

Abstract
Background:
Hereditary inclusion body myopathy (HIBM) continues to be underrecognized clinically despite a characteristic topography of weakness with total sparing of quadriceps muscles and patient being wheelchair bound. We report seven patients of HIBM from four families in North India.
Methods and Results:
Seven patients from four different families were diagnosed to have HIBM. There was no consanguinity in any of the families. While one patient had two affected siblings, another had one affected siblings and the family history was noncontributory in two patients. Two of the siblings were available for examination and confirmed clinically to be suffering from HIBM. Among the seven patients, only one was still ambulatory at the time of diagnosis.
Discussion:
This is the first case report of occurrence of HIBM in North Indian population. Despite its unique clinical presentation, HIBM is frequently misdiagnosed resulting in unnecessary diagnostic and therapeutic interventions. A high index of suspicion of this rare myopathy along with proper clinical examination may go a long way in accurate prognostication and management of these patients.
Keywords: Hereditary inclusion body myopathy (HIBM), myopathy, rimmed vacuoles, sialic acid
Introduction
Hereditary inclusion body myopathy (HIBM) is an adult-onset muscle disorder characterized by severe progressive muscle weakness and typical histopathology findings on muscle biopsy.[1] Though it has a unique topography of weakness, most patients go unrecognized due to apparent lack of knowledge regarding this rare condition. Despite lack of curative treatment, early diagnosis helps in prognostication of these patients and to avoid unnecessary therapeutic measures. We report seven cases of HIBM in four families from a tertiary care institute in Northern India. Six out of the seven cases were correct diagnosed, only when they were wheelchair bound.
Case Series
Family 1 (three cases)
A 41-year-old North Indian Sikh gentleman presented to us with insidious onset and gradually progressive quadriparesis of 10 years’ duration. There was no sensory, bladder, or bowel complaints. There were no complaints pertaining to cranial musculature except for neck weakness. The weakness started in the lower limbs and gradually progressed to the upper limbs. He was wheelchair bound for the past 2 years. He was independent in daily activities involving upper limbs. He was treated at another center with steroids without any relief of symptoms. Two out of four of his siblings (one elder and one younger sister) [Figure 1] had similar problem and were wheelchair bound. There was no history of consanguinity. On examination he had cushinoid features. There was severe weakness of all the muscles of lower limbs (Medical Research Council (MRC) Grade 1) except for the quadriceps which were normal [Table 1]. In upper limbs, there was severe weakness of shoulder girdle muscles with MRC Grade 2 power in deltoids, while in biceps and triceps, it was Grade 3 and 4, respectively. There was severe weakness of long finger flexors; while small muscles of hands and wrist and finger extensors, though week, were relatively spared. Sensory examination was normal. Other than quadriceps jerk, all other jerks were absent. CPK was mildly elevated (two times above upper limit of normal). Electromyography (EMG) showed myopathic changes. In view of characteristic clinical features, a diagnosis of HIBM was made and he underwent left triceps muscle biopsy which was consistent with diagnosis of HIBM. His two affected siblings were examined later and both of them had topography of weakness similar to the index case. He was started on physiotherapy and rehabilitation exercises and was counseled regarding the illness.
Figure 1.
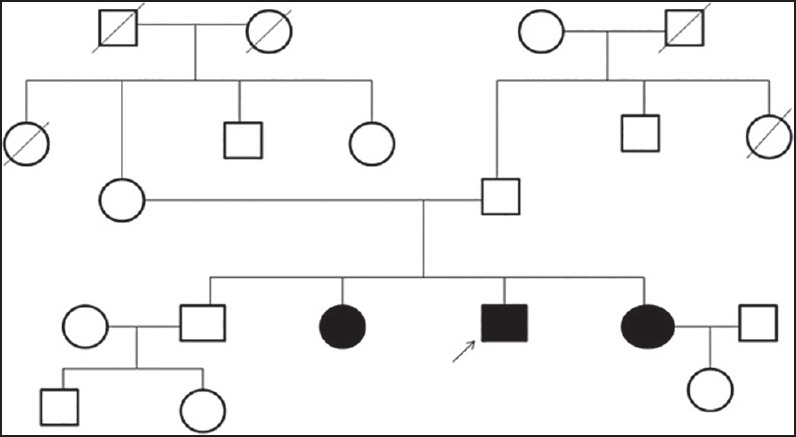
Family tree of first family showing affected siblings without involvement of other family members
Table 1.
