

Summary
Recent epidemiologic data suggest that sickle cell trait (HbAS; AS) is a risk factor for venous thromboembolism. We conducted an exploratory study of healthy subjects with AS under baseline conditions to determine whether a chronic basal hyperactivation of coagulation exists, and if so, what mechanism(s) contribute to this state. Eighteen healthy AS individuals were compared to 22 African-American controls with a normal haemoglobin profile (HbAA; AA) and 17 patients with sickle cell disease (HbSS; SS). Plasma thrombin-antithrombin complexes and D-dimer levels were elevated in AS relative to AA patients (P = 0.0385 and P = 0.017, respectively), and as expected, were much higher in SS versus AA (P < 0.0001 for both). Thrombin generation in platelet poor plasma was indistinguishable between AA and AS subjects, whereas a paradoxical decrease in endogenous thrombin potential was observed in SS (P ≤ 0.0001). Whole blood tissue factor was elevated in SS compared to AA (P = 0.005), but did not differ between AA and AS. Plasma microparticle tissue factor activity was non-significantly elevated in AS (P = 0.051), but was clearly elevated in SS patients (P = 0.004) when compared to AA controls. Further studies in larger cohorts of subjects with sickle cell trait are needed to confirm the results of this preliminary investigation.
Keywords: sickle, venous thrombosis, tissue factor, coagulation, thrombin
As many as 200 million people worldwide are heterozygous carriers of a mutation in the HBB gene (HBB E6V, also termed HbS mutation or ‘sickle gene’), resulting in a condition known as sickle cell trait (HbAS, hereafter termed AS) (Piel et al, 2013) (Dr. S. Grosse, personal communication, US Centers for Disease Control and Prevention, Atlanta, GA). Although traditionally viewed as a benign carrier state, AS may be a risk factor for certain adverse outcomes, including venous thromboembolism (VTE) (Tsaras et al, 2009; Key & Derebail, 2010; Key et al, 2015). In a large case–control study of VTE in African-American subjects, we reported that AS was associated with an increased risk of VTE, with an odds ratio (OR) of about 2; more strikingly, AS appeared to confer a particularly high risk for pulmonary embolism (PE) (OR ≈ 4) (Austin et al, 2007). In addition, a possible interaction between AS and hormonal contraceptive usage may further heighten the risk of VTE in women of reproductive age (Austin et al, 2009). Other studies have also reported an association between AS and risk of VTE, particularly PE (Heller et al, 1979; Bucknor et al, 2014; Folsom et al, 2015). This association was deemed a high priority area for further research in a recent National Institutes of Health conference on sickle cell trait (Goldsmith et al, 2012).
It has previously been reported that plasma levels of the ‘pre-thrombotic’ biomarkers thrombin-antithrombin complexes (TAT), prothrombin fragments 1 + 2 and D-dimers are elevated in subjects with AS compared to race-matched controls (Westerman et al, 2002). One additional study demonstrated elevated TAT complex levels in AS compared to non race-matched controls (Helley et al, 1997). However, comparison with race-matched controls is essential as both pre-thrombotic markers, such as D-dimer (Lutsey et al, 2006), and plasma thrombin generation (Roberts et al, 2013) are higher in healthy individuals of African descent.
In the present report, we sought to confirm the presence of a pre-thrombotic state in healthy subjects with AS using plasma markers of coagulation activation. Simultaneously, we investigated possible mechanisms that might account for the hypercoagulable state in AS subjects compared to healthy HbAA (AA) controls, as well as subjects with sickle cell disease (SCD; HbSS; SS), in whom a complex and multi-causal state of activation of coagulation has been well described (Ataga & Key, 2007; Lim et al, 2013). The choice of analytes was influenced by these prior studies, which have focused on both the trigger for coagulation activation in SS, such as circulating tissue factor (TF, also termed coagulation Factor III) (Key et al, 1998; Colella et al, 2012; Setty et al, 2012), as well as ‘downstream’ enhancement of thrombin generation capacity (Hemker et al, 2006; Gerotziafas et al, 2012; Lim et al, 2013; Noubouossie et al, 2013).
Methods
Study subjects
This exploratory cohort study was conducted at the University of North Carolina at Chapel Hill. Institutional Review Board approval was obtained and written informed consent was obtained from all study participants. The study included only African-Americans between the ages of 18 and 65 years. Ethnicity/race was self-declared.
Subjects were recruited into three study categories: Group 1 or controls (AA) included subjects without any haemoglobinopathy; Group 2 included subjects with sickle cell trait (AS); and Group 3 included subjects with SCD (SS) with either HbSS or HbS-βthal0 genotype. Exclusion criteria included the presence of a prior history of VTE, current pregnancy or use of hormonal contraceptive therapy and current use of anticoagulants or anti-platelet agents (the latter within the past 7 days). Subjects with SCD were also excluded if they had experienced a recent (<4 weeks) pain crisis or and/or hospitalization, had recently (<3 months) undergone surgery, or if they had received blood transfusion within the last 3 months. No subject in any group had diabetes mellitus or hypertension, cancer or an acute or chronic inflammatory disorder.
Phlebotomy and plasma preparation
After discarding the first 3 ml of blood, blood was drawn into appropriate commercial vacutainer or pre-prepared tubes and rapidly mixed. Samples obtained from a less than fully satisfactory venipuncture were discarded. For D-dimer and TAT analysis, blood was drawn into 3.2% sodium citrate. For thrombin generation and clot formation analyses, blood was drawn into 3.2% sodium citrate/18.5 μg/ml corn trypsin inhibitor to prevent contact activation (Luddington & Baglin, 2004). In each case, platelet-free plasma was prepared by centrifugation, followed by a second centrifugation step at 13 000 g for 2 min. Plasma aliquots were stored at −80°C until batched analysis could be performed. Frozen plasma samples were thawed rapidly at 37°C immediately prior to assay and discarded after use.
