

Abstract
Curcumin (1) down-regulates the expression as well as phosphorylation of epidermal growth factor receptor (EGFR) in lung adenocarcinoma cells expressing gefitinib-resistant EGFR. Thirty-seven newly synthesized curcumin analogues including dimethoxycurcumin (2, DMC) were evaluated for their effects on EGFR expression as well as phosphorylation in two gefitinib-resistant lung adenocarcinoma cell lines, CL1-5 (EGFRwt) and H1975 (EGFRL858R+T790M). Based on the identified structure-activity relationships, methoxy substitution at C-3', C-4', or both positions favored inhibitory activity (compounds 1, 2, 5, 8–15, 17, 36), while compounds with more polar substituents were generally less active in both cell lines. Compound 36 with a fluorine substituent at C-6' and its protonated counterpart 2 did not lose activity, suggesting halogen tolerance. In addition, a conjugated linker was essential for activity. Among all evaluated curcumin derivatives, compound 2 showed the best inhibitory effects on both wild-type and mutant EGFR by efficiently inducing gefitinib-insensitive EGFR degradation. Compound 23 also reduced gefitinib-induced gastrointestinal damage in the non-transformed intestinal epithelial cell line IEC-18.
Keywords: curcumin, EGFR, gastrointestinal tract, lung adenocarcinoma, tyrosine kinase inhibitor

1. Introduction
Curcumin (1), a member of the curcuminoid family, has been used as an herbal medicine for centuries and studied mainly for its versatile pharmacological effects against lung, neurological, liver, metabolic, autoimmune, cardiovascular, and other inflammatory diseases, as well as cancer.1 In our previous study, curcumin inhibited cell cycle progression and induced cell apoptosis and anti-metastasis in vascular smooth muscle2 and lung adenocarcinoma cells.3,4 In addition, curcumin was one of the hits among 598 natural products screened against different gefitinib-resistant non-small-cell lung cancer cell lines using a cell proliferation assay.5 Non-small cell lung cancer (NSCLC) is an aggressive tumor with poor prognosis and epidermal growth factor receptor (EGFR) is over-expressed in 40% to 80% of NSCLC patients, which correlates with poor overall survival and high metastasis rate.6,7 Thus, targeting EGFR signaling is a well-established strategy for treatment of NSCLC. Although patients with EGFR mutation at L858R or deletion of exon 19 respond well to EGFR tyrosine kinase inhibitors (TKIs), such as gefitinib, a large population of patients with wild-type EGFR or T790M mutation are resistant to TKIs.8,9 Therefore, new generation TKIs or adjuvant therapy conquering resistance are greatly needed. Our previous studies showed that curcumin can down-regulate EGFR phosphorylation dose-dependently in lung adenocarcinoma cell lines by increasing EGFR degradation in both wild-type and T790M mutated EGFR.5 Furthermore, the combination of curcumin and gefitinib potentiated anti-proliferative activity and induced apoptosis in gefitinib-resistant lung adenocarcinoma cell lines. In an in vivo study, administration of curcumin with gefitinib led to significant inhibition of CL1-5, A549, and H1975 xenograft tumor growth in SCID mice. Western blotting from tumor xenograft tissue showed reduction of EGFR, c-MET, cyclin D1 expression and activation of caspases-8, -9, and PARP leading to apoptosis. Interestingly, less gefitinib-induced intestinal tissue damage was also observed. Therefore, our previous findings strongly indicated that curcumin could be a potential lead to induce EGFR degradation and reduce TKIs-induced gastrointestinal (GI) damage.5 In our current study, new curcumin analogues were synthesized and evaluated for their ability to overcome EGFR-TKI resistance in lung adenocarcinoma cells together with GI protection or GI damage reversal.
2. Chemistry
The syntheses of compounds 3–15 were described previously.13–15 Scheme 1 shows the general synthetic pathway to new asymmetric curcumin analogues 16 and 17 with various substituents on the phenyl rings. Excess 2,4-pentanedione or ethyl 4-acetyl-5-oxo-hexanoate was used to avoid aldol condensation occurring at both terminals of the starting material and to produce the monoaryl intermediate 16a or 17a.10 The intermediates were subsequently condensed with an appropriate second benzaldehyde to give the target compounds 16 and 17.
