

Abstract
Tangier disease is an autosomal recessive disorder characterized by an abnormal accumulation of cholesterol esters in various organs secondary to adenotriphosphate binding cassette transporter A-1 (ABCA-1) transporter deficiency and disrupted reverse cholesterol transport. It causes neuropathy in half of the affected individuals. We present the clinical, electrophysiological, and histopathological findings in a middle aged gentleman of Tangier disease who was initially misdiagnosed leprosy and treated with antileprosy drugs. The presence of a demyelinating neuropathy on electrophysiology in a patient with predominant upper limb involvement and facial diplegia should raise the suspicion of Tangier disease. The characteristic lipid profile of Tangier disease was noted in this patient viz. extremely low high density lipoprotein (HDL), elevated triglyceride (TG), and reduced apolipoprotein A1. Estimation of serum lipids should form a part of routine evaluation in order to avoid misdiagnosis.
Keywords: Lipid profile, neuropathy, Tangier disease
Introduction
Neurological disorders arising from disordered lipid metabolism/transport are distinctly rare and include Tangier disease, abetalipoproteinemia, Refsum disease, Apo A1 related amyloidosis, and cerebrotendinous xanthomatosis. Tangier disease is an autosomal recessive disorder characterized by an abnormal accumulation of cholesterol esters in various organs secondary to adenotriphosphate binding cassette transporter A-1 (ABCA-1) transporter deficiency and disrupted reverse cholesterol transport.[1,2] First described by Fredrickson in 1960, in two siblings from Tangier Island off the Chesapeake Bay, it is known by several names including alphalipoprotein neuropathy, alpha high density lipoprotein (HDL) deficiency disease, analphalipoproteinemia, cholesterol thesaurismosis, familial HDL deficiency disease, familial hypoalphalipoproteinemia, HDL lipoprotein deficiency disease, familial HDL lipoprotein deficiency disease, Tangier disease neuropathy, and Tangier hereditary neuropathy. These names describe the characteristic biochemical abnormality in Tangier disease namely, markedly reduced to absent HDL, reduced low density lipoprotein (LDL), low or absent apolipoprotein A1, mild-moderate elevation of triglycerides (TG), and occasionally reduced total cholesterol. Neuropathy occurs in 50% of the patients with Tangier disease; there are two clinical phenotypes, namely, adult onset pseudosyringomyelia and relapsing-remitting mononeuritis multiplex.[2,3]
In this report, we highlight the clinical, electrophysiological, and histopathological findings in a middle-aged man with Tangier disease.
Case Report
A 43-year-old gentleman presented with incomplete eye closure of 4 years duration, associated with labial dysarthria. Over the last 3 years, he had noticed progressive difficulty in gripping objects with either hand, with thinning of forearms, and painless burns over the distal upper extremities. He then developed numbness of the head and face, trunk, and upper limbs since a year. For these symptoms, he was prescribed presumptive anti-Hansen's treatment at another hospital. There was no history of visual or hearing impairment, positive sensory symptoms, or symptoms suggesting dysautonomia. There was no family history of neuropathic illness.