Materials
Factor VIIa and factor X were from Enzyme Research Laboratories, Inc. (South Bend, IN, USA). Chromogenic substrate Pefachrome FXa 8595 was from Centerchem, Inc. (Norwalk, CT, USA). Re-lipidated recombinant human TF (Innovin®) was from Siemens Healthcare Diagnostics Products GmbH (Marburg, Germany). Immunbind TF enzyme-linked immunosorbent assay (ELISA) kits were from American Diagnostica, Inc (Stamford, CT, USA). An inhibitory monoclonal antibody (HTF1) to TF was a kind gift from Ronald Bach, PhD (Minneapolis VA Medical Center, Minneapolis, MN, USA). Corn trypsin inhibitor was from Hematologic Technologies, Inc. (Essex Junction, VT, USA). Unilamiellar phospholipid vesicles [15% phosphatidylserine (PS), 41% phosphatidylcholine (PC) 41% and phosphatidylethanolamine (PE) 44%] were prepared as described (Hope et al, 1985).
Haematology profile
Routine haematological assays were performed using standard methods at the McClendon Clinical Laboratories at the University of North Carolina Memorial Hospital. Because the laboratory reports markedly low or undetectable serum haptoglobin values as ‘<220 mg/l’, this parameter was treated as a categorical variable, and observed values were divided into those that were below (<220 mg/l), within (220–1390 mg/l) and above (>1390 mg/l) the laboratory’s quoted normal reference range.
Plasma D-dimer and thrombin-antithrombin (TAT) complexes
D-dimer on plasma samples was measured in the clinical laboratory using the HemosIL® D-Dimer HS immuno-turbidic assay on an ACL TOP® coagulation analyser (Instrumentation Laboratory, Bedford, MA, USA) (Salvagno et al, 2008). Because the laboratory reports markedly low D-dimer values as ‘<150 μg/ml’, plasma D-dimer values were treated as a categorical variable, dividing observed values into those that were below (<150 μg/ml), within (150–229 μg/ml) and above (>229 μg/ml) the laboratory’s quoted normal reference range. Plasma TAT levels were determined by using an ELISA assay kit from Affinity Biologicals (Lancaster, ON, Canada).
Thrombin generation and fibrin formation in plasma
Thrombin generation was measured fluorometrically using Calibrated Automated Thrombography (CAT), as previously described (Machlus et al, 2009), using the TF and phospholipid reagents (final concentration, 1 pmol/l and 4 μmol/l, respectively) supplied by the manufacturer (Thrombinoscope BV, Maastricht, Netherlands). Parameters of interest included: (i) lag time; (ii) time to peak thrombin generation; (iii) peak thrombin level; and (iv) area under the curve [‘endogenous thrombin potential’ (ETP)].
Fibrin formation was measured turbidimetrically by continuous measurement of absorbance at 405 nm in a Spectramax microplate reader (Molecular Devices, Sunnyvale, CA, USA). Here, platelet-free plasma samples were re-calcified, and clotting was initiated by addition of Innovin® (1:60 000 dilution, final) and phospholipid vesicles (125 μmol/l, final), as previously described (Gray et al, 2011). Endpoints of interest included: (i) onset of fibrin formation; (ii) rate of fibrin formation; and (iii) absolute change in turbidity from baseline.
Microparticle tissue factor activity
As previously described (Key & Mackman, 2010), microparticles (MPs) were isolated from citrate anticoagulated platelet-free plasma by centrifugation at 20 000 g for 30 min. The pellet was re-suspended in HBSA (20 mmol/l HEPES, pH 7.4, 150 mmol/l NaCl, and 1 mg/ml bovine serum albumin) and re-centrifuged at 20 000 g for 30 min, before final re-suspension in 250 μl HBSA. MPs were incubated with factor VIIa (100 pmol/l, final) and factor X (135 nmol/l, final), in the presence of calcium (5 mmol/l, final) and anti-TF antibody (HTF1, 10 μg/ml final) or IgG control. Factor Xa generation was measured in aliquots by chromogenic substrate cleavage as described. Specific TF activity (fmol/l) was determined by subtracting the rate of factor Xa generated in the presence of HTF1 from the rate of factor Xa generated in the presence of IgG controls with reference to a standard curve generated using lipidated recombinant human TF (In-novin®). Effective TF concentration in each lot of Innovin® was calculated as previously described (Johnson et al, 2009).
Whole blood tissue factor activity
As previously described (Key et al, 1998), frozen whole blood samples were thawed at 37°C for 3 min, after which 250 μl was added to 900 μl TBS-EDTA (20 mmol/l Tris-HCl, pH 7.5, 150 mmol/l NaCl, 100 mmol/l EDTA) and mixed by vortexing. Following three freeze-thaw cycles, sample pellets were prepared by high-speed centrifugation (100 000 g at 4°C for 60 min) and washed three times in TBS-EDTA before final suspension of the pellet in 250 μl HBSA. One hundred microlitre was incubated with HTF1 (4 μg/ml) or control antibody for 60 min. Forty microlitre of each sample was then added to a 96-well plate in duplicate. Forty microlitre of factor mix (2 nmol/l factor VIIa and 300 nmol/l factor X in HBSA with 5 mmol/l calcium) was then added to each well. Plates were incubated at 37°C for 2 h before adding 10 μl of EDTA. Forty microlitre of the chromogenic substrate, Pefachrome FXa 8595 (final concentration 0.3 mmol/l) was added to each well. The rate of factor Xa cleavage of the chromogenic substrate at room temperature in each well was monitored in a ThermoMax® plate reader (Molecular Devices, Sunnyvale, CA, USA). Innovin® at various concentrations was used as the TF standard. TF-dependent procoagulant activity (pmol/l) was defined as the difference between the rates of factor Xa cleavage of substrate in the absence/presence of HTF1.
Inflammatory markers and cytokines
Cytokine and chemokine levels in citrated plasma were determined by colourimetric bead assay using the Luminex system and human-specific bead sets (R&D Systems, Minneapolis, MN, USA). Results were interpolated from 5-parameter-fit standard curves generated using the relevant recombinant human proteins (R&D Systems).
Statistical analysis
The primary goal of the study was to explore differences in coagulation and inflammatory parameters between subjects with AS and matched controls (AA). Patients with SS were included as a positive control and to examine whether any gene dosing effects were evident. The study had planned to enrol 20 subjects from each subtype (AA, AS and SS). As this was an exploratory study, sample size was estimated from prior studies that used similar assays in individuals with SS disease (Westerman et al, 2002; Ataga et al, 2008).