Scheme 1.

The general synthetic scheme for asymmetric curcumin analogues
Curcumin analogues 18 and 26 with an ethyl acrylate group at the C4 position were synthesized in three steps from 1 and 16, respectively, as shown in Scheme 2. Compounds 1 and 16 were protected as tetrahydropyranyl (THP) ethers by using dihydropyran (DHP) in the presence of pyridium p-tolenesulfonate (PPTS) in dry dichloromethane.11 The resulting THP ethers 18a and 26a were alkylated at the C4 position using sodium hydride in anhydrous tetrahydrofuran, followed by the addition of ethyl propiolate to afford C4-substituted compounds 18b and 26b, respectively. Deprotection of 18b and 26b with PPTS in EtOH afforded the target compounds 18 and 26, respectively.
Scheme 2.

The synthesis of 18 and 26
Hydrolysis of 3, 17, 20, 22, 29, and 31 in 10% aq HCl afforded the desired products 19, 25, 27, 24, 30, and 32, respectively (Scheme 3). For curcumin analogues 20, 22, 29, and 31, commercially unavailable benzaldehydes 20a and 29a were first synthesized by reaction of vanillin with methyl bromoacetate (for 20a) or ethyl bromobutyrate (for 29a) in the presence of K2CO3 in dimethylformamide (DMF) as shown in Scheme 4. The yields of intermediates 20a and 29a were 93% and 99%, respectively. Symmetric curcumin analogues, including 20–23, 29, 31, and 33–37 with varied substituents on the phenyl rings, were then synthesized by the general synthetic pathway shown in Scheme 5. 2,4-Pentanedione or ethyl 4-acetyl-5-oxo-hexanoate was condensed with an appropriately substituted benzaldehyde in EtOAc at 70 °C using a slightly modified method of Pederson et al.12 Boric anhydride was used to form a boron complex with 2,4-pentanedione or ethyl 4-acetyl-5-oxo-hexanoate in order to prevent Knoevenagel condensation10 (Scheme 5).
Scheme 3.

The synthesis of curcumin analogues 19, 24, 25, 27, 30 and 32
Scheme 4.

The synthesis of vanillin derivatives 20a and 29a
Scheme 5.

The general synthetic strategies for symmetric curcumin analogues
Curcumin analogue 28 with an amide moiety in the C4 substituent of the aromatic rings was prepared in 35% yield by reaction of 27 with aniline in the presence of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI) and 4-dimethylaminopyridine (DMAP) (Scheme 6).
Scheme 6.

The synthesis of 28
3. Results and Discussion
3.1. Effects of curcumin and its derivatives on EGFR in gefitinib-insensitive lung adenocarcinoma cell lines
Thirty-seven synthesized curcumin analogues including curcumin (1) and dimethoxycurcumin (2, ASC-JC9) were evaluated for their ability to inhibit phosphorylation of EGFR or induce EGFR degradation acutely under EGF stimulated conditions. Two gefitinib-insensitive lung adenocarcinoma cell lines, CL1-5 (EGFRwt) and H1975 (EGFRL858R+T790M), were selected to determine which analogues could interfere with gefinitib-insensitive EGFR. Cells were serum-starved for 24 h prior to compound treatment and EGF stimulation. Cells were pre-treated with compound for 1 h and then stimulated with EGF (20 ng/mL) for 20 min. Expression level and Tyr1068-phosphorylation of EGFR were evaluated by Western blot analysis using specific antibodies to pan-EGFR and Tyr1068-phosphorylated EGFR (pEGFR), respectively. Under these conditions, wild-type EGFR (CL1-5) was efficiently phosphorylated by EGF stimulation without pre-treatment with test compound (Figure 1A). In H1975 cells expressing mutant EGFR, pEGFR was detectable without EGF stimulation (Figure 1B). Among all tested compounds, 15 compounds (1, 2, 3, 5, 8–15, 17, 22, and 36) reduced total EGFR and pEGFR levels in CL1-5 cells carrying wild-type EGFR. In gefitinib-resistant H1975 cells carrying double mutations on EGFR (L858R and T790M), 14 compounds (1, 2, 5, 8–17, and 36) reduced total EGFR expression level. Interestingly, total EGFR was retained in H1975 cells treated with 23, while pEGFR was undetectable. A similar effect was observed in CL1-5 cells treated with 34. Thus, compounds 23 and 34 were unique in inhibiting kinase activity of gefitinib-resistant EGFR without inducing EGFR degradation. The effects and structures of curcumin derivatives on EGFR are summarized in Table 1, and structure-activity relationship (SAR) correlations are discussed below.
