

Abstract
Background:
Recently, there has been tremendous interest in the sinus microbiome and how it relates to disease. However, a lack of a standardized sample collection and DNA extraction methods makes comparison of results across studies nearly impossible. Furthermore, current techniques fail to identify which components of the microbiome are actually alive within the host at the time of sampling.
Objective:
To develop and optimize a method to differentiate which bacterial species in the human sinus microbiome are live versus dead.
Methods:
Duplicate samples from the middle meatus of patients with healthy sinus tissue and those patients with chronic rhinosinusitis were collected by using brushes (n = 12), swabs (n = 27), and tissue biopsy (n = 8) methods. One sample from each pair was either deoxyribonuclease I- or control-treated before DNA extraction. The relative bacterial versus human composition of each sample was determined. A 16S ribosomal RNA gene analysis was performed on a six-paired sample from patients with healthy sinus tissue.
Results:
We found that swabs and brushes collected a higher percentage of bacterial DNA than did tissue biopsy. We also determined that as much as 50% of the bacteria collected in these samples was already dead at the time of collection. The 16S ribosomal RNA gene analysis found significant changes in the relative abundance of taxa identified in the live versus dead bacterial communities of healthy human sinuses.
Conclusions:
Our findings indicated that swabs provided the best quality microbiome samples and that a large portion of the bacteria identified in the sinus were deoxyribonuclease I sensitive. These results highlighted the need for improved techniques such as those presented here, which can differentiate between living and dead bacteria in a sample, a potentially critical distinction when examining changes in sinus innate immune function because both components play important, but distinct, functions. Further studies will determine how these living and dead bacterial populations shift in different disease states and after clinical intervention.
Keywords: Chronic rhinosinusitis, microbiome, DNase, 16S rRNA gene analysis, middle meatus, innate immunity, tissue biopsy, swabs, healthy human sinuses
Chronic rhinosinusitis (CRS) is a significant public health problem that affects >15% of the U.S. population and costs an estimated $8.6 billion annually in direct health costs and lost productivity.1,2 CRS is a highly heterogeneous disease characterized by chronic inflammation of the sinus mucosa and persistent infection of the sinonasal cavity (>12 weeks).3 Classic culture-based microbiologic studies are the standard of care and have established the role of chronic bacterial infection in the pathophysiology of CRS. Previously, the sinuses were believed to be sterile in a healthy, nondiseased state. However, with the advent of more sensitive culture-independent techniques, such as 16S ribosomal RNA (rRNA) gene analysis, we now understand that the sinuses have a “microbiome,” or collection of microbes, that play a critical role in the maintenance of health and the development of disease.4–6 Determining the healthy “normal” sinus microbiome is critical to understanding how innate immune defense in the upper airway functions and is necessary as a baseline by which we can begin to define dysbiosis in disease states.
Recent studies have begun to address the question of which bacterial species constitute a healthy sinus microbiome,7 how it is modified in CRS,7–10 and which microbes are associated with recalcitrant disease.11 However, direct comparison between studies is difficult. One limitation is the lack of consistent sinus sampling methods; various studies used brushes, swabs, lavage, and tissue biopsy, all with differing results.7,9–12 Another is that the high level of person-to-person heterogeneity seen in these studies can easily obscure potential differences between healthy and disease-state microbiomes.10,13 Also, our current 16S rRNA gene analysis protocols are unable to differentiate whether the bacterial species detected are alive or dead. This last point is frequently overlooked but critical to our understanding of the airway microbiome because the functional difference between metabolically active, replicating organisms, and dead bacteria or cell-free DNA is enormous.14,15
In this study, we compared the quantity and quality of bacterial DNA collected by using the three most common techniques: brush, tissue biopsy, and swab. We then developed a novel deoxyribonuclease I (DNase I) dependent technique to differentiate between DNA collected from live versus dead bacteria, and to determine what percentage of the bacteria in the sinuses is alive. Also, by using this DNase-dependent technique combined with 16S rRNA gene analysis, we established what the distribution of live versus dead bacterial taxa was in a cohort of disease-free subjects. Combining this additional layer of information with traditional 16S rRNA sequencing provided a more complete picture of the sinus microbiome because the functional difference between viable and dead bacteria, or extracellular DNA, cannot be ignored.
