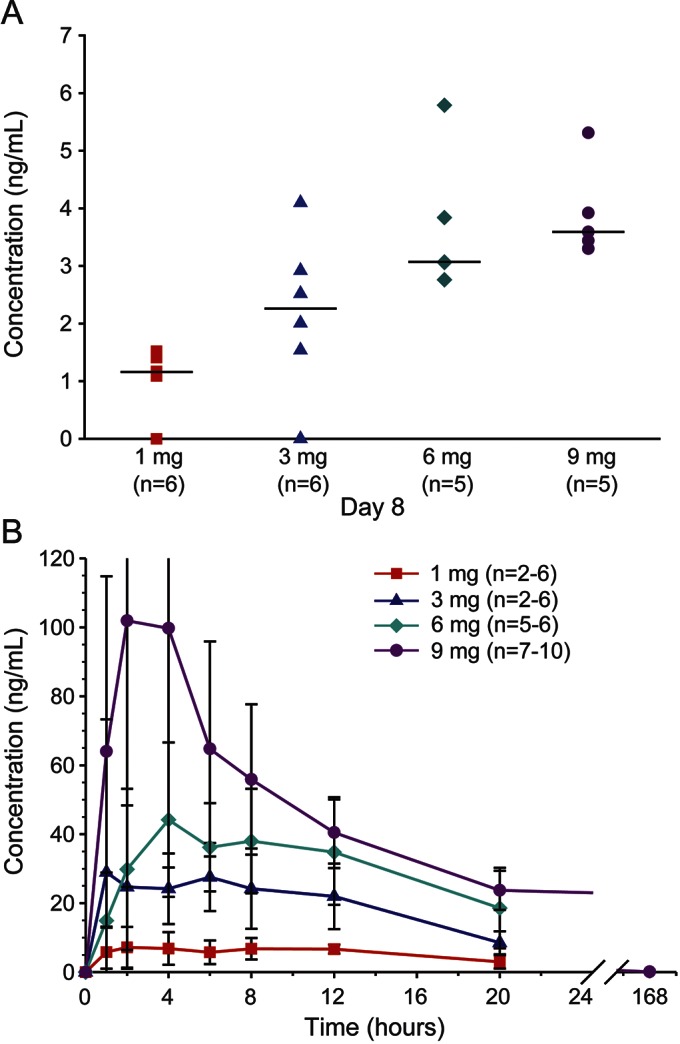
Measured nusinersen concentrations for each dose group are shown. (A) CSF at 7 days postdose. (B) Plasma over 24 hours (all groups) or 7 days (6- and 9-mg groups) postdose (mean ± SEM). As anticipated, plasma levels were below the limit of detection of the assay at day 8 postdose.
Analysis of CSF samples for SMN protein levels indicated no change in the 1- or 3-mg groups, but that SMN protein levels more than doubled at 9–14 months postdose compared with baseline in the 6- and 9-mg groups (6-mg group: baseline, 0.27 ± 0.03 pg/mL; 9–14 months, 0.56 ± 0.12 pg/mL; not statistically significant; 118% mean increase; 9-mg group: baseline, 0.31 ± 0.18 pg/mL; 9–14 months, 0.59 ± 0.22 pg/mL; p = 0.06; 161% mean increase).
Clinical outcomes.
There were no significant changes in HFMSE assessments (figure 3) from baseline at day 29, day 85, or 9–14 months postdose in the 1-, 3-, or 6-mg groups (mean change: 1-mg group, +1.0 point at day 29 and –1.7 point at 9–14 months; 3-mg group, +1.0 point at day 29 and +0.5 point at 9–14 months; 6-mg group, +0.7 point at day 85 and +2.5 at 9–14 months). In contrast, the 9-mg group demonstrated improved HFMSE scores from baseline at day 85 (mean increase of 3.1 points or 17.6% increase; p = 0.016), with 7/10 (70%) participants exhibiting an increase of 3–7 points. Observed improvements in HFMSE scores of ≥3 points were equally distributed by severity; 5/7 type 2 participants and 2/3 type 3 participants showed this level of improvement. Similarly, HFMSE score improvement was distributed broadly across the age range, with 3 children aged <5 years and 4 children aged ≥5 years showing improvement. At long-term follow-up, mean change in HFMSE score from baseline to 9–14 months was 5.8 points (32.8% increase; p = 0.008; n = 8). Six of 8 participants had an increase of ≥3 points. No participants in the 9-mg cohort declined in HFMSE score, and range of improvement was 1–14 points.