Figure 1.

Effects of curcumin derivatives on EGFR in gefitinib-insensitive cells. The gefitinib-insensitive lung adenocarcinoma cell lines were pre-treated with 10 μM compound, 1–37 as indicated, for 1 h prior to stimulation for 30 min with 20 ng/mL EGF. Cell lysates from (A) CL1-5 carrying wild-type EGFR, or (B) H1975 carrying L858R and T790M double mutations on EGFR, were analyzed by Western blot using antibodies to Tyr1068-phosphorylated EGFR (pEGFR), pan-EGFR (EGFR), and β-actin as a loading control.
Table 1.
Structures of curcumin derivatives
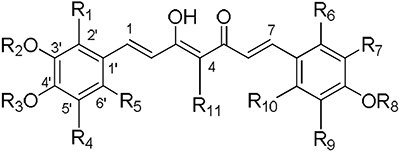
|
Effects on EGFR expressiona |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cmpd | R1 | R2 | R3 | R4 | R5 | R6 | R7 | R8 | R9 | R10 | R11 | CL1-5 | H1975 |
| 1 | H | Me | H | H | H | H | OMe | H | H | H | H | + | + |
| 2 | H | Me | Me | H | H | H | OMe | Me | H | H | H | + | +++ |
| 3 | H | Me | H | H | H | H | OMe | H | H | H | (CH2)2CO2Et | + | − |
| 4 | H | Me | Me | H | H | H | OMe | Me | H | H | CH=CHCO2Et | − | − |
| 5 | H | Me | H | H | H | H | OMe | Me | H | H | H | + | + |
| 6 | H | Me | H | H | H | H | H | H | H | H | H | − | − |
| 7 |
|
− | − | ||||||||||
| 8 | OMe | Me | Me | H | H | OMe | OMe | Me | H | H | H | + | + |
| 9 | H | Me | Me | H | OMe | H | OMe | Me | H | OMe | H | + | + |
| 10 | H | Me | Me | H | H | H | OH | Me | H | H | H | + | ++ |
| 11 | OMe | Me | Me | H | H | H | OMe | Me | H | H | H | + | + |
| 12 | H | Me | Me | H | OMe | H | OMe | Me | H | H | H | + | ++ |
| 13 | H | Me | H | H | H | H | OMe | Me | H | H | (CH2)2CO2Et | + | ++ |
| 14 | H | Me | H | H | H | H | OMe | H | H | H | COMe | + | + |
| 15 | H | Me | Me | H | H | H | OMe | Me | H | H | Me | + | + |
| 16 | H | Me | H | H | H | H | OH | H | H | H | H | − | ++ |
| 17 | H | H | H | H | H | H | OMe | H | H | H | (CH2)2CO2Et | + | + |
| 18 | H | Me | H | H | H | H | OMe | H | H | H | CH=CHCO2Et | − | − |
| 19 | H | Me | H | H | H | H | OMe | H | H | H | (CH2)2CO2H | − | − |
| 20 | H | Me | CH2CO2Me | H | H | H | OMe | CH2CO2Me | H | H | H | − | − |
| 21 | H | Me | CH2CO2Me | H | H | H | OMe | CH2CO2Me | H | H | (CH2)2CO2Et | − | − |
| 22 | H | H | H | H | H | H | OH | H | H | H | (CH2)2CO2Et | + | − |
| 23 | H | H | H | H | H | H | OH | H | H | H | H | + | + b |
| 24 | H | H | H | H | H | H | OH | H | H | H | (CH2)2CO2H | − | − |
| 25 | H | H | H | H | H | H | OMe | H | H | H | (CH2)2CO2H | − | − |
| 26 | H | Me | H | H | H | H | OH | H | H | H | CH=CHCO2Et | − | − |
| 27 | H | Me | CH2CO2H | H | H | H | OMe | CH2CO2H | H | H | H | − | − |
| 28 | H | Me | CH2CONHPh | H | H | H | OMe | CH2CONHPh | H | H | H | − | − |
| 29 | H | Me | (CH2)3CO2Et | H | H | H | OMe | (CH2)3CO2Et | H | H | H | − | − |
| 30 | H | Me | (CH2)3CO2H | H | H | H | OMe | (CH2)3CO2H | H | H | H | − | − |
| 31 | H | Me | (CH2)3CO2Et | H | H | H | OMe | (CH2)3CO2Et | H | H | (CH2)2CO2Et | − | − |
| 32 | H | Me | (CH2)3CO2H | H | H | H | OMe | (CH2)3CO2H | H | H | (CH2)2CO2H | − | − |
