

Abstract
Background
Brainstem gliomas are rare in adults and overall have superior survival outcomes compared to pediatric brainstem gliomas.
Patients and methods
We conducted a retrospective data and tissue analysis of all adult patients (≥18 years old) with World Health Organization (WHO) Grade II, III, and IV brainstem gliomas in the University of Texas MD Anderson Cancer Center institutional database from 1990 to 2012.
Results
We identified 143 cases in adults ages 18 and over. There were 28 glioblastomas, 43 anaplastic astrocytomas, 15 diffuse astrocytomas, and 11 gliomas not otherwise specified, and in 46 cases the diagnosis was made radiographically. 128 (89.5%) cases were classified radiographically as diffuse and of the focal tumors, 9 of the 15 were WHO Grade III or IV tumors. Increasing tumor grade and contrast enhancement were associated with significantly reduced overall survival. The median overall survival for the entire cohort was 32.1 months similar to previously published studies. Two of 25 grade II and III tumors, and 1 of 17 glioblastomas had IDH1 mutations on immunohistochemical testing. Nine cases had sufficient tissue for mutation profiling, 1 case had a BRAF V600E mutation and 2 had 2 PIK3CA mutations.
Conclusions
Survival outcomes for adult WHO Grade II to IV brainstem gliomas were similar to supratentorial IDH1 wild-type tumors of similar grade and histology. Potentially actionable mutations can be identified from small biopsy samples in a subset of adult brainstem gliomas.
Keywords: Brainstem glioma, Diffuse intrinsic pontine glioma, Isocitrate dehydrogenase mutation, Astrocytoma, Glioblastoma, Oligodendroglioma
1. Introduction
According the latest Central Brain Tumor Registry of the United States results, the brainstem (BS) is the location for 1.6% of all primary central nervous system (CNS) neoplasms, 3.6% of malignant CNS neoplasms, and 4.2% of CNS gliomas [1]. Among primary glial neoplasms, a BS localization accounts for 20% or more of gliomas in pediatric patients while accounting for less than 2% of gliomas in adults [2,3]. The World Health Organization (WHO) Grade II to IV infiltrating gliomas including astrocytic, oligodendroglial, mixed gliomas and glioblastomas and the grade I circumscribed, glial neoplasms, such as pilocytic astrocytomas, all may occur in the brainstem and have variable radiographic appearances. Infiltrating brainstem gliomas, classically located in the pons and referred to as diffuse infiltrating pontine gliomas (DIPGs), have a uniformly poor prognosis in pediatric patients [4]. Despite similar radiographic presentations and histologic characteristics, multiple studies to date have shown that BS located gliomas in adults have better outcomes compared to pediatric DIPGs [5–7]. The average survival in pediatric DIPGs is uniform across studies with an average survival of 10–12 months with survival beyond 3 years being a rare event [2,4]; adult studies of infiltrating, BS gliomas suggest an average survival of 30–40 months, although survival decreases with age [5–11].
Management of brainstem gliomas was often based on MRI characteristics alone [12], and due to the overall rarity of these tumors in adults and limited available tissue for study, little is known about the molecular events related to tumorigenesis and tumor progression. In this study, we reviewed the clinical, imaging, and pathologic features of brainstem gliomas in adult patients at our institution. The purpose of this study was to correlate clinical, radiographic, pathologic, and molecular data in adult, WHO Grade II to IV, infiltrating BS gliomas.
1.1. Patients and methods
We conducted a retrospective data and tissue analysis of all adult patients (≥18 years old) with WHO Grade II, III, and IV BS gliomas, or suspected BS gliomas radiographically, in the University of Texas MD Anderson Cancer Center institutional database from 1990 to 2012 under a protocol with waiver of consent approved by the institutional review board. Patients had either a biopsy or surgical resection with a pathological diagnosis of the following WHO Grade II–IV neoplasms: diffuse astrocytoma, anaplastic astrocytoma, oligodendroglioma or oligoastrocytoma (Grade II or III), glioblastoma, or gliosarcoma. We included cases where no biopsy was performed or if biopsy was inconclusive (glioma not otherwise specified) but a diagnosis of a presumed, WHO Grade II, III, or IV BS glioma was made based on radiographic features. We collected demographic, treatment, and survival data from the database. Additional clinical, neuropathologic, and neuroimaging data were obtained from the institutional electronic medical records.
For inclusion in this study, the majority of the tumor, >50%, had to involve the brainstem at diagnosis, thus excluding primarily spinal cord, cerebellar, or supratentorial tumors with extension to the brainstem. Multifocal tumors with both supratentorial and brainstem components were also excluded. Radiographic classification was based on a classification scheme similar to previously published schemes [12]. Brainstem gliomas were considered focal if the tumor had sharply defined margins and occupied less than one half of the involved BS segment. The tumor could extend to a single adjacent rostral or caudal segment (5 mm on standard MRI imaging) or 1 segment both rostrally and caudally, so long as it maintained sharp demarcation. Diffuse tumors were one or all of the following: 1) poorly demarcated from the involved brainstem segment, 2) involved more than one-half of the involved brainstem segment and 3) extended beyond one rostral or caudal segment. We included but did not classify tumors as exophytic or non-exophytic due to the difficulties in differentiating exophytic tumors, such as in the medulla, and our exclusion of pilocytic astrocytomas and other rare glial and non-glial neoplasms. If imaging was not available classification was based on the neuroradiology reports and the available clinic notes in the electronic medical record.