Figure 3. Changes in Hammersmith Functional Motor Scale Expanded (HFMSE) scores by treatment assignment.
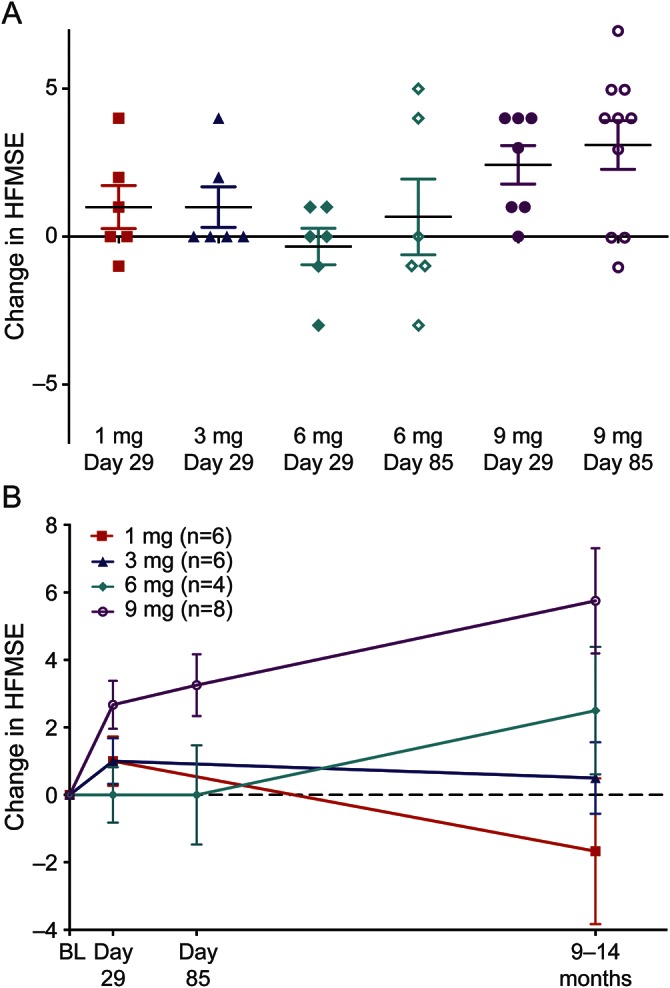
(A) Individual change in HFMSE score from baseline for all participants. (B) Mean change in HFMSE score from baseline in participants followed through 9–14 months postdose. Bars represent mean ± SEM for each dose group. BL = baseline.
A slight improvement for the PedsQL Generic Core Scales was observed compared with baseline in the 9-mg group at day 85 (mean percent change: 9.8% [patient] and 8.4% [parent], not statistically significant). Similarly, a slight improvement in the PedsQL Neuromuscular Model was observed compared with baseline in the 9-mg group at day 85, with the change being greater for the patient compared with the parent report (mean percent change: 17.7% [patient] and 4.6% [parent], not statistically significant). No meaningful changes were observed in the other dose groups.
