

Abstract
Background:
Hereditary hemoglobin (Hb) disorders are the most commonly encountered single gene disorders in India. Proper timely identification of these disorders is of paramount importance to prevent thalassemia major and clinically severe hemoglobinopathy as well as for epidemiologic purposes.
Aims:
Our aim was to determine the prevalence of thalassemia and hemoglobinopathy in patients of a tertiary care hospital of West Bengal, India.
Materials and Methods:
This prospective study was conducted on 119,336 cases over a period of 10 years. After taking clinical history and familial history, complete hemogram report was obtained by an automated cell counter. High-performance liquid chromatography (HPLC) was performed on the samples with Bio-Rad Variant using beta thalassemia short program. Confirmatory tests were performed whenever required.
Results:
A normal Hb pattern was observed in 104,804 (87.83%) cases and abnormalities were detected in 14,532 (12.17%) patients. β (beta) thalassemia trait was the commonest abnormality found in 5,488 (4.60%) patients. HbE trait was found in 3,604 (3.02%) patients, β thalassemia major/intermedia in 1,981 (1.66%) cases, and Eβ thalassemia in 1,384 (1.16 %) cases. Other variants detected included HbE disease, sickle-cell disease, sickle β thalassemia, HbD-Punjab trait, HbQ-India trait, α-thal trait, double heterozygous state of HbS and HbE, double heterozygous state of HbS and HbD, HbJ-Meerut, hereditary persistence of fetal hemoglobin (HPFH), HbH, delta β-thal trait, and Hb Lepore.
Conclusion:
In view of the high prevalence of hemoglobinopathy in this region, a routine premarital screening program is needed for the identification and prevention of high-risk marriages and thus, prevention of the psychosocial trauma of bearing a transfusion-dependent child for life.
Keywords: Hemoglobinopathy, high-performance liquid chromatography (HPLC), thalassemia
Introduction
Thalassemia is a heterogeneous group of inherited disorders of hemoglobin (Hb) characterized by reduced or absent production of globin chains. It is the commonest single gene disorder in the world first noted in the Mediterranean population and causing a significant morbidity and mortality in India and abroad.[1,2] Inherited disorders of Hb due to the structural alteration of globin chain are called hemoglobinopathies. The prevalence of thalassemia and hemoglobinopathy varies with geographic locations. It has been estimated that in India, 0.37 per 1,000 fetuses have Hb disorder.[3]
Thalassemia and hemoglobinopathy are the major health concern in the Indian subcontinent as the prevalence rate of beta thalassemia mutations is as high as 17% in some populations.[4] Hb E-β thalassemia and sickle-cell anemia are also common Hb disorders, which are reported to be quite prevalent in many parts of India.[5,6,7,8] The other significant hemoglobinopathies prevalent in India are HbE, sickle-cell anemia, and HbD.[6,7,8,9]
High-performance liquid chromatography (HPLC) is one of the best methods for the screening and detection of various hemoglobinopathies and it provides rapid, reproducible, and precise results.[10] Detail clinical history including family history and different hematologic parameters such as complete blood count, reticulocyte count, and red cell morphology are often required to confirm the diagnosis.
The present study attempted to find out the occurrence of different Hb variants in the population of West Bengal in India and adjacent areas so that a definite plan of action regarding the diagnostic, preventive, and therapeutic strategies can be formulated to minimize more serious disorders in future generations.
Materials and Methods
This study was conducted in the department of pathology of a tertiary care teaching medical institution of West Bengal, India over a period of 10 years from June 2005 to May 2015. Patients who came for a premarital checkup voluntarily, patients with abnormal hemograms suggestive of hemolytic anemia, and those patients who had a positive family history of thalassemia were included in the study. Transfusion-dependent children and adults were also included. However, patients with history of blood transfusion within the last 1 month were excluded. A signed consent form was obtained from all the patients included in the study. Detailed clinical history and family history were obtained from each patient. Past history of blood transfusion, if present, was noted.