Adjuvant radiotherapy (RT) was defined as RT given as part of the initial treatment following surgical resection within 6 months of surgery. Extent of resection was categorized as gross total resection (GTR), subtotal resection (STR), stereotactic biopsy, or biopsy not otherwise specified as recorded in the institutional database. Extent of resection was based mainly on the neurosurgeon’s or neuro-oncologist’s assessment and post-operative imaging when available.
Overall survival (OS) was defined as the period from date of diagnosis, defined in most cases by date of first MRI or CT scan showing a brainstem mass, until death. OS was calculated using the Kaplan Meier method. The log-rank test was used to determine significance of differences in OS among analyzed subgroups. Comparisons of proportions were done using Fisher’s exact test. Statistical calculations were performed using GraphPad Prism version 6.00 for Mac (GraphPad Software, CA).
Slides from cases with available formalin-fixed paraffin-embedded tissue were retrieved from the pathology archives and reviewed to confirm diagnosis and to choose a representative block for molecular studies. Cases with available tissue were tested using IDH1 (R132H) immunohistochemistry (IHC). In cases with sufficient tissue, tumor genomic DNA was extracted from deparaffinized tissue scrolls using magnetic beads (ChargeSwitch, Invitrogen, CA) and submitted to mass spectrometry array mutation profiling (MassARRAY system, Sequenom, CA) of 132 hotspot codons in 39 cancer-related genes (AKT1, AKT2, AKT3, ALK, BCOR, BRAF, CDK4, CSMD1, CTNNB1, EGFR, EPHA3, FBXO4, FBXW7, FGFR1, FGFR2, FGFR3, FOXL2, GNA11, GNAQ, GNAS, GRM3, IDH1, IDH2, KIT, KRAS, MAP2K2, MET, MGA, NRAS, PDGFRA, PIK3CA, PPP2R1A, RAF1, RET, RPL22, SFRS9, SMO, SRC, and TGM2). Sequenom™ testing is a recently developed technology with very high sensitivity and a specificity approaching 100% [22]. Validation of this clinical assay on 255 cancer cases at our institution yielded a 100% concordance with Sanger sequencing approach (unpublished observation).
2. Results
We identified 143 adult patients diagnosed with infiltrating BS gliomas. Demographic, clinical, pathologic, and radiographic features are summarized in Table 1. There were 86 males and 57 females. The median age was 36. The median Karnofsky performance score (KPS) at diagnosis was 90. Two patients had a diagnosis of neurofibromatosis type 1 (NF1). Seventeen tumors were located primarily in the midbrain, 90 in the pons, and 36 in the medulla.
Table 1.
Demographics, brainstem location, and pathology.
| Total (N) | 143 |
|---|---|
| Males, N (%) | 86 (60) |
| Females | 57 (40) |
| Median age, yrs | 36 |
| 18–21, N (%) | 11 (8) |
| 22–39 | 75 (52) |
| 40–59 | 46 (32) |
| 60 and over | 11 (8) |
| Median KPS at Dxa | 90 |
| KPS 100, N (%) | 22 (15) |
| 90 | 59 (41) |
| 80 | 31 (22) |
| 70 | 16 (11) |
| 60 | 5 (4) |
| 50 or below | 7 (5) |
| UNK in 3 cases (2%)a | |
| Brainstem location N (%): | |
| Midbrain | 17 (12) |
| Pons | 90 (63) |
| Medulla | 36 (25) |
| Pathologic diagnosesa: | |
| Glioblastoma | 28 (20) |
| Anaplastic astrocytoma | 43 (30) |
| Diffuse astrocytomas | 15 (10) |
| Glioma NOS | 11 (8) |
| No biopsy | 43 (30) |
3 cases (2%) biopsy inconclusive.
Sixty cases were stereotactically biopsied, 26 cases were classified as having a biopsy not otherwise specified, and 14 a sub-total resection (STR). Pathologic diagnoses were as follows: 28 glioblastoma (GB), 43 anaplastic astrocytoma (AA), 15 diffuse astrocytoma (DA), 11 glioma not otherwise specified (glioma NOS, and 1 case with an oligoastrocytic morphology), in 3 cases biopsy was inconclusive and not diagnostic of neoplasia (but BS glioma was suspected radiographically). Forty-three cases (30%) were not biopsied and a diagnosis of a BS glioma was made radiographically. The median age of this subgroup was 30 years with the majority of patients (33 out of 43) being less than 40 years or age. Thirty-two of the tumors were located in the pons, 6 in the midbrain and 5 in the medulla. Twenty-seven were non-enhancing at diagnosis, 8 enhanced with contrast whereas these were data not available on the remaining 8 cases. Forty-one (95%) of these cases were classified radiographically as diffuse and 2 (5%) as focal. Thirty-eight of these cases received external beam radiotherapy at a median dose of 5400 Gy. In the remaining 5 cases it was unknown whether RT treatment was rendered, but RT was recommended near home in 4 of these cases.
Overall, 56 tumors enhanced with contrast at diagnosis (N = 18 AA, 21 GB, 4 DA, 8 no biopsy, 3 glioma NOS, 2 biopsy inconclusive), 58 were non-enhancing at diagnosis (N = 14 AA, 4 GB, 9 DA, 27 no biopsy, 3 glioma NOS, 1 biopsy inconclusive), and data on contrast enhancement was not available on 29 patients. Fifteen (10.5%) tumors were classified radiologically as focal and 128 (89.5%) as diffuse. Of the 15 focal tumors, there were 7 AAs, 2 GBs, and 4 gliomas NOS, and 2 cases were not biopsied. Eight of the 15 focal tumors enhanced with contrast at diagnosis and 2 were non-enhancing (data not available on 5 patients).