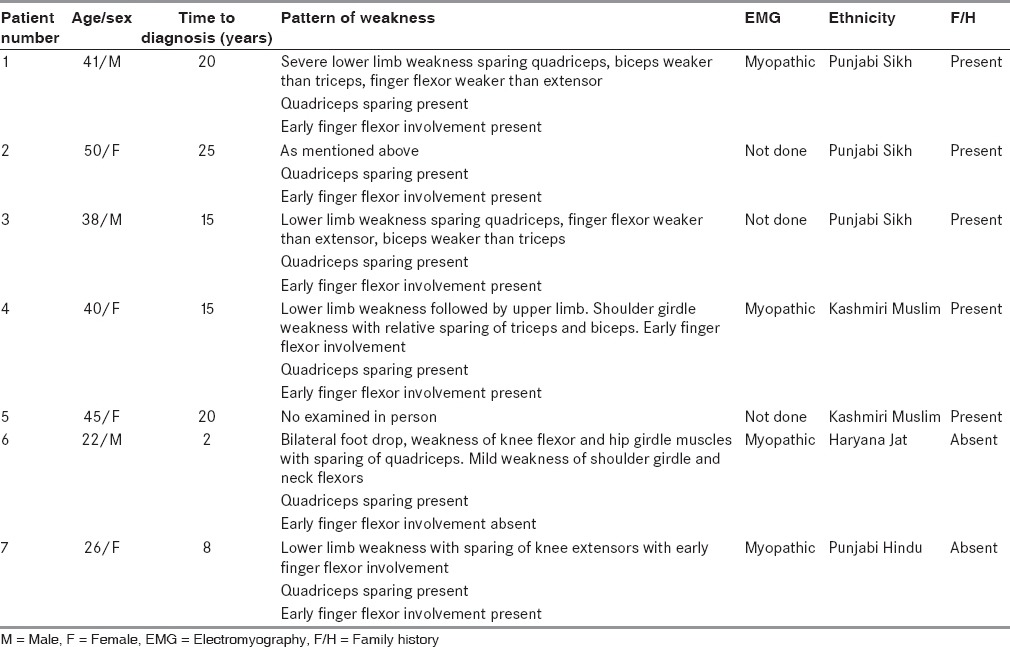
Demographic profile of patients (N = 7)
Family 2 (two cases)
A 40-year-old lady from Kashmir presented to us with gradually progressive quadriparesis of 15 years’ duration. She had onset of weakness in the legs followed by involvement of upper limbs several years later. Her elder sister suffered from similar problem and was wheelchair bound. There was no history of consanguinity. She was seen elsewhere, diagnosed as chronic inflammatory demyelinating neuropathy and myeloradiculopathy, and received steroids without any improvement. On examination, the topography of weakness was similar to previous patient with total sparing of quadriceps [Table 1], severe weakness of finger flexors compared to finger extensors, and severe weakness of shoulder girdle muscles with relative sparing of biceps (MRC Grade 3) and triceps (MRC Grade 4). Creatine kinase and nerve conduction studies were normal. EMG revealed findings consistent with myopathy. A possibility of HIBM was considered and she underwent triceps muscle biopsy which revealed features consistent with HIBM [Figure 2]. She was counseled about the nature of the disease and was given physiotherapy and rehabilitation exercises. Her sister was not examined personally due to technical problems of travel and transport.
Figure 2.