Descriptive analyses were performed to explore the distribution of each variable or analyte. Data for continuous variables were expressed as median values with interquartile ranges (IQR), and compared between groups (AA vs. AS and AA vs. SS) using the Mann–Whitney U test. For categorical data (D-dimers and haptoglobin), groups were compared using Fisher’s exact test. Spearman rank correlation was used to determine correlation between variables; in the case of a categorical variable (such as D-dimers), a rank of 1 was given to values falling below the reference range, a rank of 2 to values within the range, and a rank of 3 to values above the quoted reference range. Significance was established at the 0.05 level; no adjustments were made for multiple comparisons, because this was an exploratory study.
Results
Study subject characteristics
Fifty-seven African-American subjects were recruited: 22 AA, 18 AS and 17 SS. Age was comparable amongst the groups with median ages of 37.5 (IQR: 32–45), 38.5 (IQR: 33–45) and 41 (IQR: 24.5–47.5) years for AA, AS, and SS, respectively. Ninety-one per cent of the AA controls were females, while females accounted for 56% of the AS subjects and 53% of the SS subjects.
As expected, haemoglobin levels were similar for AA and AS subjects, but were substantially lower among those with SS, in whom anaemia was accompanied by evidence of accelerated haemolysis – specifically, higher serum lactate dehydrogenase (LDH) and lower haptoglobin values (Table I). Reticulocyte counts were not obtained. There was a trend towards lower serum haptoglobin and, to a lesser extent, higher serum LDH values in AS compared to control subjects. Total white blood cell counts were also higher in the SS group; this was accounted for by an increase in both absolute neutrophil and absolute monocyte counts. There was a trend towards elevated absolute monocyte counts in AS subjects. Platelet counts were higher in the SS group, but it is noteworthy that they were also marginally higher (P = 0.0474) in AS subjects (median: 325 × 109/l; IQR: 290–364 × 109/l) compared to AA controls (median: 285 × 109/l, IQR: 255–327 × 109/l).
Table I.
Haematological data in study subjects.
| Assay | AA (n = 22) | AS (n = 17) | SS (n = 16) | P-value* | P-value† | |
|---|---|---|---|---|---|---|
| WBC (×109/l) | 6.1 (5.0–7.4) | 6.8 (5.8–7.1) | 8.8 (6.7–12.4) | 0.5422 | 0.0105 | |
| AMC (×109/l) | 0.3 (0.3–0.4) | 0.4 (0.3–0.5) | 0.6 (0.4–0.8) | 0.0600 | 0.0023 | |
| ANC (×109/l) | 3.3 (2.2–4.0) | 4.1 (3.2–4.4) | 4.1 (3.2–6.0) | 0.1563 | 0.0397 | |
| Platelet count (×109/l) | 285 (255–327) | 325 (290–364) | 399 (308–512) | 0.0474 | 0.0052 | |
| Hb (g/l) | 125 (121, 131) | 124 (120–137) | 86 (82–100) | 0.7984 | <0.0001 | |
| LDH (iu/l) | 463 (436–525) | 518 (454–558) | 1022 (905–1144) | 0.2549 | <0.0001 | |
| Haptoglobin (mg/l) | ||||||
| <220 | 0 (0%) | 2 (12%) | 14 (93%) | 0.0553 | <0.0001 | |
| 220–1390 | 10 (45%) | 11 (65%) | 1 (7%) | |||
| >1390 | 12 (55%) | 4 (24%) | 0 (0%) | |||
Data are presented as median (interquartile range) or n (%).
AA, HbAA individuals (controls); AS, HbAS individuals (sickle cell trait); SS, HbSS individuals (sickle cell disease); WBC, white blood cell count; Hb, haemoglobin concentration; LDH, lactate dehydrogenase; ANC, absolute neutrophil count; AMC, absolute monocyte count.
Comparison of AA to AS.
Comparison of AA to SS.
Coagulation is activated in vivo in both SS and AS
D-dimer levels were elevated in every patient with SS, with a highly significant increase compared to controls (P < 0.0001) (Table II). However, D-dimer levels were also elevated in AS relative to AA (P = 0.017). It should be noted that D-dimers were above the laboratory’s reference range in 5 of 22 (23%) subjects in the AA control group. This probably reflects the fact that D-dimer levels are elevated in African-Americans compared to white Americans (Lutsey et al, 2006), who comprise the large majority of healthy subjects from whom the reference ranges are derived in the clinical laboratory.
Table II.
Coagulation data in study subjects.
| Analyte | AA (n = 22) | AS (n = 17) | SS (n = 16) | P-value* | P-value† | |
|---|---|---|---|---|---|---|
| D-Dimer (μg/ml) | ||||||
| <150 | 14 (64%) | 3 (18%) | 0 (0%) | 0.0171 | <0.0001 | |
| 150–229 | 3 (14%) | 4 (24%) | 0 (0%) | |||
| >229 | 5 (23%) | 10 (29%) | 16 (100%) | |||
| TAT (nmol/l) | 0.02 (0.02–0.02) | 0.03 (0.02–0.97) | 3.04 (0.03–8.78) | 0.0385 | <0.0001 | |
| Whole blood tissue factor activity (pmol/l) | 0.08 (0.04–0.14) | 0.09 (0.04–0.11) | 0.23 (0.09–0.33) | 0.7337 | 0.0048 | |
| Microparticle tissue factor activity (fmol/l) | 2.2 (0–3.5) | 3.8 (2.8–6.9) | 5.5 (4.5–9.3) | 0.0513 | 0.0042 | |
Data are presented as median (IQR) or n (%).
AA, HbAA individuals (controls); AS, HbAS individuals (sickle cell trait); SS, HbSS individuals (sickle cell disease); TAT, thrombin-antithrombin complex.
Comparison of AA to AS.
Comparison of AA to SS.
Plasma TAT complexes showed similar results, with a marked increase in SS (P < 0.0001) and a more modest but significant increase in AS (P = 0.0385) (Table II). Notably, in the case of both the AS and SS groups, the distribution of values included a substantial number of subjects whose TAT levels overlapped with AA controls, and the overall differences between the groups and controls was driven by the population of outliers (data not shown). For the entire group of study subjects, TAT complexes and D-dimers were moderately well correlated, with a Spearman correlation coefficient of 0.55 (P < 0.0001).