| 33 | H | Me | Me | H | H | H | OMe | Me | H | H | (CH2)2CO2Et | − | − |
| 34 | H | Me | Me | OH | H | H | OMe | Me | OH | H | (CH2)2CO2Et | + b | + |
| 35 | H | Me | H | OMe | H | H | OMe | H | OMe | H | (CH2)2CO2Et | − | − |
| 36 | H | Me | Me | H | F | H | OMe | Me | H | F | H | + | ++ |
| 37 | H | Me | Me | OH | H | H | OMe | Me | OH | H | H | − | − |
Effects of compounds on EGFR expression in H1975 cells can be ranked as negative (−), + (good), ++ (better), or +++ (best), while effects in CL1-5 cells can be observed “all (+) or none (−)” based on our analysis.
EGFR was not affected, pEGFR was reduced.
3.2. SAR of curcumin analogs on phosphorylation and expression of EGFR
Among the 26 symmetric curcumin analogues, 15 compounds did not affect EGFR and pEGFR expression. On the other hand, among the 11 asymmetric curcumin analogues, four compounds did not affect EGFR and pEGFR expression. Therefore, symmetry was not a definitive factor for activity.
Next, the effects of phenyl ring modification on the expression of EGFR and pEGFR were investigated. Overall, compounds 2, 11–13, 22, and 36 exhibited better inhibitory effects than 1 in CL1-5 cells, and compounds 2, 9–13, 16 and 36 exhibited better inhibitory effects than 1 in H1975 cells. Compound 2 with four methoxy substituents at C-3' and C-4' as well as compounds 8 and 11 with an additional one or two methoxy substituents at C-2' down-regulated EGFR and pEGFR in both cell lines. Overall, all active compounds bore methoxy or hydroxy groups on both C3' positions, while compound 6 with one methoxy group and one hydrogen atom on these positions showed no inhibitory activity. Replacing one or both 3'-methoxy groups with 3'-hydroxy moieties could affect the inhibitory activity as seen by comparison of 1 with 16 (active in H1975 cells, inactive in CL1-5 cells) and 23 (see Section 3.1). Thus, both C-3' positions should be substituted, preferably with methoxy groups for optimal effects on EGFR. Replacing the 4'-OH substituents of active compound 1 with methoxy group(s) resulted in 2 and 5, which also were active. In contrast, compounds 20, 21, and 27–32 with bulkier 4'-alkyloxy groups containing ester, carboxy, or amide moieties did not down-regulate EGFR or pEGFR in either cell line. These data suggest that methoxy or hydroxy groups on C-4' are favorable for inhibitory activity. Introducing hydroxy groups at both C-5' positions of active 2 gave 37, which showed no inhibition. Similarly, compound 3 without 5'-substituents down-regulated EGFR and pEGFR in CL1-5 cells, while 35 with two 5'-methoxy groups did not. Indeed, many of the inactive compounds were unsubstituted at C-5'. Compounds with a methoxy (9, 12) or fluorine (36) group on C-6' exhibited activity, although all of them showed weaker effects than their parent compound 2. These results suggest that the EGFR inhibitory activity might not be affected significantly by modification of the C-6' position.