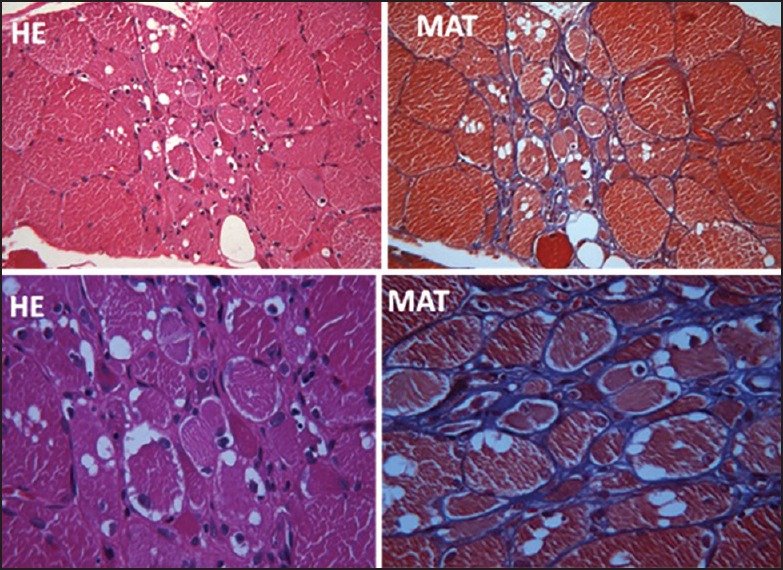
Low and high magnification of transversely cut skeletal muscle tissue showing vacuoles in left triceps muscle (second family)
Patient 6
This young gentleman (22 years) presented to us with gradually progressive difficulty in walking of 2 years’ duration, without any sensory/bladder/bowel problems. Family history was noncontributory. He had severe weakness of foot dorsiflexion (Grade 1), while power was Grade 3 + in plantar flexors of foot, flexors of thigh, hamstring, and in adductors of thigh [Table 1]. Power was grade 3 in extensors and abductors of thigh. Quadriceps was normal. There was mild weakness of neck flexors and shoulder girdle muscles. Power in rest of the muscles was normal. Deep tendon reflexes were sluggish. Sensory examination was normal. Creatine kinase and nerve conduction studies were normal, while EMG revealed evidence of myopathy. Muscle biopsy done from hamstring muscle on the left side revealed evidence of HIBM. He was counseled about the disease and given physiotherapy.
Patient 7
This young lady (26 years) presented to us with gradually progressive difficulty in walking of 8 years’ duration, without any sensory/bladder/bowel problems. Family history was negative. On examination, power was Grade 1 in foot dorsiflexors and Grade 2 in all other muscles of lower limbs except quadriceps which were normal [Table 1]. She had mild weakness of neck flexors. There was severe weakness of shoulder girdle muscles and long finger flexors. Deep tendon reflexes were sluggish. Sensory examination was normal. EMG revealed evidence of myopathy. Muscle biopsy done from left bicep muscle revealed evidence of HIBM. She was counseled about the disease and given physiotherapy.
Discussion
HIBMs include several autosomal recessive and dominant syndromes characterized by progressive course and characteristics muscle histopathology. The commonest form is the quadriceps sparing HIBM also known as h-IBM, IBM2, Nonaka myopathy, GNE myopathy, or distal myopathy with rimmed vacuoles (DMRV). It was first described in Jews of Iranian origin.[1,2] HIBM results from mutations in UDP-N-acetylglucosamine2-epimerase/N-acetylmannosamine kinase (GNE) gene on chromosome 9p13-p12, that codes for a bifunctional 722 amino acid enzyme (UDP-GlcNac epimerase/ManNac Kinase; GNE/MNK), which catalyzes a rate-limiting step during biosynthesis of sialic acid (5-N-acetylneuraminic acid), found on glycoconjugates as terminal sugar.[3,4]
GNE/MNK is a soluble protein, localizing to the cytoplasm, the Golgi region, and the nucleus. The N-terminal portion of GNE/MNK (UDP-GlyNAc 2-epimerase) catalyzes epimerization of UDP-GlyNAc to ManNAc with release of UDP. The C-terminal portion (ManNAc kinase) phosphorylates ManNAc to ManNAc-6-P and phosphenol pyruvate. ManNAc-6-P is then further condensed to sialic acid, active form of which, cytidine monophosphate (CMP)-sialic acid acts as a substrate for sialylation of glycoconjugates in Golgi complex by sialyltransferases. It is believed that decreased production of sialic acid due to mutations in GNE gene results in muscle degeneration in HIBM. This degeneration occurs rather slowly as is evidenced by the facts that clinical symptoms of HIBM usually start in adult life and muscles biopsied from patients with HIBM do show residual GNE/MNK enzyme activity. Though there is experimental evidence in support of above hypothesis, the exact cellular mechanisms resulting in muscle degeneration are still not known.[5,6,7] Thus, in HIBM, specific protein such as alpha dystroglycan may be inadequately glycosialylated resulting in degeneration of muscle.