Thrombin generation capacity and fibrin formation in AS or SS plasma are not increased
We next examined whether the capacity for thrombin generation is increased in the plasma of AS and SS subjects ex vivo, as elevated plasma TAT complexes point to the presence of accelerated thrombin generation in vivo. By CAT assay, neither lag time nor peak thrombin generation differed between the three groups (Table III). However, time to peak thrombin generation was shorter in SS vs. AA (P = 0.0001) (Fig 1). Unexpectedly, ETP was lower in patients with SS vs. AA (P ≤ 0.0001). Subjects with AA and AS did not differ in any CAT parameter.
Table III.
Thrombin generation and plasma clot formation parameters.
| Parameter | AA (n = 21) | AS (n = 16) | SS (n = 17) | P-value* | P-value† |
|---|---|---|---|---|---|
| Thrombin generation | |||||
| Lag time (min) | 5.3 (4.8–6.3) | 5.2 (4.7–5.7) | 5.1 (4.2–5.5) | 0.4433 | 0.1052 |
| Time to peak (min) | 9.9 (9.2–10.7) | 10.4 (9.2–11.4) | 8.1 (7.7–9.1) | 0.6693 | 0.0001 |
| Thrombin peak (nmol/l) | 163.6 (128.8–206.6) | 168.3 (130.6–233.1) | 203.2 (173.1–232.8) | 0.6826 | 0.0887 |
| ETP (nmol/l × min) | 1496.0 (1431.5–1770.8) | 1623.4 (1395.1–1851.5) | 1217.5 (1170.0–1329.5) | 0.4573 | <0.0001 |
| Clotting | |||||
| Onset (min) | 15.0 (12.0–16.5) | 12.3 (10.4–15.4) | 11.6 (8.9–14.8) | 0.2802 | 0.0418 |
| Rate (mOD/min) | 78.6 (52.4–99.3) | 60.8 (40.7–83.7) | 83.8 (55.4–120.9) | 0.2060 | 0.2943 |
| Change in turbidity | 0.70 (0.57–0.78) | 0.68 (0.61–0.76) | 0.67 (0.56–0.77) | 0.9638 | 0.7277 |
Data are presented as median (IQR).
AA, HbAA individuals (controls); AS, HbAS individuals (sickle cell trait); SS, HbSS individuals (sickle cell disease); ETP, endogenous thrombin potential; mOD, mean optical density.
Comparison of AA to AS.
Comparison of AA to SS.
Fig 1.
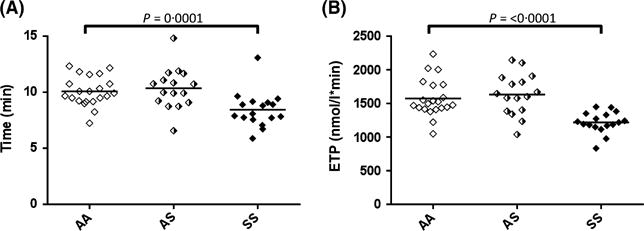
Calibrated Automated Thrombography© (CAT). CAT assays on platelet poor plasma was performed using 1 pmol/l tissue factor and 4 μmol/l phospholipid, supplied by the manufacturer. (A) Time to peak thrombin generation. (B) Endogenous thrombin potential (area under the curve). Data comparing these parameters are shown for control (AA), sickle cell trait (AS) and sickle cell disease (SS) subjects.
Fibrin formation in plasma was also measured using a turbidimetric assay. The onset of fibrin formation was more rapid in SS compared to AA (P = 0.0418), although the subsequent rate of fibrin formation did not differ (Fig 2, Table III). The more rapid onset of fibrin formation in SS is consistent with the shorter time to peak thrombin generation in the CAT assay (Fig 1). Neither the rate, nor the change in turbidity (an indicator of fibrin network structure) differed significantly between AA and SS individuals. No changes were noted in any of the measured parameters when AA and AS subjects were compared, also consistent with the thrombin generation assay data.
Fig 2.
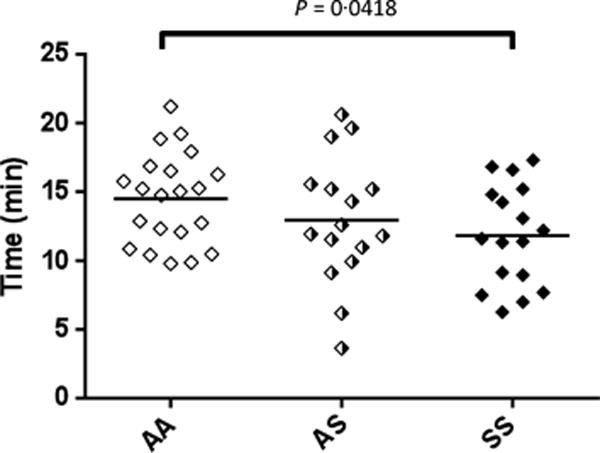
Fibrin formation. Fibrin formation in platelet poor plasma was measured using a turbidimetric assay as described in Methods. Data shown are for onset time for clot formation for control (AA), sickle cell trait (AS) and sickle cell disease (SS) subjects.
Microparticle TF procoagulant activity in AS
As described in Methods, circulating TF activity was measured using two separate assays – one for TF procoagulant activity in whole blood (WB-TF), and one for MP-associated TF (MP-TF) activity in cell free plasma. As previously reported (Key et al, 1998), WB-TF activity was elevated in SS disease compared to AA controls (P = 0.005). However, WB-TF did not differ between AA and AS subjects (P = 0.734) (Fig 3A). MP-TF procoagulant activity showed a trend towards being elevated in AS subjects (P = 0.051), and was clearly increased in SS subjects (P = 0.004) compared to controls (Fig 3B).
Fig 3.
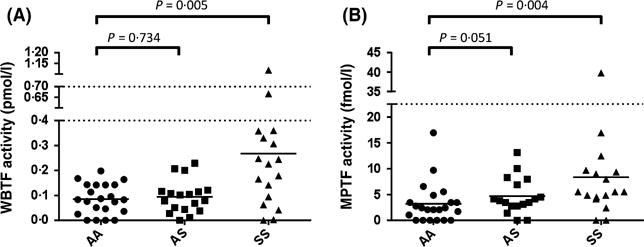
Measurement of circulating tissue factor activity. (A) Whole blood tissue factor (WBTF) activity was measured as described in Methods, and the concentration expressed as pmol/l. This assay is sensitive to all membrane-bound tissue factor in blood. (B) Microparticle tissue factor (MPTF) activity was measured on platelet-free plasma, as described in Methods, and the concentration expressed as fmol/l. Both figures show the data for control (AA), sickle cell trait (AS) and sickle cell disease (SS) subjects.