Blood samples were collected in ethylene diamine tetrachloride acetate (EDTA) vials and analyzed with Sysmex, United States of America (USA) automated cell counter for complete blood counts. Clotted blood samples were also collected for liver function tests and determination of serum iron, ferritin, cobalamin, and folate levels. For each patient, a peripheral blood smear (PBS) was prepared and stained with Leishman stain and observed microscopically for red cells morphology for the supporting of diagnosis of hemoglobinopathies. Hb variant analysis was studied for various hemoglobinopathies and variants. The tests were performed on Biorad Variant using beta thalassemia short program (Bio-Rad Laboratories, California, USA). The instrument utilized the principle of HPLC. With HPLC, the positively charged Hb fractions were separated based on their ionic interactions with a negatively charged stationary phase in a chromatography column followed by their elution by a mobile phase with phosphate buffers differing in pH and ionic strength. The adsorbed positively charged Hb molecules were eluted from the column into the liquid phase at a rate related to their affinity for the stationary phase. Hbs were identified by their retention time and quantified by computing the area under the corresponding peak in the elution profile.[11] Confirmatory tests such as acid and alkaline electrophoresis, sickling test, solubility test, and brilliant cresyl blue test for HbH inclusions were performed as and when required.
Results
During the study period of 9 years, a total of 119,336 cases were studied. The patient population comprised 66,351 (55.6%) antenatal patients, 41,290 (34.6%) patients with other hematological or nonhematological disorders, and 11,695 (9.8%) family studies. The age of the patients ranged between 5 months and 72 years. The mean age was found to be 25.8 years. Of these, 14,532 (12.17%) cases displayed abnormal Hb fractions on HPLC [Figure 1].
Figure 1.
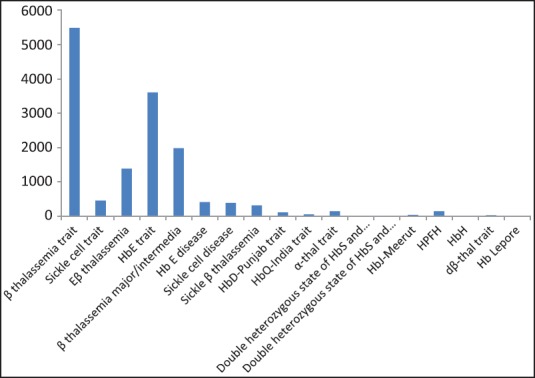
The distribution of hemoglobinopathies among positive cases Total no. of cases analyzed (n) = 119,336 patients with hemoglobinopathies (n) = 14,532 (12.17%)
The most common Hb abnormality detected was β (beta) thalassemia trait present in 5,488 (4.60%) patients. Hb E trait was found in 3,604 (3.02%) cases followed by β thalassemia major/intermedia 1,981 (1.66%) and Eβ thalassemia in 1,384 (1.16%) patients. The distribution of different Hb patterns in the study population has been shown in Table 1. For each of these groups, the hematologic parameters and the percentage of various Hbs detected in HPLC have been shown in Table 2. Interpretation of results of HPLC was done on the basis of retention time, percentage of Hb, and peak characteristics.
Table 1.