Overall, the substitutions on the phenyl rings of the curcumin derivatives affected the extent of the down-regulation of EGFR. Substitution at C-3' and C-4' appeared critical for down-regulation of gefitinib-resistant EGFR. Methoxy or hydroxy substituents at these positions were favorable to activity, while other alkyloxy groups were not. The activity was tolerant of methoxy, hydroxy, or fluorine groups at C-2' and C-6', while hydrogen seemed preferable at C-5.
Finally, the effect of the linker connecting the two phenyl rings was studied. Saturation of the linker in 2 gave 7, which exhibited no activity, indicating that an unsaturated, conjugated linker, such as 3-hydroxy-1,4,6-heptatrien-5-one, between the two phenyl rings is required for activity. Compounds with acetyl (14) and methyl (15) groups at the midpoint (C-4) of the linker reduced the phosphorylation and expression of EGFR in both cell lines. Certain compounds with a propionyl ethyl ester (3, 13, 17, 22, and 34) at the C4 position were also active in at least one cell line, while others (33, 35) were not. Interestingly, hydrolysis of the propionic ethyl ester at C4 to a more hydrophilic propionic acid abolished activity (19 vs 3, 24 vs 22, 25 vs 17) in both cell lines. Moreover, introduction of an unsaturated acrylic ester at C4 also destroyed the activity (4 vs 2, 18 vs 1, 26 vs 16), even though the conjugation in the linker was extended.
3.3. Curcumin analogs protect gastrointestinal tract from gefitinib-induced damage
According to our previous findings, curcumin showed the potential to reduce TKIs-induced gastrointestinal (GI) damage. Thus, we investigated the effects of the curcumin analogues on protection of GI tract from gefitinib-induced damages using the non-transformed intestinal epithelial cell line IEC-18 (Figure 2). At 5 μM, the curcumin derivatives reduced geftinib-induced cytotoxicity in IEC-18 (IC50 of geftinib is 40 μM) and compound 23 restored cell viability up to 80%.
Figure 2.

Reducing gefitinib-induced cytotoxicity by curcumin derivatives. Non-transformed rat small intestinal cell line IEC-18 was treated with 40 μM gefitinib with 5 μM compound 1 (curcumin), 2 (dimethyl curcumin), 5, 10, or 23, for 24 h. Cytotoxicity was evaluated by MTT assay and expressed as proliferative index (%) standardized by untreated cells (100%). Effects of compounds were evaluated by student t-test and represented p<0.05 (*) or p<0.01 (**).
4. Conclusion
In general, increasing the molecular polarity was not favorable to inhibition of EGFR phosphorylation activity or induction of EGFR degradation in CL1-5 (EGFRwt) and H1975 (EGFRL858R+T790M) cell lines. Hence, introduction of polar groups, such as hydroxy or carboxy, around the biphenyl moiety decreased activity (e.g., 19, 24, 25, 27, 30, 32), implicating a less polar binding environment. The presence of a bulky group at the C4' position (i.e., R3, R8 moiety) did not favor the inhibitory effect (e.g., 28–32), indicating a decreasing binding affinity with bulky substituents. Addition of a less polar side chain at the C4 position (i.e., R11 group) of the linker) could affect the activity, but depending on the overall molecular substitution, was not always positive. The presence of methoxy groups at C3' and C4' positions (i.e., 1, 2, 5, 8–17, 34, 36) appeared consistently favorable toward EGFR inhibitory activity. From the testing results, compound 36 with a fluorine substituent at C-6' showed comparable activity to its protonated counterpart 2 (DMC), suggesting halogen tolerance. A conjugated linker bridging the two phenyl groups was essential for activity, as compound 7 with a saturated linker exhibited diminished activity.