MP-TF activity levels and TAT complexes were only weakly correlated for the entire group (AA, AS and SS), with a coefficient of 0.30 (P = 0.0295). In addition, the correlation between MP-TF and D-dimers was only 0.28 (P = 0.0381), but this may have been influenced by the fact that D-dimer was treated as a categorical variable with only three levels. WB-TF did not correlate with TAT or D-dimer as evidenced by coefficients of 0.01 (P = 0.9215) and 0.16 (P = 0.2442), respectively.
Inflammatory markers and cytokine levels are not elevated in AS
Several markers, including soluble CD40 ligand (P = 0.0276), tumour necrosis factor (TNF α; (P = 0.0003) and soluble vascular cell adhesion molecule 1 (VCAM1; P < 0.0001) were elevated in SS compared to AA controls, whereas epithelial-derived neutrophil-activating peptide 78 [ENA78 (CXCL5); P = 0.0276] and granulocyte colony-stimulating factor (P = 0.0304) were significantly reduced (Table S1). C-reactive protein, an acute phase response marker that has been reported to be elevated in SS patients (Singhal et al, 1993), was not increased in this cohort. The inflammatory markers C-reactive protein, TNF α, interleukin (IL) 1-α, IL1β, IL6, and IL8 also did not differ between AA and AS subjects. Soluble P-selectin levels trended higher in both AS (P = 0.0551) and SS (P = 0.0577) subjects, but without an apparent ‘gene dosing effect’ observed in these groups.
Discussion
Elevated levels of plasma D-dimers, and to a lesser extent, TAT complexes in this study are consistent with a previous report that coagulation is more activated in healthy subjects with AS compared to race-matched controls (Westerman et al, 2002). However, further larger scale studies will be required to definitely establish this association. We also included a cohort of patients with SS, and observed the expected pronounced elevation in the pre-thrombotic markers in plasma (TAT complexes and D-dimers) compared to controls. Whether or not activation of coagulation in SS patients contributes to vaso-occlusive complications, such as pain crisis, stroke, or pulmonary hypertension remains an open question (Hillery & Panepinto, 2004; Ataga & Key, 2007; Pakbaz & Wun, 2014), but several pieces of evidence now support the conclusion that the risk of VTE is increased not only in SCD (Stein et al, 2006; Novelli et al, 2012; Naik et al, 2014), but also in sickle trait (Austin et al, 2007; Bucknor et al, 2014; Folsom et al, 2015). We examined whether thrombin-generating capacity in AS and SS plasma is increased in parallel with the increased levels of circulating TAT and D-dimers. Paradoxically, ETP was reduced in individuals with SS (Fig 1). Several groups have addressed the question whether thrombin generation in plasma is elevated in SCD, with mixed and sometimes opposite conclusions (Lim et al, 2013). Notably, an analogous discrepancy between elevated TAT and/or D-dimer in vivo and normal or decreased ETP ex vivo has been described elsewhere, for example in normal pregnancy (Eichinger et al, 1999). When performed in cell-free plasma, ETP is dependent on the net balance between levels of procoagulant and anticoagulant factors (Machlus et al, 2009), and therefore the combination of elevated procoagulants, such as FVIII and reduced anticoagulants (such as protein C and protein S) in SCD might be expected to result in a net increase in thrombin generation potential. Beta thalassaemia syndromes, particularly after splenectomy, represent another haemoglobinopathy that is associated with in vivo evidence of coagulation hyperactivation and a clinical risk of thrombotic events (Ataga et al, 2007). Tripodi et al (2009) demonstrated that while whole blood thrombelastometry end points were suggestive of a hypercoagulable state in β-thalassaemia, plasma thrombin generation profiles were normal. The authors concluded that abnormalities in blood cell number or function – as opposed to plasma proteins – contribute to the hypercoagulability in β-thalassaemia. The apparent discrepancy between increased plasma TAT levels and reduced ETP in this study may be indicative of a similar situation in SCD.
Some of the well-recognized complications of SCT include hyposthenuria, renal papillary necrosis, renal medullary carcinoma, splenic infarction at high altitude, glaucoma following hyphema of the anterior chamber of the eye, exertional rhabdomyolysis and exercise-related sudden death (Tsaras et al, 2009; Hooper et al, 2010; Key & Derebail, 2010). Common to all these scenarios is the likelihood that sickling of erythrocytes occurs in a vascular bed that may be either continuously or intermittently exposed to conditions that favour sickle haemoglobin polymerization, including hypoxia (e.g. in the anterior chamber of the eye or in the splenic microvasculature at high altitude), acidosis or hyperosmolarity (e.g. in the renal medulla).
We evaluated whether some of the mechanisms believed to contribute to activation of coagulation in SS (Ataga & Key, 2007) might also be at play in AS. By analogy to SCD, we evaluated whether circulating TF levels are elevated in individuals with SCT. We, and others have previously reported that cell-associated TF levels are elevated in patients with SCD (Key et al, 1998; Colella et al, 2012; Setty et al, 2012). The present study confirmed that finding, but failed to demonstrate any increase in WB-TF in AS subjects (Fig 3A). The finding of increased MP-TF activity in SS subjects (Fig 3B) is consistent with our previous observation (Key et al, 1998), but may be at odds with the study by van Beers et al (2009). These authors also found that erythrocyte and platelet MP numbers were elevated in SCD, but reported that thrombin generation was dependent on factor XI, but not factor VII, thereby implicating the ‘intrinsic’ coagulation pathway. This discrepant observation probably reflects technical differences, most notably the presence of plasma in the assay reported by van Beers et al (2009). In this study, we were surprised to find marginal, albeit insignificantly elevated levels of MP-TF procoagulant activity in subjects with AS (Fig 3B). However, the absent or modest correlations between WB-TF and MP-TF activities (respectively) question whether circulating TF is the dominant mechanism contributing to activation of coagulation in vivo. Indeed, the role of circulating MP-TF as a mediator of coagulation activation and thrombosis in human disease states has been extensively studied, although with mixed conclusions to date (Owens & Mackman, 2011; Geddings & Mackman, 2013; Lacroix et al, 2013).