External beam radiotherapy was completed in 118 cases. The median dose of RT was 5400 cGy (range 3000 to 7200 cGy). In 3 cases RT was not completed and in 2 cases RT was not started due to rapid disease progression. In 20 cases it was not known if RT was performed, but in 10 of these cases RT was recommended as the primary treatment modality near the patient’s home.
Fifty-four patients were treated with cytotoxic chemotherapy in the adjuvant setting; of these cases, 27 received concurrent plus adjuvant chemotherapy with 25 of these patients receiving the Stupp regimen with concurrent plus adjuvant temozolomide (n = 12 GB, 4 AA, 3 DA, 2 glioma NOS, 4 no biopsy) [13]. Twenty-one patients received cytotoxic chemotherapy and 17 patients received bevacizumab at tumor recurrence. Of the 17 patients treated with bevacizumab, 14 had sufficient data available to calculate PFS; the median PFS was 2.0 months. 3 of 14 patients (21%) were progression free for 6 months or longer while treated with bevacizumab with the best outcome in one patient who was progression free for 9 months. 12 patients were treated on clinical trials at our institution: 8 at tumor recurrence, 3 patients with the trial agent given concurrent with RT, and 1 patient received the trial agent as adjuvant treatment after radiotherapy.
Survival outcomes are outlined in Table 2 and displayed in Fig. 1. The median OS was 32.1 months for all patients, and median progression free survival was 18.6 months overall. The median OS by WHO Grade and histology are as follows: 77.0 months for DA, 21.1 months for AA, and 14.8 months for GB (p = 0.0104, log-rank test for trend). Gliomas NOS had a median OS of 96.2 months and non-biopsied cases had a median OS of 40.0 months. Contrast enhancing tumors had a median OS of 15.3 months compared to non-enhancing tumors which had a median OS of 61.5 months (p = 0.0007, log-rank test). Gliomas NOS and non-biopsied cases are not included in the calculated survival outcomes of DA, AA, and GB, but are included in the calculation of OS for the entire cohort and in the OS calculation and comparison of contrast-enhancing versus non-enhancing tumors.
Table 2.
Survival outcomes for adult brainstem gliomas.
| Entire cohort, N = 143, median OS (months) | 32.1 |
|---|---|
| Median OS by WHO Grade and histology (months): | |
| Glioblastoma | 14.8* |
| Anaplastic astrocytoma | 21.1 |
| Diffuse astrocytoma | 77.1 |
| Glioma NOS | 96.2 |
| No biopsy | 40.0 |
| Median OS by imaging features (months): | |
| Enhancing | 15.3** |
| Non-enhancing | 61.5 |
| Median OS by age (months): | |
| 18–21 years*** | 25.9# |
| 22–39 | 34.7 |
| 40–59 | 34.3 |
| 60 and over | 14.2 |
| Median OS by anatomic site (months): | |
| Midbrain | 66.0+ |
| Pons | 25.3 |
| Medulla | 51.3 |
p = 0.0104, log-rank test for trend, comparing OS for DA, AA, and GB.
p = 0.0007.
p = 0.2015 log-rank test for trend.
p = 0.20 log-rank test for trend comparing all ages.
p = 0.6434 log-rank test for trend comparing all sites, p = 0.0816, log-rank test, comparing pontine to other tumors sites.
Fig. 1.
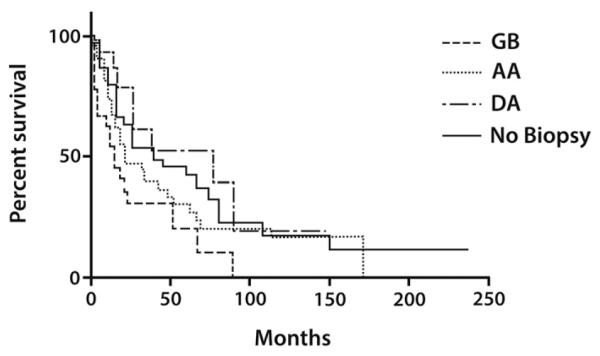
The median overall survival by WHO Grade and histology are as follows: 77.0 months for diffuse astrocytoma (DA), 21.1 months for anaplastic astrocytoma (AA), and 14.8 months for glioblastoma (GB), p = 0.0104, log-rank test for trend. In cases where the diagnosis was made radiographically, annotated as no biopsy on Fig. 1, the median OS was 40.0 months.
Survival by age group is as follows: 25.9 months for 18–21 years, 34.7 months for 22–39 years, 34.3 months for 40–59 years, and 14.2 months for patients 60 and over (p = 0.2015 log-rank test for trend). The difference in OS was non-significant when comparing patients 60 and over to all other age groups (p = 0.2865, log-rank test). The median age at tumor diagnosis by histology is as follows: DA 39.8 years, AA 35.8 years, GB 44.0 years, glioma NOS 37.1 years, and non-biopsied cases 30.3 years. Of 15 DA cases, 2 were diagnosed between ages 18 and 21 years, 5 between 22 and 39 years, 6 between ages 40 and 59 years, and 2 at ages 60 and over. Of 43 AA cases, 2 were diagnosed between ages 18 and 21 years, 24 between 22 and 39 years, 14 between ages 40 and 59 years, and 3 at ages 60 and over. Of 28 GB cases, 0 were diagnosed between ages 18 and 21 years, 10 between 22 and 39 years, 14 between ages 40 and 59 years, and 4 at ages 60 and over. Eighty-three percent of non-biopsied cases were diagnosed before age 40; 6 cases were diagnosed between 18 and 21 years and 27 between 22 and 39 years.