Distribution of hemoglobin patterns in the study population
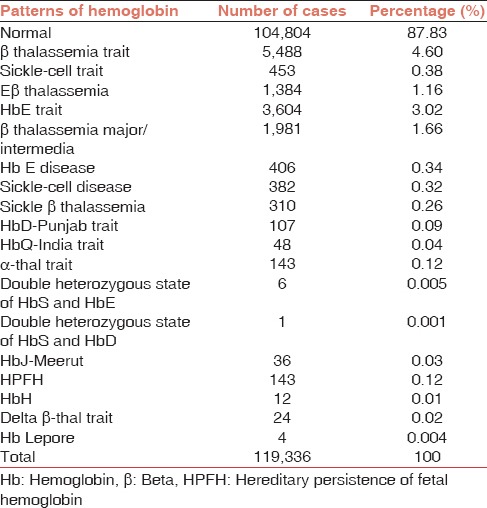
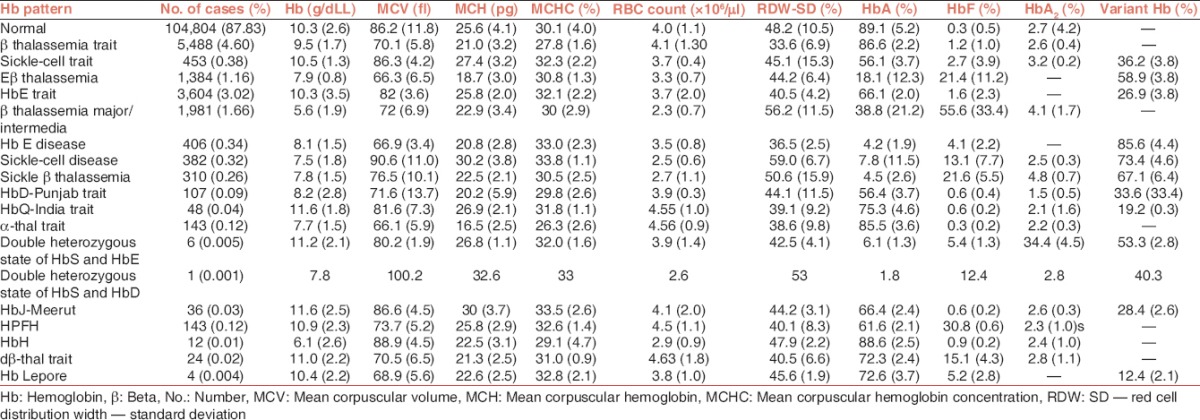
Table 2.
Hematologic parameters of the patients stratified according to the hemoglobin patterns (values mentioned are mean ± standard deviation)
Twelve (0.01%) cases of HbH were detected, which were suspected on the basis of a significant peak that appeared in each case during the first minute of elution. Confirmatory tests were performed, which included Hb electrophoresis and the test for HbH inclusion bodies. In the next window, i.e., p1 window (retention time: 0·63-0·85 min) no Hb variant was detected. High Hb level in the F window having retention time 0·98-1·2 min was detected in three different cases, namely, β thalassemia major (1·66%), Eβ thalassemia (1·16%), sickle-β thalassemia (·26%), sickle-cell disease (·32%), and hereditary persistence of fetal hemoglobin (HPFH) (0.12%).
A significant peak was obtained in the p2 window (retention time: 1·24-1·4 min) in 267 patients. As HbA1c is known to elute in this window, these patients were later tested for blood sugar levels. In 258 (96.6%) cases, the patients were found to be diabetics. The rest did not undergo follow-up. In 36 (0.03%) patients, a significant peak was found in the p3 window. Hb electrophoresis at alkaline pH in these cases showed a fast-moving band anodal to HbA. Thus, these cases were diagnosed as HbJ-Meerut [Figure 2].
Figure 2.
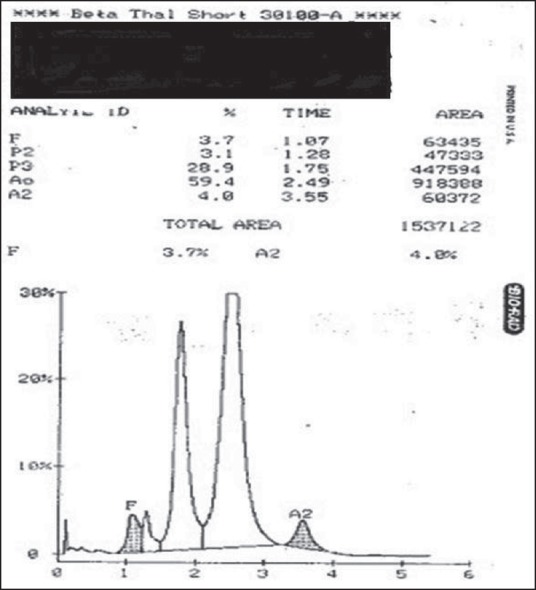
J chain hemoglobinopathy showing P3 peak 28.9% and A 59.4%
A0 window has a retention time between 1.9 min and 3.1 min. Apart from HbA, no other Hb variant was found to elute in this window. HbA2, HbE and Hb Lepore were found to elute in A2 window (retention time: 3.3-3.9 min). HbE trait was detected in 3.02% cases, Eβ thalassemia in 1.16% cases, and HbE disease in 0.34% patients. Hb Lepore was noted in four (0.004%) patients. It was confirmed by Hb-electrophoresis at alkaline pH, which showed major band at AF, another around the SD region.