Overall, compound 2 (DMC) showed the best inhibitory effects on both wild-type and mutant EGFR. Compound 2 efficiently induced gefitinib-insensitive EGFR degradation. However, compound 23 inhibited EGFR phosphorylation in both CL1-5 and H1975 cells without induction of EGFR degradation. In addition, compound 23 reduced the cytotoxicity of gefinitib against a small intestinal cell line, suggesting that 23 may inhibit gefitinib-insensitive EGFR activation and protect small intestinal cells from gefitinib-induced cytotoxicity.
5. Experimental section
5.1. Chemistry
Melting points were determined on a Fisher-John melting point apparatus and are uncorrected. Proton nuclear magnetic resonance (1H NMR) and 13C NMR spectra were measured on Varian Gemini 300, Inova 400, or JEOL AL-400 spectrometers with tetramethylsilane (TMS) as the internal standard. Chemical shifts are reported in δ (ppm). Mass spectra (MS) were obtained on a Shimadzu LCMS-2010 and JEOL JMS-700. CombiFlash® chromatographic system (Isco Companion) with a Grace silica gel cartridge was used for general separation and purification. Precoated silica gel plates (Kieselgel 60, F254, 0.25 mm) were used for thin layer chromatography (TLC) analysis. All reagents and solvents were purchased from Aldrich, Fisher, and other venders. Some chemicals were used after purification, and others were used as purchased.
Synthesis of 16 and 17
2,4-Pentanedione (3 equiv; for compound 16a) or ethyl 4-acetyl-5-oxo-hexanoate (3 equiv; for compound 17a) and boric anhydride (2 equiv) dissolved in EtOAc were stirred for 1 h at 70 °C. Vanillin (for compound 16a) or 3,4-dihydroxybenzaldehyde (for compound 17a) and tributylborate (1 equiv) dissolved in EtOAc were added, and the mixture was stirred for 30 min at 70 °C. n-Butylamine (1 equiv) dissolved in EtOAc was added dropwise over 15 min at 85 °C. Stirring was continued for 3 h at 100 °C. The mixture was then hydrolyzed by adding 0.1N HCl and heating at 50 °C for 30 min. The organic layer was separated, and the aqueous layer was extracted three times with EtOAc. The combined organic layers were washed with H2O and brine, and then dried over anhydrous Na2SO4, and the solvent was removed in vacuo. The crude products were purified by CombiFlash® column chromatography eluting with n-hexane-EtOAc gradient to give the desired 16a or 17a.
16a: 76% yield (started with 26.3 mmol of vanillin), amorphous, 1H-NMR (δ, CDCl3): 2.17 (3H, s, COCH3), 3.94 (3H, s, OCH3), 5.63 (1H, s, C=CH), 6.32 and 7.53 (each 1H, d, J = 15.8 Hz, CH=CH), 6.92 (1H, d, J = 8.2 Hz, Ar-H), 7.02 (1H, d, J = 1.9 Hz, Ar-H), 7.09 (1H, dd, J = 8.2, 1.9 Hz, Ar-H), ESI-MS (negative, m/z): 233.05 [M-H]−.
17a: 13% yield (started with 65.0 mmol of 3,4-dihydroxybenzaldehyde), amorphous; 1H-NMR (δ, CDCl3): 1.25 (3H, t, J = 7.0 Hz, CH2CH3), 2.19 (3H, s, COCH3), 2.23 (1H, t, J = 7.0 Hz, CH2), 2.33, 2.47 and 2.76 (each 1H, m, CH2), 4.14 (2H, q, J = 7.0 Hz, CH2CH3), 6.64 and 7.59 (each 1H, d, J = 15.8 Hz, CH=CH), 6.89 and 7.04 (each 1H, d, J = 8.2 Hz, Ar-H), 7.12 (1H, s, Ar-H), ESI-MS (negative, m/z): 319.15 [M-H]−.