Midbrain tumors had a median OS of 66.0 months, pontine tumors 25.3 months, and medullary tumors 51.3 months (p = 0.2148 log-rank test for trend). There was a trend towards reduced OS in pontine tumors when compared to midbrain and medullary tumors (p = 0.0816, log-rank test). Twelve GBs received concurrent chemoradiotherapy and adjuvant temozolomide, the Stupp regimen, and 16 GBs did not receive the Stupp regimen. Median KPS was 90 and median age at diagnosis was 41.5 years in the patients treated with the Stupp regimen compared to a median KPS of 80 and median age of 47.6 years in those patients not treated with this regimen. All 12 cases treated with the Stupp regimen were diagnosed after 2005, and 10 of the 16 cases not treated with this regimen were diagnosed in the pre-temozolomide era before 2005. Glioblastomas which received the Stupp regimen had a median OS of 23.1 months compared to 4.0 months for those that did not (p = 0.0369, log-rank test).
Forty-three cases had IDH1 (R132H) IHC performed. The results from the grade II and III tumors in this cohort were published previously [14]. Forty cases were IDH1 wild-type and 3 cases had IDH1 R132H mutations. The IDH1 mutated cases included 1 DA, 1 AA, and 1 GB. Nine cases had sufficient tissue for Sequenom testing. Two cases had PIK3CA mutations and 1 case had a BRAF V600E mutation. Both patients with PIK3CA mutations (H1047R), 1 with GB and 1 AA, have had prolonged survival with both patients alive and stable at 40 and 164 months of follow-up respectively. The clinical details of the cases with the mutations specified above are summarized in Appendix 1. IDH1 wild-type tumors (N = 40) had a median OS of 21.4 months. Median OS for IDH1 wild-type tumors by pathologic diagnosis is as follows: GBs (N = 16) 12.4 months, AAs (N = 14) 21.4 months, and DAs (N = 9) 89.8 months (p = 0.0609, log-rank test for trend); there was 1 case of IDH1 wild-type glioma NOS. Two of the patients with an IDH1 mutation were stable at last follow-up (7 and 14 months from diagnosis); the third patient with an IDH1 mutation died at 12 months from diagnosis due to progressive tumor.
3. Discussion
This is the largest single institution series of adults with infiltrating BS gliomas reported to date, and similar to previous studies we found a male predominance, the pons as the most common anatomic site, and a diagnosis made radiographically in 30% of cases. The median OS of 32.1 months is similar to previous studies [5–7]. Increasing tumor grade and contrast enhancement were associated with significantly decreased OS; outcomes were inferior in older patients and pontine-localized tumors, although these results were not statistically significant[7,15,16]. Exclusion of pilocytic astrocytomas likely contributed to the lack of statistically significant differences in survival outcomes observed based on BS location and age, given the younger age, prolonged survival, and frequent brainstem localization within the medulla and midbrain of these tumors.
The subset of tumors diagnosed radiographically (30% of cases) were mainly diffusely, infiltrating, non-enhancing, pontine masses, and were diagnosed in patients 40 years and younger. The median OS of 40 months in these cases is similar to survival outcomes in non-biopsied patients from prior studies which report a range of survival outcomes from 38 months to 7.3 years [5,7,11]. In a study of 104 histologically confirmed adult BS gliomas the median OS was 18.8 months, a decreased OS relative to our cohort and other studies which included non-biopsied cases [15].
When feasible, biopsy is necessary in focal and enhancing BS masses given a radiologic differential diagnosis including grade I–IV gliomas, glioneuronal and ependymal neoplasms, and non-tumor entities such as inflammatory and demyelinating disorders [17]. We cannot exclude the possibility that a small proportion of the non-biopsied cases in our series were grade I, glioneuronal, or ependymal neoplasms, or non-tumor entities thus influencing survival outcomes. The 11 gliomas NOS had the longest OS time (96.2 months); although this subgroup is relatively small, the prolonged OS suggests that this group may represent a mixture of both pilocytic and diffuse astrocytomas. In our cohort, OS in the non-biopsied cases was intermediate between DAs and AAs suggesting that the majority of diffusely infiltrating, non-enhancing brainstem gliomas in young adults are usually either WHO Grade II or III astrocytomas. This would differ from pediatrics where most DIPGs are GBs or AAs, although survival does not tend to differ based on histology [4].