METHODS
Subjects and Sample Collection
Study participants were patients who were undergoing endoscopic sinus surgery at the Banner University Medical Center, Tucson, Arizona. This study was evaluated and approved by the institutional review board at The University of Arizona (approval 1502660530). Each subject was fully informed and provided written consent before participating. Demographic information is presented in Table 1. Duplicate brush, swab, and tissue samples were collected from patients at the start of the surgery. Sterile cytology brushes (HistoBrush; Puritan Medical, Guilford, ME) and double-tip CultureSwabs EZII (BD Diagnostics, Sparks, MD) were used to sample the middle meatus due to ease of access and the representative nature of this area.16 Tissue was excised from the uncinate process. Samples were immediately placed in sterile collection tubes, labeled with deidentified study identification numbers, and snap-frozen in a CoolBox 30 (BioCision LLC, San Rafael, CA). Samples were stored at −80°C.
Table 1.
Subject demographics for global microbiome analysis (n = 32)

NR = not reported; BMI = body mass index.
*A combination of oral and/or nasal antibiotics and steroids.
A subcohort of six subjects who were undergoing endoscopic sinus surgery for reasons other than chronic sinus infections were selected for bacterial rRNA gene analysis by using paired control-treated and DNase-treated samples collected in parallel with double-tip swabs. Reasons for surgery in these patients included nasal septal deviation or pituitary tumors. All the patients reported no recent antibiotic use (<6 weeks) at the time of surgery. None of the subjects received intraoperative antibiotic therapy. One patient was currently using a nasal steroid spray. A clinical lack of sinus disease was confirmed via radiographic computed tomography and endoscopic examination by the participating surgeon (E. C. and A. C.).
Extraction of Genomic DNA
Duplicate samples were placed in separate, sterile tubes and immerged in sufficient diluted DNase I buffer per manufacture standard protocol (Roche, Basel, Switzerland); 10 U/μL DNase I (Roche) was added to one sample in each pair. Hereafter, both samples were run in parallel through the same protocol to minimize variation. The samples were incubated at 37°C for 10 minutes to allow for DNase I-mediated degradation of free DNA, followed by 75°C for 10 minutes to deactivate the DNase I.15 Samples were immediately transferred to bead beating tubes (PowerSoil DNA Kit; MoBio Labs, Carlsbad, CA), and any remaining solution was centrifuged at 21,000 rcf for 5 minutes to collect remaining cells and to ensure complete removal of the DNase buffers. DNA was then extracted per the manufacturer's standard protocol. Samples were quantified by using a NanoDrop Lite (Thermo Fisher Scientific, Waltham, MA) and stored at −80°C.
Quantitative Polymerase Chain Reaction
Quantitative polymerase chain reaction (qPCR) was performed on all 32 patient samples by using bacterial and human-specific primers to quantify the respective components of each DNA sample. Each reaction contained 20 ng of DNA, 250 nM each of forward/reverse primers, and 5 μL of PerfeCTa SYBRGreen FastMix (Quanta BioSciences, Inc., Gaithersburg, MD) in a 10-μL total volume. Human IntraYd6-Alu primers used were the following: F 5′-GAGATCGAGACCACGGTGAAA-3′, R 5′-TTTGAGACGGAGTCTCGTT-3′; program: 12 minutes at 95°C, followed by 40 cycles of 95°C for 15 seconds and 61°C for 1 minute.17 Bacterial 16S rRNA primers used were the following: F 5′-ACTCCTACGGGAGGCAGCAGT-3′, R 5′-ATTACCGCGGCTGCTGGC-3′; program: 10 minutes at 95°C, followed by 40 cycles of 95°C for 30 seconds and 68°C for 1 minute.18 All reactions were performed in triplicate by using a Rotogene RG-3000 (Qiagen, Hilden, Germany). Standard curves were generated by using known concentrations of Human Male Genomic (EMD Millipore, Billerica, MA) and Escherichia coli (Affymetrix, Santa Clara, CA) DNA. Average cycle times (Ct) values were determined and then compared with the standard curve to calculate the concentration of human and bacterial genomic DNA in each sample. Statistical analysis via a paired t-test was performed by using GraphPad Prism (v.5) (GraphPad Software, La Jolla, CA), with p < 0.05 accepted as significant.