DISCUSSION
Nusinersen was well-tolerated when given as a single intrathecal bolus injection to children with SMA and no safety concerns were identified. No serious AEs or discontinuations due to AEs were reported. The most commonly reported AEs were headache, post-LP headache, and backache; however, none of these events was considered to be related to nusinersen. No clinically significant abnormalities were noted on neurologic examinations or laboratory safety assessments, including CSF safety assessments at 7 or 28 days and 9–14 months postdose. These results are consistent with previous but limited clinical studies on direct CNS delivery of antisense oligonucleotide drugs, which appeared to be similarly well-tolerated.16,17 In this study, the LP injection procedure itself was well-tolerated and was shown to be feasible in children with SMA. Incidence rate of post-LP headache overall was 10.9%, which is consistent with the expected post-LP headache rate of ∼10% in children from published literature,18–20 further taking into account that patients in the study underwent 2 LPs (except for 1 patient who had only 1 LP) and that patients with SMA often have scoliosis or spinal deformities, making LPs more challenging. Eighteen of the children with SMA underwent 3 repeated LPs, with no increase in AEs or tolerability issues reported, further supporting the feasibility of this approach. The presence of hardware from scoliosis surgery in some patients with SMA, however, may limit the feasibility of intrathecal injections in the broader population of patients with SMA.
CSF and plasma drug concentrations of nusinersen were measurable and dose-dependent. No readily apparent correlations were observed between age or total body weight and CSF concentrations, consistent with literature reports indicating that CSF volume is similar in all children aged >2 years,21 and suggesting that fixed doses are appropriate in this pediatric population, with no dose scaling necessary. CSF and plasma drug levels were reasonably consistent with predicted values (within 2- to 3-fold) based on preclinical (monkey to human) CSF volume scaling. Dose-dependent mean peak plasma levels were observed within a few hours after dosing and then typically declined slowly ≤20 hours after intrathecal dosing. The slow decline may be because of CSF turnover and transfer of any remaining drug not taken up into CNS tissues from CSF into systemic circulation, coupled with the expected rapid and extensive distribution of systemically available drug from the plasma to systemic tissues. A consistent finding of a long drug half-life in CNS tissue for nusinersen has been observed in mice (145–191 days) and monkeys (139 ± 54 days)22 and was confirmed in humans by the observed CSF half-life of between 4–6 months in children with SMA. This observation is critical in that it provides support for infrequent administration of nusinersen for maintenance of drug levels (i.e., once every 4–6 months) following drug loading of the target tissue.
Although preliminary and requiring confirmation by a larger controlled study, improvement in patients' motor function as evidenced by increases in HFMSE score was observed following a single intrathecal dose of 9 mg nusinersen, with a 3.1-point mean increase from baseline at 3 months and a 5.8-point mean increase at 9–14 months. This improvement in motor function is not expected to occur as part of the disease process, as the predicted natural history trajectory of the HFMSE in type 2 and type 3 patients with SMA indicates stability over 1 year23 and a small decline over 2 years (−1 point).24 Though trials in patients with SMA evaluating other investigational therapies have ultimately been unsuccessful and several previous open-label trials have shown strong placebo effects, variations on the HFMSE have been previously used as the primary outcome measures in SMA clinical studies.25,26 It has been projected, including as an assumption in previous trials, that a change of 3 points over a 6-month period would constitute a clinically meaningful change in this endpoint,26 and thus, the 5.8-point increase observed at 9–14 months in the 9-mg group in this study clearly falls within the meaningful range. Consistent with the intended biological mechanism of nusinersen, an increase in CSF SMN protein concentrations correlated well with the improvement in motor function. In addition, the study findings reaffirm the sensitivity of the HFMSE to detect change in functional motor outcome and inform the length of time that is required to observe improvement in motor function after treatment.
Nusinersen dose levels for this first-in-human study were selected based on preclinical pharmacokinetic observations from monkey studies and consideration of the target tissue concentration anticipated for drug pharmacology from SMA transgenic mouse models.22 As nonhuman primates do not express the SMN2 gene, it was not possible to measure pharmacology in that animal species. Based upon pharmacology and pharmacokinetic results in SMN2 transgenic mice, the target tissue concentration to produce 50%–90% SMN2 exon 7 inclusion was estimated to be between 1 and 10 μg/g in spinal cord tissues.10 The lowest dose selected for this clinical study (1 mg) was predicted to achieve the low end of this range, while the highest dose (9 mg) was predicted to achieve spinal cord tissue concentrations at the upper end of this range. However, no dose-limiting safety issues were identified in this study, suggesting that doses higher than the 9 mg tested in this study should be considered.