The majority of the tumors in our series were treated with RT at the highest tolerated brainstem dose at diagnosis. Chemotherapy was used mainly in the adjuvant setting in our study, with the most frequent regimen being the Stupp regimen, with both concurrent and adjuvant temozolomide [13]. The subset of GB patients treated after 2005 with the Stupp regimen had significantly improved OS outcomes compared to those GB patients who did not receive this regimen, 23.1 versus 4.0 months, although differences in age, KPS, and other selection biases not readily defined in a retrospective study may have contributed to this result. In a retrospective study of 15 adult patients, defined as low grade BS gliomas, treatment with temozolomide at the time of tumor progression resulted in radiographic responses in 40% of patients and median PFS and OS times of 9.5 and 14.4 months respectively [18]. In the subgroup of patients treated with bevacizumab for tumor recurrence in our study, only 21% of evaluable patients were progression free at 6 months. This is inferior to the PFS results seen in supratentorial, recurrent GBs where 40% or more of patients treated with bevacizumab are progression free at 6 months [19]. In a case series of 3 progressive adult BS gliomas, salvage treatment with bevacizumab resulted in radiographic responses, reduction in steroid requirements, and progression free survival times ranging from 8 to 24 months [20]. Taking into account the limitations of the available data, temozolomide and bevacizumab may be reasonable salvage regimens in carefully selected adult BS glioma patients. Due to varied chemotherapy regimens, treatment setting (adjuvant versus recurrence), and different tumor grades treated in our study, whether chemotherapy improved outcomes cannot be determined. Prospective studies with larger numbers of patients are needed to clearly define a role for adjuvant chemotherapy and optimal salvage regimens. It is worth noting that only 8% of patients were treated on clinical trial; this is not unexpected as many clinical trials do not include infratentorial tumors.
Of the 100 cases that underwent either a biopsy (86%) or STR (14%), biopsy was diagnostic of neoplasia in 97% and in 86% of cases tumor grade could be determined based on the tissue specimen. This diagnostic yield is similar to prior studies [7]. Our analysis is limited as we were not able to determine the complication rate associated with brainstem biopsy in our series. In a study of 46 radiographically suspected BS gliomas in adults, only 61% were proven BS gliomas pathologically suggesting that a heterogeneous group of tumors can mimic brainstem gliomas [21]. This heterogeneity is particularly important in enhancing brainstem masses. In a study of 96 adults with BS masses, only 67.8% of diffuse enhancing BS masses and 34.7% of focal enhancing masses were infiltrating glial neoplasms [17]. We did not collect data on non-tumor entities, non-glial neoplasms, and pathologically diagnosed WHO Grade I and other circumscribed glial neoplasms. Eight of the 15 (53%) focal tumors were enhancing and 9 of the 15 (60%) of the focal tumors in our study were AAs or GBs lending further support to the importance of biopsy in enhancing and focal BS masses as low and high-grade tumors are in the differential diagnosis. Similar to previous studies [15,21], our results suggest that WHO Grade correlates with OS and tissue diagnosis may be important for counseling on prognosis and have implications for developing an individualized treatment plan.
IDH1 mutations are present in approximately 70 to 80% of supratentorial, WHO Grade II and III astrocytic, oligodendoglial, and mixed tumors and less than 10% of supratentorial GBs [22,23]. As we previously reported [14] and updated in this paper, IDH1 mutations are rare in infratentorial WHO Grade II and III gliomas, occurring in only 8% (2 of 25) of cases. IDH1 mutations were also rarely found in brainstem GBs in our study (1 of 17, 6%) of cases; this rate is similar to that seen with supratentorial GB [23,24]. IDH1 mutation may be of limited diagnostic utility in brainstem gliomas, given that its absence does not decrease the likelihood of WHO Grade II or III infiltrating glioma. We did not perform sequencing to determine the presence of IDH1 and IDH2 mutations in all of the IDH1 wild-type cases based on IHC. A recent study found IDH1 R132H mutations in only 1 of 17 adult brainstem gliomas, but 2 out of 7 cases with negative IDH1 R132H IHC had rare IDH1 mutations, IDH1 R132C and IDH1 R132G, suggesting that these rare IDH1 mutations may occur more frequently in adult BS gliomas [10]. We did not assess O6- methylguanine methyltransferase (MGMT) promoter methylation, a known prognostic biomarker in supratentorial, WHO Grade II to IV gliomas [25–27]. Whether MGMT promoter methylation is of prognostic significance in adult BS gliomas is currently unknown.
In our study, the WHO Grade II, III, and IV brainstem-located, diffuse astrocytic tumors had similar survival outcomes to supratentorial, IDH1 wild-type WHO Grade II, III and IV tumors [24,28–30]. Extrapolating from the median OS, the subset of younger patients with diffuse, non-enhancing tumors, diagnosed radiographically, are likely to represent mainly IDH1 wild-type WHO Grade II and III astrocytomas. It should be noted that the adult brainstem gliomas in our cohort had similar outcomes to supratentorial IDH1 wild-type tumors despite surgical management with biopsy, or no biopsy, whereas supratentorial tumors are more likely to undergo surgical resection with maximal tumor resection associated with significantly improved outcomes [31,32]. There was only 1 tumor with oligo-like histology in our cohort, and mixed oligoastrocytic tumors were seen in only 2 of 104 adult brainstem gliomas in another study [15]. There is only 1 previously reported case of a brainstem oligodendroglioma with 1p/19q chromosomal co-deletion [10,33]. Given the rarity of IDH1 mutations in adult BS gliomas it is not unexpected that 1p/19q chromosomal deletion is also rare given that the majority of oligodendroglial tumors with 1p/19q co-deletion are also IDH1 mutated [34]. The low rate of IDH1 mutation in WHO Grade II and III adult brainstem gliomas and similar survival outcomes to supratentorial IDH1 wild-type tumors despite management without maximal surgical resection may suggest a unique cell or origin.