16S rRNA Gene Sequencing and Analysis
The 16S rDNA V4 region was amplified by PCR and sequenced in the MiSeq platform (Illumina, San Diego, CA) by using the 2 × 250-bp paired-end protocol, which yields pair-end reads. The primers used for amplification contain adapters for MiSeq sequencing and single-end barcodes, which allows pooling and direct sequencing of PCR products.19 The read pairs were demultiplexed based on the unique molecular barcodes, and reads were merged by using USEARCH v7.0.1090,20 which allows zero mismatches and a minimum overlap of 50 bases. Merged reads were trimmed at the first base with Q5. A quality filter was applied to the resulting merged reads, and reads that contained >0.05 expected errors were discarded.
The 16S rRNA gene sequences were clustered into operational taxonomic units (OTU) at a similarity cutoff value of 97% by using the UPARSE algorithm.21 OTUs were mapped to an optimized version of the SILVA Database,22 which contained only the 16S v4 region to determine taxonomies. Abundances were recovered by mapping the demultiplexed reads to the UPARSE OTUs. A custom script constructed a rarefied OTU table from the output files generated in the previous two steps for downstream analyses of α-diversity, β-diversity,23 and phylogenetic trends.
RESULTS
Comparison of Collection Methods
Microbiome samples were collected from the middle meatus of 32 patients who were undergoing endoscopic sinus surgery by using three common sinus sampling methods: cytology brushes, double-tip foam swabs, or tissue biopsy. Demographic information for this cohort is listed in Table 1. Total DNA yields and quality were comparable among all three methods. To determine how much of the DNA extracted from each of our collection techniques originated from bacteria rather than the host, we performed quantitative PCR by using bacterial- or human-specific primer sets.17,18 We found that 4–5% of the DNA extracted from our brushes and swabs was of bacterial origin, whereas only ∼2% of the DNA from the tissue samples was bacterial (Table 2).
Table 2.
Comparison of sinus microbiome sampling techniques

Analysis of Live versus Dead Bacterial Content
To determine which bacterial species in our samples were alive versus dead at the time of collection, we split our paired samples into control- or DNase-treated conditions. All steps in the subsequent sample treatment and DNA extraction were identical, with the exception of the presence of the DNase enzyme in one of the two duplicate samples (Fig. 1). This method allowed us to directly compare total bacterial DNA in a sample, the method used in most microbiome studies to date, with only that bacterial DNA protected within living bacteria from an essentially identical sample. Control experiments were performed by using live or heat-killed Pseudomonas aeruginosa strain K subjected to the same control or DNase treatment described above. No decrease in DNA quantity was seen after DNase treatment of the live P. aeruginosa strain K samples (control, 84.0 ng/μL; DNase, 109.6 ng/μL); however, little to no intact DNA was extracted from the heat-killed P. aeruginosa strain K after DNase treatment, whereas the control-treated sample remained unchanged (control, 89.2 ng/μL; DNase, 2.8 ng/μL). In our patient samples, we found that as much as 50% of the total DNA resulted from dead or damaged cells (Fig. 2).
Figure 1.