The favorable risk-benefit profile from this first-in-human clinical study of nusinersen in children with SMA provides encouragement for further development of nusinersen for the treatment of SMA, for which no therapeutic options currently exist. Ongoing multiple-dose studies, which include incorporation of higher doses, are currently being conducted in both infants and children with SMA.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the patients who participated in this study and their parents/guardians and family members; and Cure SMA (formerly the Families of SMA) and Spinal Muscular Atrophy Foundation. Contributors: The authors also thank the people who contributed to this study: Rosangel Cruz (Columbia clinical site, coordinator); Sally Dunaway (Columbia clinical site, physical therapist); Nicole Holuba (Columbia clinical site, coordinator); Jonathan Marra (Columbia clinical site, coordinator); Douglas Sproule (Columbia clinical site, subinvestigator); Heather Allen (University of Utah, coordinator); Nicole Rausch (University of Utah, coordinator); Sandra Reyna (University of Utah, subinvestigator); Ai Sakonju (University of Utah, subinvestigator); Abby Smart (University of Utah, coordinator); Donata Viazzo-Trussel (University of Utah, physical therapist); Robert Graham (Boston Children's Hospital, subinvestigator); Wendy Liew (Boston Children's Hospital, subinvestigator); Rebecca Parad (Boston Children's Hospital, coordinator); Amy Pasternak (Boston Children's Hospital, physical therapist); Elizabeth Shriber (Boston Children's Hospital, subinvestigator); Heather Szelag (Boston Children's Hospital, coordinator); Diane Castro (University of Texas Southwestern Medical School, subinvestigator); Leslie Nelson (University of Texas Southwestern Medical School, physical therapist); Katie Alexander (Ionis Pharmaceuticals, Inc., clinical operations); Shannon Fine (Ionis Pharmaceuticals, Inc., clinical operations); Shannon Hall (Ionis Pharmaceuticals, Inc., pharmacokinetics analyses); Viola Kam (Ionis Pharmaceuticals, Inc., project manager); Katherine Kwoh (Ionis Pharmaceuticals, Inc., data manager); Dan Schultz (Ionis Pharmaceuticals, Inc., biostatistician); Celeste Vanderpool (Ionis Pharmaceuticals, Inc., clinical supplies); Shuting Xia (Ionis Pharmaceuticals, Inc., biostatistician); Mason Yamashita (Ionis Pharmaceuticals, Inc., drug safety physician); Dawn McGuire, MD (independent medical monitor); Walter Bradley (data safety and monitoring board); Anne Connolly (data safety and monitoring board); Patti Dickson (data safety and monitoring board); Stephen Reingold (data safety and monitoring board); Sarah Hamren (Singulex, Inc., CSF SMN protein assay development and sample testing); Ali Vahad (Singulex, Inc., CSF SMN protein assay development and sample testing); and Maria Hovenden (Excel Scientific Solutions with funding from Biogen, editorial support based on input from the authors).
GLOSSARY
- AE
adverse event
- AUC
area under the concentration time curve
- DSMB
data and safety monitoring board
- HFMSE
Hammersmith Functional Motor Scale Expanded
- LP
lumbar puncture
- PedsQL
Pediatric Quality of Life Inventory
- SMA
spinal muscular atrophy
- SMN
survival of motor neuron
Footnotes
Editorial, page 884
AUTHOR CONTRIBUTIONS
C.A.C., K.J.S., B.T.D., S.T.I., D.C.D., D.A.N., and K.M.B. designed the trial. C.A.C., K.J.S., B.T.D., S.T.I., J.M., and D.C.D. conducted the trial. D.A.N. and K.M.B. performed the analysis of the data. C.A.C., D.A.N., F.B., and K.M.B. interpreted the data and wrote the manuscript. All authors critically revised and approved the final manuscript.