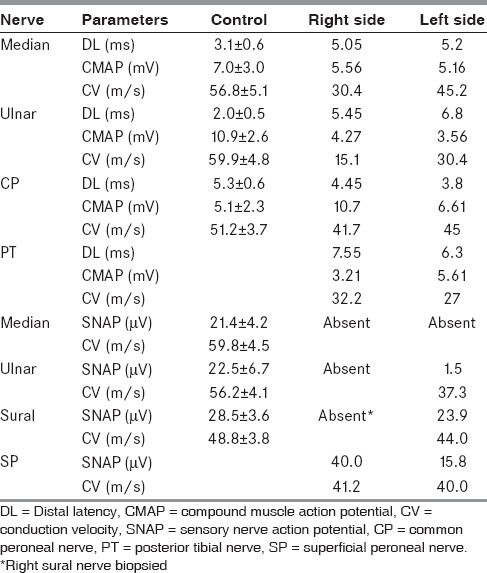
Examination showed healed thermal burns over bilateral forearms, bifacial weakness, distal hypotonia, with wasting and weakness of distal upper limbs. There was diminished touch and pin-prick over the scalp, face, trunk, upper limbs, and proximal lower limbs up to mid-thigh reminiscent of a syrinx. Kinesthetic sensations and cerebellar system were intact. Routine investigations including hemogram, erythrocyte sedimentation rate, and hepatic and renal function tests were normal. Autoantibody profile (Euroline®, immunoblot technique), serum protein electrophoresis, serum angiotensin converting enzyme (ACE) levels (20.1 U/l; ref: 20-70 U/l), human immunodeficiency virus (HIV) antibody test, abdominal ultrasound, audiometry, and other conventional autonomic function tests were found to be within normal limits. Urine examination for porphobilinogen and Bence-Jones protein yielded negative results. Lumbar cerebrospinal fluid (CSF) was normal except for mildly elevated protein (cell count: 0/mm3; protein: 58 mg/dl, ref: 20-40 mg/dl; and glucose: 61 mg/dl, ref: 40-60 mg/dl). Investigations at our center showed electrophysiological evidence of demyelinating polyneuropathy [Table 1]. Sympathetic skin responses were absent from the upper and lower limbs.
Table 1.
Summary of electrophysiological findings
The serum lipid profile was abnormal with extremely low levels of HDL (5 mg/dl; ref: 35-65 mg/dl) and elevated TG (185 mg/dl; ref: 50-150 mg/dl); and normal levels of total cholesterol (149 mg/dl; ref: 110-220 mg/dl), very low density lipoprotein (VLDL) (37 mg/dl; ref: 10-40 mg/dl), and LDL (110 mg/dl; ref: 60-160 mg/dl). Serum apolipoprotein A1 was significantly reduced (<5.38 mg/dl; ref: 110-205 mg/dl). His sural nerve had been biopsied elsewhere, this was retrieved and reviewed. It showed mild depletion of small diameter myelinated fibers in small pockets in the periphery of each fascicle. In addition, there was a striking vacuolation of the Schwann cell cytoplasm indenting the nucleus [Figure 1]. Skin biopsy revealed numerous bloated, finely vacuolated fibroblasts throughout the entire thickness of the skin, surrounding the dermal capillaries, nerve twigs, and cutaneous adnexal structures. The vacuolated granular cytoplasm was seen indenting the nucleus [Figure 1]. These vacuoles stained with Oil Red O, confirming the presence of lipid rich material [Figure 1]. Slit lamp examination for corneal fat deposits, Doppler of the neck vessels, and echocardiography revealed normal findings.
Figure 1.
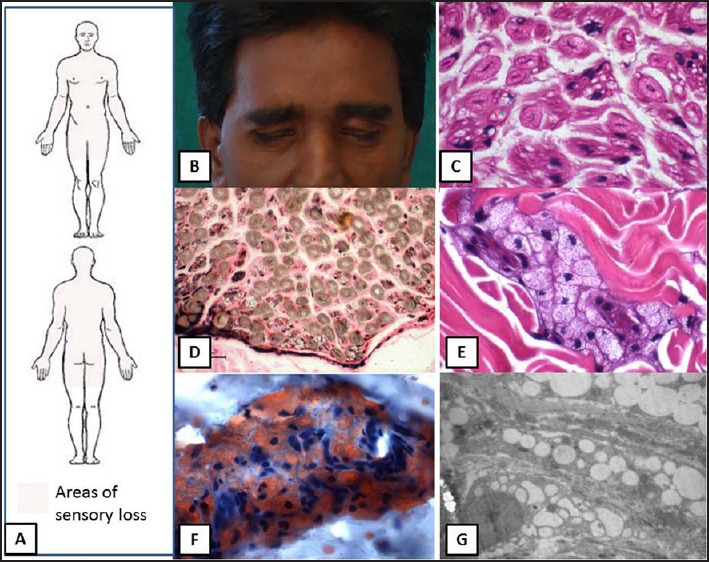
Schematic representation of the pattern of sensory loss to pain and temperature (a) and clinical photograph shows bifacial weakness. (b) sural nerve biopsy demonstrates vacuolation of the Schwann cells within the Schwann cell subunits. (c) and mild fiber loss (d) Skin biopsy reveals accumulation of bloated, finely vacuolated foam cells with granular cytoplasm around cutaneous adnexa. (e) the vacuoles contain Oil Red O positive neutral lipidic material. (f) and electron micrograph reveals numerous lipid vacuoles in the cytoplasm of fibroblasts (g)
Discussion
Tangier disease is a rare autosomal recessively inherited disorder that causes neuropathy in 50% of the cases. Other clinical manifestations include hepatosplenomegaly, ischemic heart disease or stroke, anemia, thrombocytopenia, corneal opacities hypocholesterolemia, or asymptomatic detection following familial screening.[2] The available literature is sparse; less than 100 cases are reported worldwide since the original description half a century ago. The key findings in this middle-aged male patient included
Bifacial weakness,
Distal bibrachial weakness, and
An unusual pattern of dissociated sensory loss involving the face, trunk, upper limbs, and proximal thighs.