In addition, we evaluated whether a number of pro-inflammatory cytokines are elevated in subjects with AS, as is the case in SS (Hoppe, 2014). It is well established that one mechanism contributing to the ‘cross talk’ between inflammation and coagulation is mediated by certain pro-inflammatory cytokines, such as TNF-α, and IL1 and 6 (Levi & van der Poll, 2010; Engelmann & Massberg, 2013; Sparkenbaugh & Pawlinski, 2013). However, in this study, we were unable to demonstrate any significant increase in these inflammatory markers in AS subjects. Consistent with our results, Tripette et al (2010) found no difference in baseline levels of multiple cytokines and soluble adhesion molecules [TNFα, IL6, soluble (s) Intercellular Adhesion Molecule 1 (ICAM1), sVCAM1, sP-selectin and sL-selectin] in patients with SCT, although differences were observed following a sub-maximal exercise protocol.
Several limitations of this study warrant discussion. We did not perform formal age- and sex-matching among the three cohorts, which resulted in a greater preponderance of females among the control group. Secondly, because this was designed as an exploratory study, the relatively small sample size may have limited our ability to draw definitive conclusions about the role of any single mechanism in the hyper-activation of coagulation in AS. Indeed, by analogy to the complexity of the hypercoagulability in SS, other candidate mechanisms may also contribute to the excessive activation of coagulation in subjects with AS. For example, we have recently evaluated erythrocyte PS expression in subjects with AS. While we have not observed any increase in AS subjects compared to controls at baseline in preliminary studies, we have detected a significant increase in red cell PS expression in AS subjects on haemodialysis compared to matched controls. Furthermore, red cell PS expression was highly correlated with plasma TAT levels (Brittain et al, 2010). These data suggest that a ‘second hit’, which might include extreme hypoxia in the renal medulla or the venous valvular pockets in the lower extremity (Sevitt, 1974; Hamer et al, 1981), may be required to perturb AS red cells, and thereby promote their ability to contribute to activation of coagulation. We are continuing to evaluate this and other possibilities with the ultimate goal of identifying biomarkers of thrombotic risk that may be present in sickle haemoglobinopathies.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. David Barrow, Dell Strayhorn RN FNP, Susan Jones RN, Ben Hulkower, Patrick Moody and Laura Gray for their excellent technical assistance. The authors also acknowledge funding from the North Carolina TRACS Institute (UL1TR000083), UO1HL117659 (NSK and KIA), T32HL007149 (CA, SA and MJM), K12HL087097 (MJM), R01 HL094740 (ASW) and the Doris Duke Charitable Foundation (NSK), and Yale CTSA (UL1TR000142).
Footnotes
Authorship and disclosures
NSK was the principal investigator and takes primary responsibility for the paper. CA, SA and KIA recruited the study subjects; CA, MJM, AK, FK, JEB, J-YC, and ASW performed the laboratory assays; DE was primarily responsible for statistical analysis; NSK coordinated the research; NSK and ASW wrote the paper. The authors report no potential conflicts of interest.
Additional Supporting Information may be found in the online version of this article:
Table S1. Plasma inflammatory marker and cytokine levels.
References
- Ataga KI, Key NS. Hypercoagulability in sickle cell disease: new approaches to an old problem. Hematology Am Soc Hematol Educ Program. 2007:91–96. doi: 10.1182/asheducation-2007.1.91. [DOI] [PubMed] [Google Scholar]
- Ataga KI, Cappellini MD, Rachmilewitz EA. Beta-thalassaemia and sickle cell anaemia as paradigms of hypercoagulability. British Journal of Haematology. 2007;139:3–13. doi: 10.1111/j.1365-2141.2007.06740.x. [DOI] [PubMed] [Google Scholar]
- Ataga KI, Moore CG, Hillery CA, Jones S, Whinna HC, Strayhorn D, Sohier C, Hinderliter A, Parise LV, Orringer EP. Coagulation activation and inflammation in sickle cell disease-associated pulmonary hypertension. Haematologica. 2008;93:20–26. doi: 10.3324/haematol.11763. [DOI] [PubMed] [Google Scholar]
- Austin H, Key NS, Benson JM, Lally C, Dowling NF, Whitsett C, Hooper WC. Sickle cell trait and the risk of venous thromboembolism among blacks. Blood. 2007;110:908–912. doi: 10.1182/blood-2006-11-057604. [DOI] [PubMed] [Google Scholar]
- Austin H, Lally C, Benson JM, Whitsett C, Hooper WC, Key NS. Hormonal contraception, sickle cell trait, and risk for venous thromboembolism among African American women. American Journal of Obstetrics and Gynecology. 2009;200:e621–e623. doi: 10.1016/j.ajog.2009.01.038. [DOI] [PubMed] [Google Scholar]
- van Beers EJ, Schaap MC, Berckmans RJ, Nieuwland R, Sturk A, van Doormaal FF, Meijers JC, Biemond BJ, CURAMA study group Circulating erythrocyte-derived microparticles are associated with coagulation activation in sickle cell disease. Haematologica. 2009;94:1513–1519. doi: 10.3324/haematol.2009.008938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brittain JE, Derebail VK, Mooberry MJ, Ataga KI, Kshirsagar AV, Key NS. Increased red cell phosphatidylserine exposure correlates with enhanced thrombin generation in sickle trait patients with end stage renal disease. Blood. 2010;116 Abstract 2665. [Google Scholar]
- Bucknor MD, Goo JS, Coppolino ML. The risk of potential thromboembolic, renal and cardiac complications of sickle cell trait. Hemoglobin. 2014;38:28–32. doi: 10.3109/03630269.2013.832689. [DOI] [PubMed] [Google Scholar]
- Colella MP, De Paula EV, Conran N, Machado-Neto JA, Annicchino-Bizzacchi JM, Costa FF, Saad ST, Traina F. Hydroxyurea is associated with reductions in hypercoagulability markers in sickle cell anemia. Journal of Thrombosis and Haemostasis. 2012;10:1967–1970. doi: 10.1111/j.1538-7836.2012.04861.x. [DOI] [PubMed] [Google Scholar]