Overall, we found oncogene mutations in 33% (3 of 9) of cases with sufficient tissue for Sequenom testing. The only known genetic risk factor for diffuse brainstem gliomas in pediatric patients is NF1, and notably these tumors can display an indolent clinical behavior [35]. There were 2 NF1 patients in our cohort (one previously reported [36]), and one of these patients’ tumors had a BRAF V600E mutation (Appendix 1). We found 2 PIK3CA H1047R mutations in patients with, relatively, prolonged survival although the significance of this association is uncertain. We did not assess for histone 3.3 mutations, present in the majority of pediatric DIPGs, as these mutations were not included in the Sequenom panel used at the time of this study; this is a significant limitation of these study results and needs to be addressed in future studies [37]. As previously demonstrated in pediatric DIPGs [38,39], these results demonstrate the feasibility of doing mutation profiling on brainstem biopsy samples, and the identification of potentially actionable mutations.
This study is limited by its retrospective design from a single cancer center in the United States. Varied treatment regimens and selection bias limit the interpretation of treatment and survival outcomes. The definition of focal versus diffuse tumors used for radiographic classification differs from study to study, and makes comparison of our results to other studies difficult. Only a subset of tumors had available tissue for IDH1 IHC and an even smaller number of cases (n = 9) had sufficient tissue for mutation profiling, thus limiting any conclusion as to the frequency and clinical significance of IDH1 and other oncogene mutations.
4. Conclusions
In adult BS gliomas, IDH1 mutations are rare and survival outcomes are similar to IDH1 wild-type supratentorial astrocytic tumors of similar grade and histology. Brainstem biopsy was diagnostic of tumor in 90% of cases in our study and our results suggest that determining tumor grade and histology have prognostic significance. Biopsy is necessary in contrast enhancing brainstem masses to differentiate high grade gliomas from low grade glial and non-glial neoplasms as management differs amongst these tumor types. Biopsy of non-enhancing brainstem gliomas can provide additional prognostic information. However, determining tumor grade may not significantly change management which consists of adjuvant radiotherapy in most situations [15]. Determining whether to biopsy adult brainstem gliomas should be done in a multi-disciplinary setting. Mutation profiling can be done on brainstem biopsy samples, and may identify potentially actionable mutations in a subset of patients. Given the rarity of adult BS gliomas and difficulty in obtaining tissue samples, collaborative studies with larger patient samples, optimization of surgical techniques for obtaining adequate tissue sampling, and testing using a broader, genomic approach are needed to enhance our understanding of the tumor biology of WHO Grade II-IV, BS gliomas in adults.
Supplementary Material
Acknowledgments
Funding
Supported in part by the Ferenc and Phyllis Gyorkey Endowed Chair in Pathology of UT MD Anderson Cancer Center (JMB) and by NCI K24-CA160777 (VP).
Abbreviations
- BS
brainstem
- WHO
World Health Organization
- DIPG
diffuse intrinsic pontine glioma
- IDH1
isocitrate dehydrogenase 1
- RT
radiotherapy
- GTR
gross total resection
- STR
subtotal resection
- OS
overall survival
- IHC
immunohistochemistry
- KPS
Karnofsky performance score
- NF1
neurofibromatosis type I
- GB
glioblastoma
- AA
anaplastic astrocytoma
- DA
diffuse astrocytoma
- NOS
not otherwise specified
- MGMT
O6-methylguanine methyltransferase
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.jns.2015.04.014.
Conflicts of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Disclaimer
The views expressed are those of the author(s) and do not reflect the official policy of Walter Reed National Military Medical Center, the Department of the Army, the Department of Defense, or the U.S. Government.
The identification of specific products or scientific instrumentation does not constitute endorsement or implied endorsement on the part of the author, DoD, or any component agency. While we generally excise references to products, companies, manufacturers, organizations, etc. in government produced works, the abstracts produced and other similarly situated researcher presents a special circumstance when such product inclusions become an integral part of the scientific endeavor.