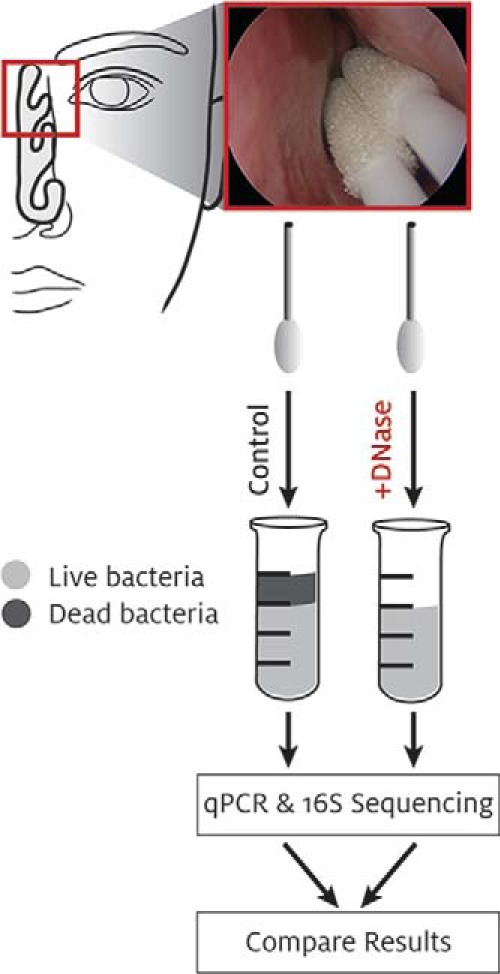
Diagram of experimental design. 1st: Sample collection. 2nd: Deoxyribonuclease I (DNase) treatment. 3rd: DNA isolation. 4th: Downstream applications. 5th: Comparative analysis.
Figure 2.
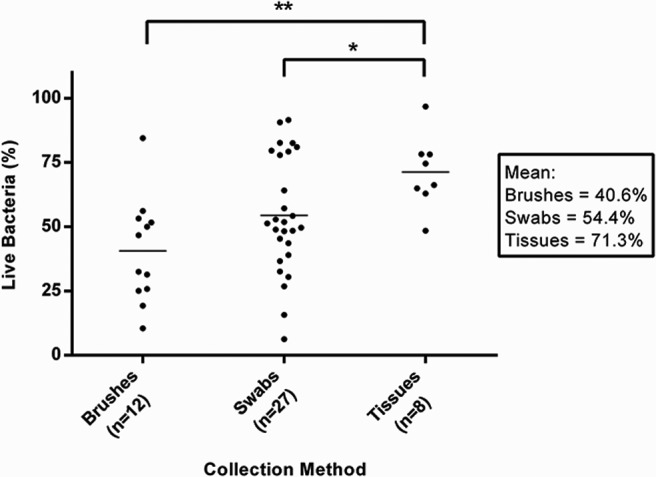
Percentage of live DNA within samples collected by using brushes, swabs, and tissue biopsy specimens. Quantity of DNA extracted from control- versus deoxyribonuclease-treated paired samples. The average percentage of DNase-insensitive (“live”) bacteria from each collection method is shown in the box on the right. **p = 0.0016. *p = 0.046.
Identification of Live versus Dead Microbiomes in the Healthy Human Sinus
To determine which bacterial taxa were affected by DNase treatment, we selected six healthy subjects without clinical sinus disease and performed 16S rRNA gene analysis on both control and DNase samples collected in parallel from the middle meatus by using the double-tip culture swabs. In total, 164 unique OTUs were identified across the 12 samples. We found that the overall bacterial diversity of our samples decreased with DNase treatment: an average of 33 unique OTUs were sequenced from our control-treated samples compared with an average of 27 unique OTUs in the DNase-treated samples. Besides the genera that appeared to be present only as dead bacteria or free DNA (i.e., Propionibacterium and Akkermansia), a number of other bacteria commonly found in the sinuses were found to be highly DNase sensitive. Twenty-five percent of the Staphylococcus detected was already dead on sampling as well as >50% of the Enterobacter, Haemophilus, Neisseria, Escherichia, and Bacteroides, all genus that contain known or suspected respiratory pathogens (Table 3).
Table 3.
Taxa abundance in sinus of healthy patients (control treated vs DNase I treated)

DNase I = deoxyribonuclease I.