The differential diagnoses in a patient with predominant bifacial weakness could be acute and chronic inflammatory demyelinating neuropathies, Lyme's disease, sarcoidosis, HIV infection, porphyria, Hansen's disease, Gelsolin familial amyloid neuropathy, Tangier's disease, and vasculitis such as Sjogren syndrome. The combination of facial diplegia and predominant involvement of upper limbs is the characteristic clinical phenotype; this coupled with the typical lipid profile and nerve biopsy observations clinched the diagnosis of Tangier disease. Yet, several cases are misdiagnosed as leprosy or other immune mediated neuropathies, including the above patient.
The principal neurological manifestation of Tangier disease is neuropathy which may be the presenting feature in nearly one-third of the patients.[3] This may manifest as a mono- or polyneuropathy, or a syringomyelia-like neuropathy; the clinical course may be relapsing-remitting in some and may in fact mimic an immune mediated neuropathy both clinically and electrophysiologically.[4] Because of the unusual trophic distribution with predominant involvement of the upper limbs and regional endemicity, the current patient was initially misdiagnosed leprosy, notwithstanding the fact that sural nerve biopsy did not show evidence of leprosy. A similar patient has already been reported by the authors.[5] A demyelinating electrophysiology is recognized in Tangier disease.[4] The current patient had clinical signs and symptoms restricted to the face, trunk, and upper limbs; although electrophysiological examination revealed a more widespread involvement. The presence of demyelinating electrophysiology correlates with the histopathological finding of lipid accumulation in the Schwann cells of peripheral nerves. The lack of ABCA-1 transporter responsible for transport of HDL cholesterol from various cells including the Schwann cells to plasma results in abnormal lipid accumulation.[6] In addition, it has been hypothesized that focal nerve ischemia secondary to lipid accumulation also disrupts myelin and contributes to demyelination, slowed conduction velocity and conduction blocks.[7]
Apart from the Schwann cells, abnormal lipids also accumulate in several tissues including tonsils, liver, spleen, lymph nodes, thymus, gastrointestinal mucosa, bone marrow, and fibroblasts of the skin in Tangier disease. Cutaneous deposition of cholesterol esters has been demonstrated in clinically uninvolved skin in patients with Tangier disease.[8] Thus, skin biopsy is a valuable tool for diagnosis; it may also serve as a model for further research into the pathogenesis, development of newer drugs, and follow-up of patients with this rare disorder.[8] Patients presenting with clinical features of polyneuropathy or mononeuritis multiplex are likely to undergo skin biopsy as a part of diagnostic evaluation for vasculitis, leprosy, sarcoidosis, and amyloidosis. The presence of lipid accumulation in these peripheral tissues should be looked for in addition to the above differential diagnosis. Subsequently, a simple biochemical test in the form of serum lipid profile clinches the diagnosis.