References
- [1].CBTRUS CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2004–2008. 2012. [DOI] [PMC free article] [PubMed]
- [2].Grimm SA, Chamberlain MC. Brainstem glioma: a review. Curr Neurol Neurosci Rep. 2013;13:346. doi: 10.1007/s11910-013-0346-3. http://dx.doi.org/10.1007/s11910-013-0346-3. [DOI] [PubMed] [Google Scholar]
- [3].Reyes-Botero G, Mokhtari K, Martin-Duverneuil N, et al. Adult brainstem gliomas. Oncologist. 2012;17:388–97. doi: 10.1634/theoncologist.2011-0335. http://dx.doi.org/10.1634/theoncologist.2011-0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Donaldson SS, Laningham F, Fisher PG. Advances toward an understanding of brainstem gliomas. J Clin Oncol. 2006;24:1266–72. doi: 10.1200/JCO.2005.04.6599. http://dx.doi.org/10.1200/JCO.2005.04.6599. [DOI] [PubMed] [Google Scholar]
- [5].Guillamo JS, Monjour A, Taillandier L, et al. Brainstem gliomas in adults: prognostic factors and classification. Brain. 2001;124:2528–39. doi: 10.1093/brain/124.12.2528. [DOI] [PubMed] [Google Scholar]
- [6].Babu R, Kranz PG, Agarwal V, et al. Malignant brainstem gliomas in adults: clinicopathological characteristics and prognostic factors. J Neurooncol. 2014 doi: 10.1007/s11060-014-1471-9. http://dx.doi.org/10.1007/s11060-014-1471-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kesari S, Kim RS, Markos V, et al. Prognostic factors in adult brainstem gliomas: a multicenter, retrospective analysis of 101 cases. J Neurooncol. 2008;88:175–83. doi: 10.1007/s11060-008-9545-1. http://dx.doi.org/10.1007/s11060-008-9545-1. [DOI] [PubMed] [Google Scholar]
- [8].Rineer J, Schreiber D, Choi K, Rotman M. Characterization and outcomes of infratentorial malignant glioma: a population-based study using the Surveillance Epidemiology and End-Results database. Radiother Oncol J Eur Soc Ther Radiol Oncol. 2010;95:321–6. doi: 10.1016/j.radonc.2010.04.007. http://dx.doi.org/10.1016/j.radonc.2010.04.007. [DOI] [PubMed] [Google Scholar]
- [9].Hundsberger T, Tonder M, Hottinger A, et al. Clinical management and outcome of histologically verified adult brainstem gliomas in Switzerland: a retrospective analysis of 21 patients. J Neurooncol. 2014;118:321–8. doi: 10.1007/s11060-014-1434-1. http://dx.doi.org/10.1007/s11060-014-1434-1. [DOI] [PubMed] [Google Scholar]
- [10].Reyes-Botero G, Giry M, Mokhtari K, et al. Molecular analysis of diffuse intrinsic brainstem gliomas in adults. J Neurooncol. 2014;116:405–11. doi: 10.1007/s11060-013-1312-2. http://dx.doi.org/10.1007/s11060-013-1312-2. [DOI] [PubMed] [Google Scholar]
- [11].Salmaggi A, Fariselli L, Milanesi I, et al. Natural history and management of brainstem gliomas in adults. A retrospective Italian study. J Neurol. 2008;255:171–7. doi: 10.1007/s00415-008-0589-0. http://dx.doi.org/10.1007/s00415-008-0589-0. [DOI] [PubMed] [Google Scholar]
- [12].Fischbein NJ, Prados MD, Wara W, et al. Radiologic classification of brain stem tumors: correlation of magnetic resonance imaging appearance with clinical outcome. Pediatr Neurosurg. 1996;24:9–23. doi: 10.1159/000121010. [DOI] [PubMed] [Google Scholar]
- [13].Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96. doi: 10.1056/NEJMoa043330. http://dx.doi.org/10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- [14].Ellezam B, Theeler BJ, Walbert T, et al. Low rate of R132H IDH1 mutation in infratentorial and spinal cord grade II and III diffuse gliomas. Acta Neuropathol. 2012;124:449–51. doi: 10.1007/s00401-012-1011-7. http://dx.doi.org/10.1007/s00401-012-1011-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Reithmeier T, Kuzeawu A, Hentschel B, et al. Retrospective analysis of 104 histologically proven adult brainstem gliomas: clinical symptoms, therapeutic approaches and prognostic factors. BMC Cancer. 2014;14:115. doi: 10.1186/1471-2407-14-115. http://dx.doi.org/10.1186/1471-2407-14-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Babu R, Kranz PG, Karikari IO, et al. Clinical characteristics and treatment of malignant brainstem gliomas in elderly patients. J Clin Neurosci Off J Neurosurg Soc Australas. 2013;20:1382–6. doi: 10.1016/j.jocn.2012.12.011. http://dx.doi.org/10.1016/j.jocn.2012.12.011. [DOI] [PubMed] [Google Scholar]
- [17].Dellaretti M, Touzet G, Reyns N, et al. Correlation between magnetic resonance imaging findings and histological diagnosis of intrinsic brainstem lesions in adults. Neuro Oncol. 2012;14:381–5. doi: 10.1093/neuonc/nor215. http://dx.doi.org/10.1093/neuonc/nor215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Reyes-Botero G, Laigle-Donadey F, Mokhtari K, et al. Temozolomide after radiotherapy in recurrent “low grade” diffuse brainstem glioma in adults. J Neurooncol. 2014;120:581–6. doi: 10.1007/s11060-014-1589-9. http://dx.doi.org/10.1007/s11060-014-1589-9. [DOI] [PubMed] [Google Scholar]
- [19].Friedman HS, Prados MD, Wen PY, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27:4733–40. doi: 10.1200/JCO.2008.19.8721. http://dx.doi.org/10.1200/JCO.2008.19.8721. [DOI] [PubMed] [Google Scholar]
- [20].Reithmeier T, Lopez WOC, Spehl TS, et al. Bevacizumab as salvage therapy for progressive brain stem gliomas. Clin Neurol Neurosurg. 2013;115:165–9. doi: 10.1016/j.clineuro.2012.04.027. http://dx.doi.org/10.1016/j.clineuro.2012.04.027. [DOI] [PubMed] [Google Scholar]