DISCUSSION
In this study, we compared collection of sinus microbiome samples by using the three most common methodologies in the literature: swabs, brushes, and tissue biopsy. Although total DNA yields and quality were equivalent among the three methods, which was consistent with one previous publication that compared maxillary sinus brushings with tissue biopsy,24 we found that significantly more bacterial DNA was collected by using the swabs and brushes (Table 1). This difference was most likely due to the much larger sinus epithelia surface area that can be sampled by using these methods. Contributing to this difference could also be that tissue biopsy specimens include subcutaneous epithelial cells, whereas brushes typically include only surface epithelial cells. From a practical standpoint, we found that the double-tip culture swabs were gentle enough to allow for sample collection in the clinic without the use of analgesics and allowed for parallel collection of two virtually identical samples from the same location, critical for our DNase studies.
Up to 50% of the DNA identified in these samples was derived from dead or damaged cells. Of the limited number of studies performed to date that looked at live versus dead bacteria the majority was performed in isolated bacterial populations or environmental samples, which makes it difficult to compare.25,26 However, Maurice et al.27 found that >17% of bacteria in human fecal samples were propidium iodide-sensitive, and Pezzulo et al.15 found that 63% of bacteria from porcine bronchoalveolar lavage fluid samples were DNase sensitive. These striking differences indicated that the fraction of dead bacteria observed in a given microbial community may vary greatly, depending on disease state, location, sampling method, and other factors yet to be elucidated. They also highlight the importance of looking at both the total and live bacteria-derived DNA to avoid greatly overestimating the living components of the microbiome.
Although interest in improved understanding of the airway microbiome has increased dramatically in the past 5 years, the majority of studies overlook this issue of “live versus dead.” Recent studies that investigated the gut microbiome used a technique known as viability PCR to try and identify only those bacterial species that are alive. Viability PCR depends on cell membrane impermeability to exclude a dye, such as propidium monoazide or propidium iodide, from gaining access to the nucleus.15,25 Damaged or dead cells that have lost membrane integrity will take up the dye, which is then covalently cross-linked to the cell's DNA. PCR amplification of this bound DNA is blocked, and, therefore, downstream detection by sequencing is prevented. However, these techniques are complicated because excess dye is difficult to remove, which requires multiple washing, target transfer steps, and potentially introduces contamination, result in bacterial target loss, and may give false-negative results due to carryover of unbound dye.26
As an alternative to these dye-based assays, two groups recently published studies that used the enzyme DNase to differentiate live from dead bacteria.15,26 DNase is an endonuclease that nonspecifically cleaves both single- and double-stranded DNA. As with phorbol 12-myristate 13-acetate and propidium iodide, DNase can only act on accessible DNA, which is not protected within an intact cell membrane. However, this technique differs in that, rather than modifying the DNA to block PCR amplification, the enzyme directly cleaves available DNA into fragments too small for PCR detection. This cleavage minimizes the possibility of false-positives by destroying DNA from dead bacteria rather than just shielding it from PCR amplification. DNase also has the advantage of being susceptible to proteolytic cleavage by pronase or irreversible heat inactivation, which thus removes the risk of false-negative results, the need for multiple wash steps, and potential loss of target.15,26 Although DNase has been shown to have a negative effect on bacterial biofilm formation after long-term treatment, even these experiments, which used higher concentrations of DNase and far longer incubations than those used here, did not report cell death, just a slowing of replication.28 Furthermore, the sturdy nature of bacterial cell envelopes, which can remain intact hours or even days after cell death,25,29,30 combined with the very short treatment time required (10 minutes before DNase inactivation and cell lysis) does not provide sufficient time for the cells to permeate and render themselves susceptible to DNase degradation.
In a cohort of six healthy subjects, we found significant changes in the sinus microbiome post-DNase treatment (Table 3). Overall, our results from the control-treated samples were consistent with primary taxa observed by Ramakrishnan et al.7 in their profiling of the healthy adult sinus. However, some of the bacterial taxa detected both in the cohort of Ramakrishnan et al.7 and in our own cohort turned out to be from entirely DNase-sensitive organisms, and highly significant changes were seen in the relative abundance of a number of clinically relevant taxa. Due to the strict quality controls implemented both in the wet laboratory and data analysis stages, we did not believe this difference was due to PCR bias, although we acknowledge that this is a concern with any 16S rRNA study.