- Eichinger S, Weltermann A, Philipp K, Hafner E, Kaider A, Kittl EM, Brenner B, Mannhalter C, Lechner K, Kyrle PA. Prospective evaluation of hemostatic system activation and thrombin potential in healthy pregnant women with and without factor V Leiden. Thrombosis and Haemostasis. 1999;82:1232–1236. [PubMed] [Google Scholar]
- Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nature Reviews Immunology. 2013;13:34–45. doi: 10.1038/nri3345. [DOI] [PubMed] [Google Scholar]
- Folsom AR, Tang W, Roetker NS, Kshirsagar AV, Derebail VK, Lutsey PL, Naik R, Pankow JS, Grove ML, Basu S, Key NS, Cushman M. Prospective study of sickle cell trait and venous thromboembolism incidence. Journal of Thrombosis and Haemostasis. 2015;13:2–9. doi: 10.1111/jth.12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geddings JE, Mackman N. Tumor-derived tissue factor-positive microparticles and venous thrombosis in cancer patients. Blood. 2013;122:1873–1880. doi: 10.1182/blood-2013-04-460139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerotziafas GT, Van Dreden P, Chaari M, Galea V, Khaterchi A, Lionnet F, Stankovic-Stojanovic K, Blanc-Brude O, Woodhams B, Maier-Redelsperger M, Girot R, Hatmi M, Elalamy I. The acceleration of the propagation phase of thrombin generation in patients with steady-state sickle cell disease is associated with circulating erythrocyte-derived microparticles. Thrombosis and Haemostasis. 2012;107:1044–1052. doi: 10.1160/TH11-10-0689. [DOI] [PubMed] [Google Scholar]
- Goldsmith JC, Bonham VL, Joiner CH, Kato GJ, Noonan AS, Steinberg MH. Framing the research agenda for sickle cell trait: building on the current understanding of clinical events and their potential implications. American Journal of Hematology. 2012;87:340–346. doi: 10.1002/ajh.22271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray LD, Hussey MA, Larson BM, Machlus KR, Campbell RA, Koch G, Ezban M, Hedner U, Wolberg AS. Recombinant factor VIIa analog NN1731 (V158D/E296V/M298Q-FVIIa) enhances fibrin formation, structure and stability in lipidated hemophilic plasma. Thrombosis Research. 2011;128:570–576. doi: 10.1016/j.thromres.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamer JD, Malone PC, Silver IA. The PO2 in venous valve pockets: its possible bearing on thrombogenesis. British Journal of Surgery. 1981;68:166–170. doi: 10.1002/bjs.1800680308. [DOI] [PubMed] [Google Scholar]
- Heller P, Best WR, Nelson RB, Becktel J. Clinical implications of sickle-cell trait and glucose-6-phosphate dehydrogenase deficiency in hospitalized black male patients. New England Journal of Medicine. 1979;300:1001–1005. doi: 10.1056/NEJM197905033001801. [DOI] [PubMed] [Google Scholar]
- Helley D, Girot R, Guillin MC, Bezeaud A. Sickle cell disease: relation between procoagulant activity of red blood cells from different phenotypes and in vivo blood coagulation activation. British Journal of Haematology. 1997;99:268–272. doi: 10.1046/j.1365-2141.1997.4173226.x. [DOI] [PubMed] [Google Scholar]
- Hemker HC, Al Dieri R, De Smedt E, Beguin S. Thrombin generation, a function test of the haemostatic-thrombotic system. Thrombosis and Haemostasis. 2006;96:553–561. [PubMed] [Google Scholar]
- Hillery CA, Panepinto JA. Pathophysiology of stroke in sickle cell disease. Microcirculation. 2004;11:195–208. doi: 10.1080/10739680490278600. [DOI] [PubMed] [Google Scholar]
- Hooper WC, Miller CH, Key NS. Complications associated with carrier status among people with blood disorders: a commentary. American Journal of Preventive Medicine. 2010;38:S456–S458. doi: 10.1016/j.amepre.2010.01.009. [DOI] [PubMed] [Google Scholar]
- Hope MJ, Bally MB, Webb G, Cullis PR. Production of large unilamellar vesicles by a rapid extrusion procedure: characterization of size distribution, trapped volume and ability to maintain a membrane potential. Biochimica et Biophysica Acta. 1985;812:55–65. doi: 10.1016/0005-2736(85)90521-8. [DOI] [PubMed] [Google Scholar]
- Hoppe CC. Inflammatory mediators of endothelial injury in sickle cell disease. Hematology/oncology Clinics of North America. 2014;28:265–286. doi: 10.1016/j.hoc.2013.11.006. [DOI] [PubMed] [Google Scholar]
- Johnson GJ, Leis LA, Bach RR. Tissue factor activity of blood mononuclear cells is increased after total knee arthroplasty. Thrombosis and Haemostasis. 2009;102:728–734. doi: 10.1160/TH09-04-0261. [DOI] [PubMed] [Google Scholar]
- Key NS, Derebail VK. Sickle-cell trait: novel clinical significance. Hematology Am Soc Hematol Educ Program. 2010;2010:418–422. doi: 10.1182/asheducation-2010.1.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Key NS, Mackman N. Tissue factor and its measurement in whole blood, plasma, and microparticles. Seminars in Thrombosis and Hemostasis. 2010;36:865–875. doi: 10.1055/s-0030-1267040. [DOI] [PubMed] [Google Scholar]
- Key NS, Slungaard A, Dandelet L, Nelson SC, Moertel C, Styles LA, Kuypers FA, Bach RR. Whole blood tissue factor procoagulant activity is elevated in patients with sickle cell disease. Blood. 1998;91:4216–4223. [PubMed] [Google Scholar]
- Key NS, Connes P, Derebail VK. Negative health implications of sickle cell trait in developed countries: from the football field to the laboratory. British Journal of Haematology. 2015;170:5–14. doi: 10.1111/bjh.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacroix R, Dubois C, Leroyer AS, Sabatier F, Dignat-George F. Revisited role of microparticles in arterial and venous thrombosis. Journal of Thrombosis and Haemostasis. 2013;11:24–35. doi: 10.1111/jth.12268. [DOI] [PubMed] [Google Scholar]
- Levi M, van der Poll T. Inflammation and coagulation. Critical Care Medicine. 2010;38:S26–S34. doi: 10.1097/CCM.0b013e3181c98d21. [DOI] [PubMed] [Google Scholar]