In this study, 107 (0.09%) cases had a significant peak in the D window (retention time:3.9-4.3 min), which was diagnosed to be HbD-Punjab trait. A statistically significant difference (P < 0∙0001) was found between the mean values of HbA2 in the normal samples (2∙7 ± 0.4) and that in HbD-Punjab trait (1∙5 ± 0∙5). HbS was found to elute in the S window (retention time: 4.3-4.7 min). In this study, sickle-cell trait was found in 453 (0.38%) cases and sickle-cell disease was noted in 382 (0.32%) patients. Later, sickling test and solubility test were performed to confirm the diagnosis of sickle-cell disease. The mean value of HbA2 in sickle-cell trait (3∙2 ± 0∙2) was found to be significantly higher (P < 0∙0001) compared to that in normal samples (2∙7 ± 0.4). In 48 (0.04%) cases, there was a significant peak with the window having retention time 4.70-4.90 min, which was consequently diagnosed as HbQ-India trait. No Hb variant was detected in the C window (retention time: 4.9-5.3 min).
Discussion
Thalassemia and hemoglobinopathy are autosomal recessive inherited disorders, primarily affecting the globin moiety of the Hb molecule. These disorders, which were mainly confined to certain areas, religions, castes, and tribes, particularly with endogamous norms of marriages, are now widely prevalent all over the world. This is because of the migration of various races over the ages and hence, being home to an assortment of sociocultural, linguistic, and ethnically diverse people.[12]
This study was conducted primarily with patients of West Bengal, India and nearby areas. In our present cross-sectional study, the prevalence of Hb disorders was found to be 12.17%, which was lesser than another study conducted in the southern part of West Bengal where the prevalence of such disorders were reported to be 25%.[13] However, our study results correlates well to the prevalence of thalassemia and hemoglobinopathy of northern India where it is reported to be 12.5%.[14] The most common Hb abnormality detected in this study was that of β thalassemia trait (4.60%). Colah R et al. reported that nearly 1.5% of the world's population was carriers of β thalassemia.[15] Several studies reveal that in most parts of India, β thalassemia trait was the commonest Hb disorder. A study conducted by Madan N and coinvestigators showed the overall gene frequency of β thalassemia trait reported in northern and western India was 4.05%.[16] The prevalence of β thalassemia trait has been reported to be as high as 10.38% in the rural parts of West Bengal and in central India, it was found to be 9∙59%.[17,18]
An important finding was the high incidence of HbE beta thalassemia (1.16%). It is said that worldwide approximately 50% of severe beta thalassemia major patients are of HbE beta thalassemia.[19] These patients presented with a variable clinical picture ranging from a condition indistinguishable from beta thalassemia major requiring blood transfusions from infancy to a mild form of thalassemia intermedia showing a mild asymptomatic anemia. The Indian Council of Medical Research (ICMR) study reported a higher incidence of 1.44% in the general population.[20] The prevalence of HbE trait was found in 3.02% cases. A study conducted in the rural areas of West Bengal reported the prevalence of HbE trait to be 3.86% and that of Eβ thalassemia to be 1.25%.[21] Interestingly, in Odisha, sickle-cell trait was the most common abnormality found.[1] In the present study, sickle-cell trait was found in 0.38% cases.
Other variants detected in this study were Hb E disease, sickle-cell disease, sickle β thalassemia, HbD-Punjab trait, HbQ-India trait, α-thal trait, double heterozygous state of HbS and HbE, double heterozygous state of HbS and HbD, HbJ-Meerut [Figure 2], hereditary persistence of fetal hemoglobin (HPFH), HbH, delta β-thal trait [Figure 3], and Hb Lepore [Figure 4]. Rao S et al. reported three cases of HbQ-India, which eluted in an unknown window (retention time: 4-4.9 min).[9]
Figure 3.