Although our DNase protocol allows for the differentiation of living versus dead bacteria in the sinus microbiome, the relative importance of this finding is not yet known. It would be incorrect to ignore dead bacteria in microbiome analysis because by-products of dead bacteria (including extracellular vesicles, lipopolysaccharides, and free DNA) are known stimulators of innate immunity and contribute to biofilm formation and to the overall picture of microbial interactions. We do, however, believe that it is critical to differentiate between these two distinct populations and believe that our protocol adds an additional layer of vital information to traditional 16S rRNA gene analysis techniques. Although our sample size was small, our study was the first, to our knowledge, to profile the live versus dead bacterial species present in the healthy human sinus. Further studies currently underway in our laboratory will identify how this healthy microbiome shifts in patients with chronic sinus disease and how these populations may further change after clinical intervention. It is also possible that different sites within the sinuses may have different bacterial communities that may be more or less stable due to how exposed they are to the outside environment. Current microbiome analyses include bacterial richness, evenness, and diversity in the sinus; however, we propose that DNase sensitivity may also be a critical factor in understanding the mechanisms of host-immune response because these mechanisms regulate the sinus microbiome in both health and disease.
ACKNOWLEDGMENTS
We thank our clinical coordinator Cynthia Thompson for invaluable assistance, Christopher Le, M.D., Audriana Hurbon, BS, and Caitlin Gluck, BS, for sample collection. Kwan Chul Kim, Ph.D., and Kosuke Kato, Ph.D., for P. aeruginosa. Gwynneth Ballentine, Ph.D., MBA, and Nadim Ajami, Ph.D., (Diversigen, Inc.) for the 16S rRNA gene sequencing and analysis, and Heddwen Brooks, Ph.D., for use of their Rotogene RG-3000. We also thank the patients who volunteered to participate in our study because, without them, this project would not have been possible.
Footnotes
Funding was provided by the National Institute of Health - National Institute of Dental and Craniofacial Research, K08 (DE021413) and the Cystic Fibrosis Foundation (CHANG131O)
Presentation at the American Rhinologic Society (Academy of Otolaryngology - Head & Neck Society), Dallas, Texas, September 25, 2015
The authors have no conflicts of interest to declare pertaining to this article
REFERENCES
- 1. Bhattacharyya N. Incremental healthcare utilization and expenditures for allergic rhinitis in the United States. Laryngoscope 121:1830–1833, 2011. [DOI] [PubMed] [Google Scholar]
- 2. Ray NF, Baraniuk JN, Thamer M, et al. Healthcare expenditures for sinusitis in 1996: Contributions of asthma, rhinitis, and other airway disorders. J Allergy Clin Immunol 103:408–414, 1999. [DOI] [PubMed] [Google Scholar]
- 3. Report of the Rhinosinusitis Task Force Committee Meeting Alexandria, Virginia, August 17, 1996 Otolaryngol Head Neck Surg 117(pt. 2):S1–S68, 1997. [PubMed] [Google Scholar]
- 4. Feazel LM, Frank DN, Ramakrishnan VR. Update on bacterial detection methods in chronic rhinosinusitis: Implications for clinicians and research scientists. Int Forum Allergy Rhinol 1:451–459, 2011. [DOI] [PubMed] [Google Scholar]
- 5. Hauser LJ, Feazel LM, Ir D, et al. Sinus culture poorly predicts resident microbiota. Int Forum Allergy Rhinol 5:3–9, 2015. [DOI] [PubMed] [Google Scholar]
- 6. Kennedy JL, Borish L. Chronic rhinosinusitis and antibiotics: The good, the bad, and the ugly. Am J Rhinol Allergy 27:467–472, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ramakrishnan VR, Feazel LM, Gitomer SA, et al. The microbiome of the middle meatus in healthy adults. PLoS One 8:e85507, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Carter JM, Amedee RG. Contrasting the microbiomes from healthy volunteers and patients with chronic rhinosinusitis. Am J Rhinol Allergy 28:182, 2014. [DOI] [PubMed] [Google Scholar]