- Lim MY, Ataga KI, Key NS. Hemostatic abnormalities in sickle cell disease. Current Opinion in Hematology. 2013;20:472–477. doi: 10.1097/MOH.0b013e328363442f. [DOI] [PubMed] [Google Scholar]
- Luddington R, Baglin T. Clinical measurement of thrombin generation by calibrated automated thrombography requires contact factor inhibition. Journal of Thrombosis and Haemostasis. 2004;2:1954–1959. doi: 10.1111/j.1538-7836.2004.00964.x. [DOI] [PubMed] [Google Scholar]
- Lutsey PL, Cushman M, Steffen LM, Green D, Barr RG, Herrington D, Ouyang P, Folsom AR. Plasma hemostatic factors and endothelial markers in four racial/ethnic groups: the MESA study. Journal of Thrombosis and Haemostasis. 2006;4:2629–2635. doi: 10.1111/j.1538-7836.2006.02237.x. [DOI] [PubMed] [Google Scholar]
- Machlus KR, Colby EA, Wu JR, Koch GG, Key NS, Wolberg AS. Effects of tissue factor, thrombomodulin and elevated clotting factor levels on thrombin generation in the calibrated automated thrombogram. Thrombosis and Haemostasis. 2009;102:936–944. doi: 10.1160/TH09-03-0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik RP, Streiff MB, Haywood C, Jr, Segal JB, Lanzkron S. Venous thromboembolism incidence in the Cooperative Study of Sickle Cell Disease. Journal of Thrombosis and Haemostasis. 2014;12:2010–2016. doi: 10.1111/jth.12744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noubouossie DC, Le PQ, Rozen L, Ziereisen F, Willems D, Demulder A, Ferster A. Thrombin generation in children with sickle cell disease: relationship with age, hemolysis, transcranial Doppler velocity, and hydroxyurea treatment. European Journal of Haematology. 2013;91:46–54. doi: 10.1111/ejh.12113. [DOI] [PubMed] [Google Scholar]
- Novelli EM, Huynh C, Gladwin MT, Moore CG, Ragni MV. Pulmonary embolism in sickle cell disease: a case–control study. Journal of Thrombosis and Haemostasis. 2012;10:760–766. doi: 10.1111/j.1538-7836.2012.04697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens AP, 3rd, Mackman N. Microparticles in hemostasis and thrombosis. Circulation Research. 2011;108:1284–1297. doi: 10.1161/CIRCRESAHA.110.233056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pakbaz Z, Wun T. Role of the hemostatic system on sickle cell disease pathophysiology and potential therapeutics. Hematology/oncology Clinics of North America. 2014;28:355–374. doi: 10.1016/j.hoc.2013.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piel FB, Patil AP, Howes RE, Nyangiri OA, Gething PW, Dewi M, Temperley WH, Williams TN, Weatherall DJ, Hay SI. Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates. Lancet. 2013;381:142–151. doi: 10.1016/S0140-6736(12)61229-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts LN, Patel RK, Chitongo P, Bonner L, Arya R. African-Caribbean ethnicity is associated with a hypercoagulable state as measured by thrombin generation. Blood Coagulation & Fibrinolysis. 2013;24:40–49. doi: 10.1097/MBC.0b013e32835a07fa. [DOI] [PubMed] [Google Scholar]
- Salvagno GL, Lippi G, Montagnana M, Poli G, Giavarina D, Manzato F, Guidi GC. Performance of the automated and rapid HemosIL D-Dimer HS on the ACL TOP analyzer. Blood Coagulation & Fibrinolysis. 2008;19:817–821. doi: 10.1097/MBC.0b013e32830f1bae. [DOI] [PubMed] [Google Scholar]
- Setty BN, Key NS, Rao AK, Gayen-Betal S, Krishnan S, Dampier CD, Stuart MJ. Tissue factor-positive monocytes in children with sickle cell disease: correlation with biomarkers of haemolysis. British Journal of Haematology. 2012;157:370–380. doi: 10.1111/j.1365-2141.2012.09065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevitt S. Organization of valve pocket thrombi and the anomalies of double thrombi and valve cusp involvement. British Journal of Surgery. 1974;61:641–649. doi: 10.1002/bjs.1800610812. [DOI] [PubMed] [Google Scholar]
- Singhal A, Doherty JF, Raynes JG, McAdam KP, Thomas PW, Serjeant BE, Serjeant GR. Is there an acute-phase response in steady-state sickle cell disease? Lancet. 1993;341:651–653. doi: 10.1016/0140-6736(93)90418-g. [DOI] [PubMed] [Google Scholar]
- Sparkenbaugh E, Pawlinski R. Interplay between coagulation and vascular inflammation in sickle cell disease. British Journal of Haematology. 2013;162:3–14. doi: 10.1111/bjh.12336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein PD, Beemath A, Meyers FA, Skaf E, Olson RE. Deep venous thrombosis and pulmonary embolism in hospitalized patients with sickle cell disease. American Journal of Medicine. 2006;119(10):897.e7–11. doi: 10.1016/j.amjmed.2006.08.015. [DOI] [PubMed] [Google Scholar]
- Tripette J, Connes P, Hedreville M, Etienne-Julan M, Marlin L, Hue O, Hardy-Dessources MD. Patterns of exercise-related inflammatory response in sickle cell trait carriers. British Journal of Sports Medicine. 2010;44:232–237. doi: 10.1136/bjsm.2008.047530. [DOI] [PubMed] [Google Scholar]
- Tripodi A, Cappellini MD, Chantarangkul V, Padovan L, Fasulo MR, Marcon A, Mannucci PM. Hypercoagulability in splenectomized thalassemic patients detected by whole-blood thromboelastometry, but not by thrombin generation in platelet-poor plasma. Haematologica. 2009;94:1520–1527. doi: 10.3324/haematol.2009.010546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsaras G, Owusu-Ansah A, Boateng FO, Amoateng-Adjepong Y. Complications associated with sickle cell trait: a brief narrative review. American Journal of Medicine. 2009;122:507–512. doi: 10.1016/j.amjmed.2008.12.020. [DOI] [PubMed] [Google Scholar]
- Westerman MP, Green D, Gilman-Sachs A, Beaman K, Freels S, Boggio L, Allen S, Schlegel R, Williamson P. Coagulation changes in individuals with sickle cell trait. American Journal of Hematology. 2002;69:89–94. doi: 10.1002/ajh.10021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.