HIBM has been described from all over the world including Indian, Chinese, Thai, Japanese, and African descent. The clinical course of HIBM is characterized by progressive proximal and distal weakness and wasting of both upper and lower limbs usually beginning after 20 years of age.[7] The typical clinical feature which aids in diagnosis is sparing of quadriceps. Foot dorsiflexors are involved early followed by forearm flexors, girdle, and axial muscles. In our cohort we found weakness of long finger flexors and wrist flexors more than finger and wrist extensors. In our view, this is also a unique feature of HIBM and helps in clinical localization even more precisely. Ocular, pharyngeal, and respiratory muscles are spared. Though muscles of shoulder girdle are affected severely; biceps, triceps, and to a lesser extent deltoid are involved to a lesser degree. The progressive weakness eventually forces to patient to be wheelchair bound usually 2-3 decades after the onset. The creatine kinase levels are normal or mildly elevated and nerve conduction studies are normal. Diagnosis is based on clinical findings as well as histopathology of a muscle biopsy.[7,8,9,10,11,12] The unique histological features include the presence of red rimmed vacuoles on Gomori's trichrome stain, small fibers in groups, and occasional amyloid deposits best seen with Congo-red stain. The vacuoles contain concentric membranous bodies and typical inclusion bodies in the cytoplasm and nucleus. These inclusions are composed of double twisted filaments with a diameter of 15-18 nm. The vacuoles are thought to represent autophagic vacuoles as suggested by high acid phosphatase activity, reactivity with lysosomal markers, and presence of multilammelar bodies on electron microscopy. In contrast to the sporadic inclusion body myositis, HIBM lacks inflammatory infiltrates.[1,10,13,14,15]
Regarding management, no therapies are yet available. Dietary treatment include increased intake of magnesium (a cofactor for GNE/MNK enzyme); reduced consumption of alcohol (promotes hydrolysis of sialoconjugates); and avoidance of excessive zinc, selenium, and copper. Intravenous immunoglobulin (IVIg; 8 μmol of sialic acid/g) has been tried with an aim to administer sialic acids. However, the results are disappointing. An alternative approach has been administration of sialic acid, ManNAc, as it enters into the sialic acid biosynthetic pathway distal to the rate-limiting step. Though this approach has been found useful in experimental animals, human trials are awaited. Gene therapy aimed at delivering healthy GNE gene into the patient's cells and tissue is still in the initial stages.[7,16,17,18]
To the best of our knowledge, this is the first report of occurrence of HIBM in Punjabi Sikh and Kashmiri Muslim ethnicity, though there are other large series described from Southern India.[19,20] These studies as well as our cases illustrate the clinical difficulties encountered while dealing with HIBM. Most of the times, the diagnosis is not made correctly owing to lack of awareness about the disease resulting in unnecessary diagnostic and therapeutic interventions. It is therefore possible that this disease may actually be much more common. In addition, five out of seven of our patients received steroids which are associated with unacceptable side effects. This further stresses the need to keep this rare disorder in mind so as to avoid unnecessary diagnostic and therapeutic errors.
To conclude HIBM is a frequently misdiagnosed myopathy despite the presence of unique clinical features. Awareness of this disease and careful examination of patients that will go a long way is rapid diagnosis of this rare disorder.
Acknowledgement
We are thankful to department of photography for their kind help in photography of the patients.
Footnotes
Source of Support: Nil
Conflicts of Interest: None declared.