- 9. Abreu NA, Nagalingam NA, Song Y, et al. Sinus microbiome diversity depletion and Corynebacterium tuberculostearicum enrichment mediates rhinosinusitis. Sci Transl Med 4:151ra124, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Aurora R, Chatterjee D, Hentzleman J, et al. Contrasting the microbiomes from healthy volunteers and patients with chronic rhinosinusitis. JAMA Otolaryngol Head Neck Surg 139:1328–1338, 2013. [DOI] [PubMed] [Google Scholar]
- 11. Ramakrishnan VR, Hauser LJ, Feazel LM, et al. Sinus microbiota varies among chronic rhinosinusitis phenotypes and predicts surgical outcome. J Allergy Clin Immunol 136:334–342.e1, 2015. [DOI] [PubMed] [Google Scholar]
- 12. Liu CM, Soldanova K, Nordstrom L, et al. Medical therapy reduces microbiota diversity and evenness in surgically recalcitrant chronic rhinosinusitis. Int Forum Allergy Rhinol 3:775–781, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schwarzberg K, Le R, Bharti B, et al. The personal human oral microbiome obscures the effects of treatment on periodontal disease. PLoS One 9:e86708, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Klein DA. Bulk extraction-based microbial ecology: Three critical questions. Biol Rev 72:557–578, 2008. [Google Scholar]
- 15. Pezzulo AA, Kelly PH, Nassar BS, et al. Abundant DNase I-sensitive bacterial DNA in healthy porcine lungs and its implications for the lung microbiome. Appl Environ Microbiol 79:5936–5941, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Feazel LM, Robertson CE, Ramakrishnan VR, Frank DN. Microbiome complexity and Staphylococcus aureus in chronic rhinosinusitis. Laryngoscope 122:467–472, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Walker JA, Kilroy GE, Xing J, et al. Human DNA quantitation using Alu element-based polymerase chain reaction. Anal Biochem 315:122–128, 2003. [DOI] [PubMed] [Google Scholar]
- 18. Lazarevic V, Gaia N, Girard M, et al. Comparison of DNA extraction methods in analysis of salivary bacterial communities. PLoS One 8:e67699, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Caporaso JG, Lauber CL, Walters WA, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6:1621–1624, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461, 2010. [DOI] [PubMed] [Google Scholar]
- 21. Edgar RC. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10:996–998, 2013. [DOI] [PubMed] [Google Scholar]
- 22. Quast C, Pruesse E, Yilmaz P, et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lozupone C, Knight R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71:8228–8235, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Roediger FC, Slusher NA, Allgaier S, et al. Nucleic acid extraction efficiency and bacterial recovery from maxillary sinus mucosal samples obtained by brushing or biopsy. Am J Rhinol Allergy 24:263–265, 2010. [DOI] [PubMed] [Google Scholar]
- 25. Cangelosi GA, Meschke JS. Dead or alive: Molecular assessment of microbial viability. Appl Environ Microbiol 80:5884–5891, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Villarreal JV, Jungfer C, Obst U, Schwartz T. DNase I and proteinase K eliminate DNA from injured or dead bacteria but not from living bacteria in microbial reference systems and natural drinking water biofilms for subsequent molecular biology analyses. J Microbiol Methods 94:161–169, 2013. [DOI] [PubMed] [Google Scholar]
- 27. Maurice CF, Haiser HJ, Turnbaugh PJ. Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell 152:39–50, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tetz VV, Tetz GV. Effect of extracellular DNA destruction by DNase I on characteristics of forming biofilms. DNA Cell Biol 29:399–405, 2010. [DOI] [PubMed] [Google Scholar]
- 29. Nystrom T. Not quite dead enough: On bacterial life, culturability, senescence, and death. Arch Microbiol 176:159–164, 2001. [DOI] [PubMed] [Google Scholar]
- 30. Nystrom T. Conditional senescence in bacteria: Death of the immortals. Mol Microbiol 48:17–23, 2003. [DOI] [PubMed] [Google Scholar]