STUDY FUNDING
Funded by Ionis Pharmaceuticals, Inc.
DISCLOSURE
C. Chiriboga has served as a consultant to Roche Pharmaceuticals. She receives research support from the DOD and the Spinal Muscular Atrophy Foundation. K. Swoboda serves as a Scientific Advisory Board member for Roche Pharmaceuticals, Avexis, CureSMA, and the Alternating Hemiplegia of Childhood Foundation. She receives research support from the NIH. She has received clinical trial support from Biogen and Ionis Pharmaceuticals. B. Darras has served, ad hoc, as an Advisory Board member for Roche Pharmaceuticals, Sarepta Therapeutics, Ionis Pharmaceuticals, Inc., Cytokinetics, Inc., and Audentes Therapeutics. He has received grant/research/clinical trial support from PTC Therapeutics; Ionis Pharmaceuticals, Inc., Sarepta Therapeutics, Valerion Therapeutics, the NIH NINDS and NIAMS, Spinal Muscular Atrophy Foundation, Muscular Dystrophy Association, and the Slaney Family Fund for SMA. S. Iannaccone reports no disclosures relevant to the manuscript. J. Montes serves as a consultant to Ionis Pharmaceuticals, Inc. D. De Vivo has served as a consultant to Roche Pharmaceuticals, Ultragenyx Therapeutics, and Sarepta Therapeutics. He receives research support from the NIH, DOD, Colleen Giblin Foundation, Milestones for Children, Glut1 Deficiency Foundation, and the Spinal Muscular Atrophy Foundation. D. Norris is an employee of Ionis Pharmaceuticals, Inc. C. Bennett is an employee of Ionis Pharmaceuticals, Inc. K. Bishop was an employee of Ionis Pharmaceuticals, Inc. while this work was conducted. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Lunn MR, Wang CH. Spinal muscular atrophy. Lancet 2008;371:2120–2133. [DOI] [PubMed] [Google Scholar]
- 2.Darras BT, Markowitz JA, Monani UR, De Vivo DC. Spinal muscular atrophies. In: Darras BT, Jones HR, Jr, Ryan MM, De Vivo DC, eds. Neuromuscular Disorders of Infancy, Childhood, and Adolescence: A Clinician's Approach, 2nd ed San Diego: Academic Press; 2014:117–145. [Google Scholar]
- 3.Russman BS. Spinal muscular atrophy: clinical classification and disease heterogeneity. J Child Neurol 2007;22:946–951. [DOI] [PubMed] [Google Scholar]
- 4.Lefebvre S, Bürglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995;80:1155–1165. [DOI] [PubMed] [Google Scholar]
- 5.Cartegni L, Krainer AR. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet 2002;30:377–384. [DOI] [PubMed] [Google Scholar]
- 6.Hua Y, Sahashi K, Hung G, et al. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev 2010;24:1634–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hua Y, Vickers TA, Okunola HL, Bennett CF, Krainer AR. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet 2008;82:834–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hua Y, Sahashi K, Rigo F, et al. Peripheral SMN restoration is essential for long-term rescue of a severe SMA mouse model. Nature 2011;478:123–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Passini MA, Bu J, Richards AM, et al. Antisense oligonucelotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci Transl Med 2011;3:72ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geary RS, Yu RZ, Levin AA. Pharmacokinetics of phosphorothioate antisense oligodeoxynucleotides. Curr Opin Investig Drugs 2001;2:562–573. [PubMed] [Google Scholar]
- 11.Yu RZ, Baker B, Chappell A, Geary RS, Cheung E, Levin AA. Development of an ultrasensitive noncompetitive hybridization–ligation enzyme-linked immunosorbent assay for the determination of phosphorothioate oligodeoxynucleotide in plasma. Anal Biochem 2002;304:19–25. [DOI] [PubMed] [Google Scholar]