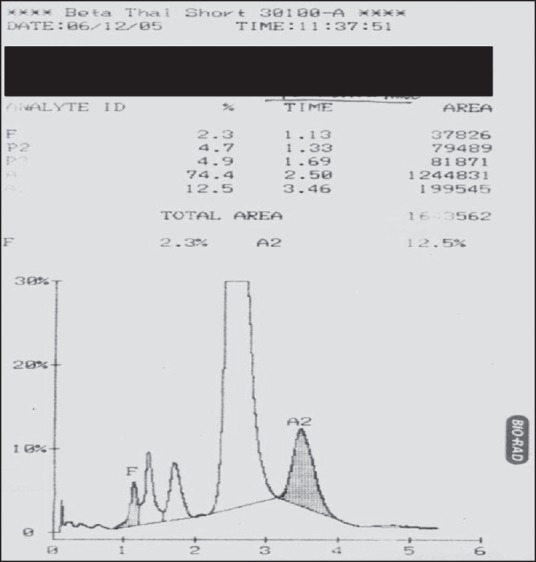
Hb Lepore: Hb A2: 12.5% (RT-3.46’), Hb F: 2.3%, Hb A0: 74.4%
Figure 4.
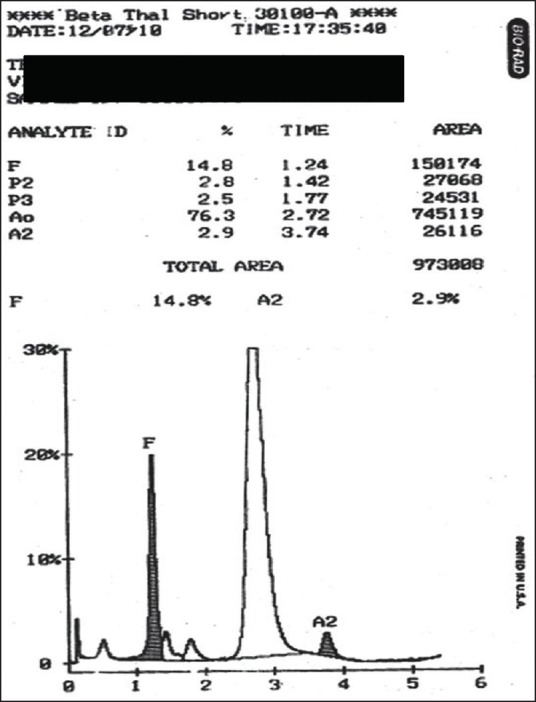
Delta beta thalassemia with hemoglobin F level of 14.8%
Delta beta thalassemia is a rare cause of elevated fetal Hb. Very few cases have been reported from India.[22] HPFH has also been reported infrequently from different regions of India.[23] In this study, the difference in the two was made on the basis of the percentage of fetal hemoglobin (HbF), the mean corpuscular volume (MCV), and the red blood cell distribution width (RDW). The ICMR multicenter study has found an incidence of 0.73% of delta beta thalassemia and 0.18% of HPFH, which we found 0.02% and 0.12%, respectively.
HbD trait and double heterozygote condition of HbSD were detected in 108 (0.091%) patients. In India, the gene frequency of HbD is relatively low with a tendency to cluster toward the northwestern part of the country.[24] Alpha thalassemia is a common problem of Southeast Asian countries with gene frequencies reaching up to 30-40% in some parts of Thailand.[25] An incidence of 3.84% in some Assam tribesmen has been reported previously.[26] In our current study, α-thal trait prevalence was detected to be 0.12%. J chain hemoglobinopathy is also a rare finding in previous studies. Srinivas U et al. in their study have reported seven cases from New Delhi in Delhi, India.[27] We found a total of 36 cases with HbJ-Meerut. These alpha chain hemoglobinopathies are clinically silent and are detected accidentally.