References
- 1.Argov Z, Yarom R. “Rimmed vacuole myopathy” sparing the quadriceps. A unique disorder in Iranian Jews. J Neurol Sci. 1984;64:33–43. doi: 10.1016/0022-510x(84)90053-4. [DOI] [PubMed] [Google Scholar]
- 2.Argov Z, Eisenberg I, Grabov-Nardini G, Sadeh M, Wirguin I, Soffer D, et al. Hereditary inclusion body myopathy: The Middle Eastern genetic cluster. Neurology. 2003;60:1519–23. doi: 10.1212/01.wnl.0000061617.71839.42. [DOI] [PubMed] [Google Scholar]
- 3.Eisenberg I, Avidan N, Potikha T, Hochner H, Chen M, Olender T, et al. The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat Genet. 2001;29:83–7. doi: 10.1038/ng718. [DOI] [PubMed] [Google Scholar]
- 4.Varki A. Sialic acids as ligands in recognition phenomena. FASEB J. 1997;11:248–55. doi: 10.1096/fasebj.11.4.9068613. [DOI] [PubMed] [Google Scholar]
- 5.Krause S, Hinderlich S, Amsili S, Horstkorte R, Wiendl H, Agrov Z, et al. Localization of UDP-GlcNAc 2-epimerase/ManAc Kinase (GNE) in the Golgi complex and the nucleus of mammalian cells. Exp Cell Res. 2005;304:365–79. doi: 10.1016/j.yexcr.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 6.Effertz K, Hinderlich S, Reuter W. Selective loss of either the epimerase or kinase activity of UDP-N-acetylglucosamine 2-epimerase/N-acetlymannosamine kinase due to site directed mutagenesis based on sequence alignments. J Biol Chem. 1999;274:28771–8. doi: 10.1074/jbc.274.40.28771. [DOI] [PubMed] [Google Scholar]
- 7.Huizing M, Krasnewich DM. Hereditary inclusion body myopathy: A decade of progress. Biochim Biophys Acta. 2009;1792:881–7. doi: 10.1016/j.bbadis.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mastaglia FL, Lamont PJ, Laing NG. Distal myopathies. Curr Opin Neurol. 2005;18:504–10. doi: 10.1097/01.wco.0000175936.23945.b6. [DOI] [PubMed] [Google Scholar]
- 9.Illa I. Distal myopathies. J Neurol. 2000;247:169–74. doi: 10.1007/s004150050557. [DOI] [PubMed] [Google Scholar]
- 10.Nonaka I, Noguchi S, Nishino I. Distal myopathy with rimmed vacuoles and hereditary inclusion body myopathy. Curr Neurol Neurosci Rep. 2005;5:61–5. doi: 10.1007/s11910-005-0025-0. [DOI] [PubMed] [Google Scholar]
- 11.Broccolini A, Gidaro T, Morosetti R, Sancricca C, Mirabella M. Hereditary inclusion-body myopathy with sparing of the quadriceps: The many tiles of an incomplete puzzle. Acta Myol. 2011;30:91–5. [PMC free article] [PubMed] [Google Scholar]
- 12.Simmons Z, Towfighi J. Sporadic inclusion body myositis and hereditary inclusion body myopathy. J Clin Neuromuscul Dis. 2002;3:122–32. doi: 10.1097/00131402-200203000-00005. [DOI] [PubMed] [Google Scholar]
- 13.Griggs RC, Askanas V, DiMauro S, Engel A, Karpati G, Mendell JR, et al. Inclusion body myositis and myopathies. Ann Neurol. 1995;38:705–13. doi: 10.1002/ana.410380504. [DOI] [PubMed] [Google Scholar]
- 14.Massa R, Weller B, Karpati G, Shoubridge E, Carpenter S. Familial inclusion body myositis among Kurdish-Iranian Jews. Arch Neurol. 1991;48:519–22. doi: 10.1001/archneur.1991.00530170083024. [DOI] [PubMed] [Google Scholar]
- 15.Mitrani-Rosenbaum S, Argov Z, Blumenfeld A, Seidman CE, Seidman JG. Hereditary inclusion body myopathy maps to chromosome 9p1-q1. Hum Mol Genet. 1996;5:159–63. doi: 10.1093/hmg/5.1.159. [DOI] [PubMed] [Google Scholar]
- 16.Darvish D. Magnesium may help patients with recessive hereditary inclusion body myopathy, a pathological review. Med Hypotheses. 2003;60:94–101. doi: 10.1016/s0306-9877(02)00339-0. [DOI] [PubMed] [Google Scholar]
- 17.Spark S, Rakocevic G, Joe G, Manoli I, Shrader J, Hoarris-Love M, et al. Intravenous immune globulin in hereditary inclusion body myopathy: A pilot study. BMC Neurol. 2007;7:3. doi: 10.1186/1471-2377-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Charter NW, Mahal LK, Koshland DE, Jr, Bertozzi CR. Biosynthetic incorporation of unnatural sialic acids into polysialic acids on neural cells. Glycobiology. 2000;10:1049–56. doi: 10.1093/glycob/10.10.1049. [DOI] [PubMed] [Google Scholar]
- 19.Nalini A, Gayathri N, Dawn R. Distal myopathy with rimmed vacuoles: Report on clinical characteristics in 23 cases. Neurol India. 2010;58:235–41. doi: 10.4103/0028-3886.63804. [DOI] [PubMed] [Google Scholar]
- 20.Nalini A, Gayathri N, Nishino I, Hayashi YK. GNE myopathy in India. Neurol India. 2013;61:371–4. doi: 10.4103/0028-3886.117609. [DOI] [PubMed] [Google Scholar]