- [21].Rachinger W, Grau S, Holtmannspötter M, et al. Serial stereotactic biopsy of brainstem lesions in adults improves diagnostic accuracy compared with MRI only. J Neurol Neurosurg Psychiatry. 2009;80:1134–9. doi: 10.1136/jnnp.2009.174250. http://dx.doi.org/10.1136/jnnp.2009.174250. [DOI] [PubMed] [Google Scholar]
- [22].Kloosterhof NK, Bralten LB, Dubbink HJ, et al. Isocitrate dehydrogenase-1 mutations: a fundamentally new understanding of diffuse glioma? Lancet Oncol. 2011;12:83–91. doi: 10.1016/S1470-2045(10)70053-X. http://dx.doi.org/10.1016/S1470-2045(10)70053-X. [DOI] [PubMed] [Google Scholar]
- [23].Lai A, Kharbanda S, Pope WB, et al. Evidence for sequenced molecular evolution of IDH1 mutant glioblastoma from a distinct cell of origin. J Clin Oncol. 2011;29:4482–90. doi: 10.1200/JCO.2010.33.8715. http://dx.doi.org/10.1200/JCO.2010.33.8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. http://dx.doi.org/10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. http://dx.doi.org/10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- [26].Van den Bent MJ, Dubbink HJ, Sanson M, et al. MGMT promoter methylation is prognostic but not predictive for outcome to adjuvant PCV chemotherapy in anaplastic oligodendroglial tumors: a report from EORTC Brain Tumor Group Study 26951. J Clin Oncol. 2009;27:5881–6. doi: 10.1200/JCO.2009.24.1034. http://dx.doi.org/10.1200/JCO.2009.24.1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Everhard S, Kaloshi G, Criniere E, et al. MGMT methylation: a marker of response to temozolomide in low-grade gliomas. Ann Neurol. 2006;60:740–3. doi: 10.1002/ana.21044. http://dx.doi.org/10.1002/ana.21044. [DOI] [PubMed] [Google Scholar]
- [28].Noushmehr H, Weisenberger DJ, Diefes K, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–22. doi: 10.1016/j.ccr.2010.03.017. http://dx.doi.org/10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hartmann C, Hentschel B, Wick W, et al. Patients with IDH1 wild type anaplastic astrocytomas exhibit worse prognosis than IDH1-mutated glioblastomas, and IDH1 mutation status accounts for the unfavorable prognostic effect of higher age: implications for classification of gliomas. Acta Neuropathol. 2010;120:707–18. doi: 10.1007/s00401-010-0781-z. http://dx.doi.org/10.1007/s00401-010-0781-z. [DOI] [PubMed] [Google Scholar]
- [30].Ahmadi R, Stockhammer F, Becker N, et al. No prognostic value of IDH1 mutations in a series of 100 WHO grade II astrocytomas. J Neurooncol. 2012;109:15–22. doi: 10.1007/s11060-012-0863-y. http://dx.doi.org/10.1007/s11060-012-0863-y. [DOI] [PubMed] [Google Scholar]
- [31].Marko NF, Weil RJ, Schroeder JL, et al. Extent of resection of glioblastoma revisited: personalized survival modeling facilitates more accurate survival prediction and supports a maximum-safe-resection approach to surgery. J Clin Oncol Off J Am Soc Clin Oncol. 2014;32:774–82. doi: 10.1200/JCO.2013.51.8886. http://dx.doi.org/10.1200/JCO.2013.51.8886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Smith JS, Chang EF, Lamborn KR, et al. Role of extent of resection in the long-term outcome of low-grade hemispheric gliomas. J Clin Oncol. 2008;26:1338–45. doi: 10.1200/JCO.2007.13.9337. http://dx.doi.org/10.1200/JCO.2007.13.9337. [DOI] [PubMed] [Google Scholar]
- [33].Hewer E, Beck J, Vassella E, Vajtai I. Anaplastic oligodendroglioma arising from the brain stem and featuring 1p/19q co-deletion. Neuropathol Off J Jpn Soc Neuropathol. 2014;34:32–8. doi: 10.1111/neup.12043. http://dx.doi.org/10.1111/neup.12043. [DOI] [PubMed] [Google Scholar]
- [34].Labussiere M, Idbaih A, Wang XW, et al. All the 1p19q codeleted gliomas are mutated on IDH1 or IDH2. Neurology. 2010;74:1886–90. doi: 10.1212/WNL.0b013e3181e1cf3a. http://dx.doi.org/10.1212/WNL.0b013e3181e1cf3a. [DOI] [PubMed] [Google Scholar]
- [35].Guillamo JS, Doz F, Delattre JY. Brain stem gliomas. Curr Opin Neurol. 2001;14:711–5. doi: 10.1097/00019052-200112000-00006. [DOI] [PubMed] [Google Scholar]
- [36].Theeler BJ, Ellezam B, Yust-Katz S, et al. Prolonged survival in adult neurofibromatosis type I patients with recurrent high-grade gliomas treated with bevacizumab. J Neurol. 2014 doi: 10.1007/s00415-014-7292-0. http://dx.doi.org/10.1007/s00415-014-7292-0. [DOI] [PubMed] [Google Scholar]
- [37].Wu G, Broniscer A, McEachron TA, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. 2012;44:251–3. doi: 10.1038/ng.1102. http://dx.doi.org/10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Grill J, Puget S, Andreiuolo F, et al. Critical oncogenic mutations in newly diagnosed pediatric diffuse intrinsic pontine glioma. Pediatr Blood Cancer. 2012;58:489–91. doi: 10.1002/pbc.24060. http://dx.doi.org/10.1002/pbc.24060. [DOI] [PubMed] [Google Scholar]
- [39].Puget S, Philippe C, Bax DA, et al. Mesenchymal transition and PDGFRA amplification/mutation are key distinct oncogenic events in pediatric diffuse intrinsic pontine gliomas. PLoS One. 2012;7:e30313. doi: 10.1371/journal.pone.0030313. http://dx.doi.org/10.1371/journal.pone.0030313. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.