HPLC has been established as a sensitive, specific, and accurate technique for the identification and quantification of different Hb fractions. However, it should be kept in mind that HPLC is limited by its inability to detect α thalassemia and normal HbA2 β thalassemia. Hb variants that elute with the same retention time also cannot be separately identified by HPLC and during interpretation of chromatograms, nutritional anemias must always be taken into account. A low level of HbA2 may be induced by iron deficiency, thus masking β thalassemia trait. Similarly, cobalamin or folate deficiency may raise HbA2 level, leading to a false diagnosis of thalassemia trait[14] and whenever necessary, HPLC must be followed by molecular studies, such as polymerase chain reaction (PCR), amplification refractory mutation system (ARMS), and other similar tests to determine specific mutations responsible for the Hb disorder.[28]
Conclusion
To conclude, HPLC forms a rapid, accurate, and reproducible tool for the early detection and management of hemoglobinopathies and variants. This is especially important in view of the high incidence of beta thalassemia trait in the Indian subcontinent. Thus, premarital and antenatal screening should be mandatory to prevent the birth of offspring with β thalassemia major. Moreover, knowledge of common Hb patterns in a particular region helps to formulate appropriate preventive and therapeutic strategies.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Balgir RS. Genetic epidemiology of three predominant abnormal hemoglobins in India. J Assoc Physicians India. 1996;44:25–8. [PubMed] [Google Scholar]
- 2.Borgna-Pignatti C, Galanello R. Thalassemias and related disorders: Quantitative disorders of hemoglobin synthesis. In: Greer JP, Foerster J, Lukens JN, Rodgers GM, Parakevas F, Glader B, editors. Wintrobes Clinical Hematology. Philadelphia: Lippincott Willams and Wilkins; 2004. pp. 1319–65. [Google Scholar]
- 3.Borgna-Pignatti C, Galanello R. Thalassemias and related disorders: Quantitative disorders of hemoglobin synthesis. In: Greer JP, Foerster J, Rodgers GM, Parakevas F, Glader B, Means RT, editors. Wintrobe's Clinical Hematology. 12th ed. Philadelphia: Lippincott Williams and Wilkins; 2009. pp. 1082–131. [Google Scholar]
- 4.Vaz FE, Thakur CB, Banerjee MK, Gangal SG. Distribution of beta-thalassemia mutations in the Indian population referred to a diagnostic center. Hemoglobin. 2000;24:181–94. doi: 10.3109/03630260008997526. [DOI] [PubMed] [Google Scholar]
- 5.Chatterjea JB. Some aspects of HbE and its genetic interaction with thalassemia. Indian J Med Res. 1965;53:377–83. [PubMed] [Google Scholar]
- 6.Gupta SC, Mehrotra TN, Mehrotra VG. Haemoglobin-E-thalassaemia in Uttar Pradesh. Indian J Med Res. 1970;58:857–62. [PubMed] [Google Scholar]
- 7.Shukla RM, Solanki BR. Sickle-cell trait in Central India. Lancet. 1985;1:297–8. doi: 10.1016/s0140-6736(58)91035-3. [DOI] [PubMed] [Google Scholar]
- 8.Kamle M, Chaturvedi P. Epidemiology of sickle cell disease in a rural hospital in central India. Indian Pediatr. 2000;37:391–6. [PubMed] [Google Scholar]
- 9.Rao S, Kar R, Gupta SK, Chopra A, Saxena R. Spectrum of hemoglobinopathies diagnosed by cation exchange-HPLC and modulating effects of nutritional deficiency anaemias from north India. Indian J Med Res. 2010;132:513–9. doi: 10.4103/0971-5916.73390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bravo-Urquuiola M, Arends A, Montilla S, Velasquez D, Garcia G, Alvarez M, et al. Advantages in the use of high performance chromatography technique for screening hemoglobinopathies in Venezuela. Invest Clin. 2004;45:309–15. [PubMed] [Google Scholar]