- 12.O'Hagen JM, Glanzman AM, McDermott MP, et al. An expanded version of the Hammersmith Functional Motor Scale for SMA II and III patients. Neuromuscul Disord 2007;17:693–697. [DOI] [PubMed] [Google Scholar]
- 13.Glanzman AM, O'Hagen JM, McDermott MP, et al. ; Pediatric Neuromuscular Clinical Research Network for Spinal Muscular Atrophy (PNCR), Muscle Study Group (MSG). Validation of the Expanded Hammersmith Functional Motor Scale in spinal muscular atrophy type II and III. J Child Neurol 2011;26:1499–1507. [DOI] [PubMed] [Google Scholar]
- 14.Varni JW, Seid M, Rode CA. The PedsQL: measurement model for the pediatric quality of life inventory. Med Care 1999;37:126–139. [DOI] [PubMed] [Google Scholar]
- 15.Iannaccone ST, Hynan LS, Morton A, Buchanan R, Limbers CA, Varni JW; AmSMART Group. The PedsQL in pediatric patients with spinal muscular atrophy: feasibility, reliability, and validity of the Pediatric Quality of Life Inventory Generic Core Scales and Neuromuscular Module. Neuromuscul Disord 2009;19:805–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jaschinski F, Rothhammer T, Jachimczak P, Seitz C, Schneider A, Schlingensiepen KH. The antisense oligonucleotide trabedersen (AP 12009) for the targeted inhibition of TGF-β2. Curr Pharm Biotechnol 2011;12:2203–2213. [DOI] [PubMed] [Google Scholar]
- 17.Miller TM, Pestronk A, David W, et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol 2013;12:435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramamoorthy C, Geiduschek JM, Bratton SL, Miser AW, Miser JS. Postdural puncture headache in pediatric oncology patients. Clin Pediatr 1998;37:247–251. [DOI] [PubMed] [Google Scholar]
- 19.Keidan I, Bielorei B, Berkenstadt H, et al. Prospective evaluation of clinical and laboratory effects of intrathecal chemotherapy on children with acute leukemia. J Pediatr Hematol Oncol 2005;27:307–310. [DOI] [PubMed] [Google Scholar]
- 20.Lee LC, Sennett M, Erickson JM. Prevention and management of post-lumbar puncture headache in pediatric oncology patients. J Pediatr Oncol Nurs 2007;24:200–207. [DOI] [PubMed] [Google Scholar]
- 21.Matsuzawa J, Matsui M, Konishi T, et al. Age-related volumetric changes of brain gray and white matter in healthy infants and children. Cereb Cortex 2001;11:335–342. [DOI] [PubMed] [Google Scholar]
- 22.Rigo F, Chun SJ, Norris DA, et al. Pharmacology of a central nervous system delivered 2'-O-methoxyethyl-modified survival of motor neuron splicing oligonucleotide in mice and nonhuman primates. J Pharmacol Exp Ther 2014;350:46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaufmann P, McDermott MP, Darras BT, et al. ; Muscle Study Group; Pediatric Neuromuscular Clinical Research Network for Spinal Muscular Atrophy. Observational study of spinal muscular atrophy type 2 and 3: functional outcomes over 1 year. Arch Neurol 2011;68:779–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaufmann P, McDermott MP, Darras BT, et al. ; Muscle Study Group (MSG), Pediatric Neuromuscular Clinical Research Network for Spinal Muscular Atrophy (PNCR). Prospective cohort study of spinal muscular atrophy types 2 and 3. Neurology 2012;79:1889–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mercuri E, Bertini E, Messina S, et al. Randomized, double-blind, placebo-controlled trial of phenylbutyrate in spinal muscular atrophy. Neurology 2007;68:51–55. [DOI] [PubMed] [Google Scholar]
- 26.Swoboda KJ, Scott CB, Crawford TO, et al. ; Project Cure Spinal Muscular Atrophy Investigators Network. SMA CARNI-VAL trial part I: double-blind, randomized, placebo-controlled trial of L-carnitine and valproic acid in spinal muscular atrophy. PLoS One 2010;5:e12140. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.