A close differential diagnosis of neuropathy associated with reduced HDL and Apo-A1 related amyloidosis, can be distinguished clinically from Tangier disease by the presence of small fiber involvement, autonomic dysfunction, renal failure, and cardiomyopathy.[9] The management of Tangier disease is essentially limited to dietary modifications with low fat content, prevention of injuries and prevention and management of cardiac complications. Newer experimental synthetic molecules including fatty acid bile acid conjugates (FABACS) such as aramchol are designed to increase the reverse cholesterol transport; but current evidence suggests that they do not overcome the critical step requiring ABCD-1 activity in reverse cholesterol transport.[10] Cholesterol ester transfer protein (CETP) inhibitors like dalctrapid and reconstituted HDL may be considered pending the development of more effective therapies.[2]
In conclusion, the presence of a demyelinating electrophysiology in a patient with predominant upper limb involvement and facial diplegia should raise the suspicion of Tangier's disease. Simple biochemical tests in the form of estimation of serum lipids should form a part of routine evaluation in these patients in order to avoid misdiagnosis. This will in turn avoid misdiagnosis and institution of inappropriate therapy.
Footnotes
Source of Support: Nil
Conflicts of Interest: None declared.
References
- 1.Rust S, Roiser M, Funke H, Real J, Amoura Z, Piette JC, et al. Tangier disease is caused by mutations in the gene encoding ATP-binding cassette transporter 1. Nat Genet. 1999;22:352–5. doi: 10.1038/11921. [DOI] [PubMed] [Google Scholar]
- 2.Puntoni M, Sbrana F, Bigazzi F, Sampietro T. Tangier disease: Epidemiology, pathophysiology, and management. Am J Cardiovasc Drugs. 2012;12:303–11. doi: 10.2165/11634140-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 3.Assman G, von Eckardstein A, Brewer HB. Familial analphalipoproteinemia: Tangier disease. In: Scriver CR, Beauder AL, Sly WS, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill Co; 2001. pp. 2937–53. [Google Scholar]
- 4.Theaudin M, Couvert P, Fournier E, Bouige D, Bruckert E, Perrotte P, et al. Lewis-Sumner syndrome and Tangier disease. Arch Neurol. 2008;65:968–70. doi: 10.1001/archneur.65.7.968. [DOI] [PubMed] [Google Scholar]
- 5.Sinha S, Mahadevan A, Lokesh L, Ashraf V, Chandrasekhar Sagar BK, Taly AB, et al. Tangier disease- a diagnostic challenge in countries endemic for leprosy. J Neurol Neurosurg Psychiatry. 2004;75:301–4. doi: 10.1136/jnnp.2003.022277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cai Z, Blumbergs PC, Cash K, Rice PJ, Manavis J, Swift J, et al. Paranodal pathology in Tangier disease with remitting-relapsing multifocal neuropathy. J Clin Neurosci. 2006;13:492–7. doi: 10.1016/j.jocn.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 7.Marbini A, Gemignani F, Ferrarini G, Maccari S, Lucci B, Bragaglia MM, et al. Tangier disease: A case with sensorimotor distal polyneuropathy and lipid accumulation in striated muscle and vasa nervorum. Acta Neuropathol. 1985;67:121–7. doi: 10.1007/BF00688132. [DOI] [PubMed] [Google Scholar]
- 8.Waldorf DS, Levy RI, Fredrickson DS. Cutaneous cholesterol ester deposition in Tangier disease. Arch Dermatol. 1967;95:161–5. [PubMed] [Google Scholar]
- 9.Joy T, Wang J, Hahn A, Hegele RA. APOA1 related amyloidosis: A case report and literature review. Clin Biochem. 2003;36:641–5. doi: 10.1016/s0009-9120(03)00110-3. [DOI] [PubMed] [Google Scholar]
- 10.Goldiner I, van der Velde AE, Vandenberghe KE, van Wijland MA, Halpern Z, Gilat T, et al. ABCA1-dependent but apoA-I-independent cholesterol efflux mediated by fatty acid-bile acid conjugates (FABACs) Biochem J. 2006;396:529–36. doi: 10.1042/BJ20051694. [DOI] [PMC free article] [PubMed] [Google Scholar]