- 11.Bain BJ. Haemoglobinopathy Diagnosis. 2nd ed. Massachusetts USA: Blackwell Publishing; 2006. pp. 26–62. [Google Scholar]
- 12.Patne SC, Shukla J. Hemoglobin E disorders in Eastern Uttar Pradesh. Indian J Pathol Microbiol. 2009;52:110–2. doi: 10.4103/0377-4929.44991. [DOI] [PubMed] [Google Scholar]
- 13.Manna AK, Dutta SK, Chatterjee A. Relative incidence of different thalassaemias and haemoglobinopathies in South Bengal. J Indian Med Assoc. 2009;107:347–9. [PubMed] [Google Scholar]
- 14.Sachdev R, Dam AR, Tyagi G. Detection of Hb variants and haemoglobinopathies in Indian population using HPLC: Report of 2600 cases. Indian J Pathol Microbiol. 2010;53:57–62. doi: 10.4103/0377-4929.59185. [DOI] [PubMed] [Google Scholar]
- 15.Colah R, Gorakshakar A, Nadkarni A. Global burden, distribution and prevention of β-thalassemias and hemoglobin E disorders. Expert Rev Hematol. 2010;3:103–17. doi: 10.1586/ehm.09.74. [DOI] [PubMed] [Google Scholar]
- 16.Madan N, Sharma S, Sood SK, Colah R, Bhatia LH. Frequency of β-thalassemia trait and other hemoglobinopathies in northern and western India. Indian J Hum Genet. 2010;16:16–25. doi: 10.4103/0971-6866.64941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dolai TK, Dutta S, Bhattacharyya M, Ghosh MK. Prevalence of hemoglobinopathies in rural Bengal, India. Hemoglobin. 2012;36:57–63. doi: 10.3109/03630269.2011.621007. [DOI] [PubMed] [Google Scholar]
- 18.Chatterjee N, Mishra A, Soni R, Kulkarni H, Mamtani M, Shrivasatava M. Bayesian estimates of the prevalence of β-thalassemia trait in voluntary blood donors of central India: A survey. Hemoglobin. 2010;34:548–60. doi: 10.3109/03630269.2010.526488. [DOI] [PubMed] [Google Scholar]
- 19.Olivieri NF, Pakbaz Z, Vichinsky E. Hb E/beta-thalassaemia: A common and clinically diverse disorder. Indian J Med Res. 2011;134:522–31. [PMC free article] [PubMed] [Google Scholar]
- 20.Mohanty D, Colah RB, Gorakshakar AC, Patel RZ, Master DC, Mahanta J, et al. Prevalence of β-thalassemia and other haemoglobinopathies in six cities in India: A multicentre study. J Community Genet. 2013;4:33–42. doi: 10.1007/s12687-012-0114-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mondal B, Maiti S, Biswas BK, Ghosh D, Paul S. Prevalence of hemoglobinopathy, ABO and rhesus blood groups in rural areas of West Bengal, India. J Res Med Sci. 2012;17:772–6. [PMC free article] [PubMed] [Google Scholar]
- 22.Verma S, Bhargava M, Mittal SK, Gupta R. Homozygous delta-beta thalassemia in a child: A rare cause of elevated fetal hemoglobin. Iran J Ped Hematol Oncol. 2013;3:222–7. [PMC free article] [PubMed] [Google Scholar]
- 23.Balgir RS. Hereditary persistence of foetal haemoglobin in a tribal family of Orissa, India. Natl Med J India. 2004;17:138–40. [PubMed] [Google Scholar]
- 24.Balgir RS. The burden of hemoglobinopathies in India and the challenges ahead. Curr Sci. 2000;79:1536–47. [Google Scholar]
- 25.Fucharoen S, Winichagoon P. Haemoglobinopathies in Southeast Asia. Indian J Med Res. 2011;134:498–506. [PMC free article] [PubMed] [Google Scholar]
- 26.Sen R, Chakrabarti S, Sengupta B, De M, Haldar A, Poddar S, et al. Alpha-thalassemia among tribal populations of Eastern India. Hemoglobin. 2005;29:277–80. doi: 10.1080/03630260500310711. [DOI] [PubMed] [Google Scholar]
- 27.Srinivas U, Mahapatra M, Pati HP. Hb J Meerut, fast-moving hemoglobin: A study of seven cases from India and a review of literature. Am J Hematol. 2007;82:666–7. doi: 10.1002/ajh.20826. [DOI] [PubMed] [Google Scholar]
- 28.Moorchung N, Phillip J, Sarkar RS, Prasad R, Dutta V. Is high pressure liquid chromatography an effective screening tool for characterization of molecular defects in hemoglobinopathies? Indian J Pathol Microbiol. 2013;56:36–9. doi: 10.4103/0377-4929.116146. [DOI] [PubMed] [Google Scholar]