16a (for compound 16) or 17a (for compound 17) and boric anhydride (0.7 equiv) dissolved in EtOAc were stirred for 1 h at 70 °C. 3,4-Dihydroxybenzaldehyde (2 equiv; for compound 16) or vanillin (1.5 equiv; for compound 17) and tributylborate (2.1 equiv) dissolved in EtOAc were added, and the mixture was stirred for 30 min at 70 °C. n-Butylamine (2 equiv) dissolved in EtOAc was added dropwise over 15 min at 85 °C. Stirring was continued for 3 h at 100 °C. The mixture was then hydrolyzed by adding 1N HCl and heating at 50 °C for 45 min. The organic layer was separated, and the aqueous layer was extracted three times with EtOAc. The combined organic layers were washed with H2O and brine, and then dried over anhydrous Na2SO4, and the solvent was removed in vacuo. The crude products were purified by CombiFlash® column chromatography eluting with n-hexane-EtOAc gradient to give the desired 16 or 17.
16: 19% yield (started with 18.1 mmol of 16a), amorphous, 1H-NMR (δ, CDCl3): 3.95 (3H, s, OCH3), 5.78 (1H, s, C=CH), 6.45, 6.48, 7.54 and 7.59 (each 1H, d, J = 15.8 Hz, CH=CH), 6.89 and 6.94 (each 1H, d, J = 8.2 Hz, Ar-H), 7.05 and 7.10 (each 1H, d, J = 1.9 Hz, Ar-H), 7.06 and 7.13 (each 1H, dd, J = 8.2, 1.9 Hz, Ar-H), ESI-MS (negative, m/z): 353.10 [M-H]−.
17: 8% yield (started with 1.7 mmol of 17a), amorphous, 1H-NMR (δ, CDCl3): 1.26 (3H, t, J = 7.2 Hz, CH2CH3), 2.55 and 2.93 (each 2H, m, CH2), 4.15 (2H, q, J = 7.2 Hz, CH2CH3), 6.89 and 6.94 (each 1H, d, J = 8.2 Hz, Ar-H), 6.93, 6.97, 7.65 and 7.70 (each 1H, d, J = 15.8 Hz, CH=CH), 7.05 and 7.17 (each 1H, dd, J = 8.2, 1.9 Hz, Ar-H), 7.08 and 7.15 (each 1H, d, J = 1.9 Hz, Ar-H), ESI-MS (positive, m/z): 455.10 [M+H]+.
Synthesis of 18 and 26
3,4-Dihydro-2H-pyran (25 equiv) and pyridinium p-toluenesulfonate (PPTS, 0.1 equiv) were added to a solution of 1 (for compound 18a) or 16 (for compound 26a) in CH2Cl2, and the mixture was stirred at rt for 3 or 4 days under N2. The mixture was diluted with CH2Cl2 and then washed with water and brine, then dried over anhydrous Na2SO4. After removal of the solvent in vacuo, the product was purified by CombiFlash® column chromatography eluting with n-hexane-EtOAc gradient to give the desired 18a or 26a.
18a: 38% yield (started with 0.27 mmol of curcumin), amorphous, 1H-NMR (δ, CDCl3): 1.69 (8H, m, CH2), 1.96 (4H, m, CH2), 3.62 and 3.93 (each 2H, m, OCH2), 3.91 (6H, s, OCH3), 5.47 (2H, t, J = 3.1 Hz, OCH), 5.82 (1H, s, C=CH), 6.50 and 7.60 (each 2H, d, J = 15.8 Hz, CH=CH), 7.09 (2H, s, Ar-H), 7.12 (2H, d, J = 8.2 Hz, Ar-H), 7.15 (2H, d, J = 8.2 Hz, Ar-H), ESI-MS (positive, m/z): 537.30 [M+H]+.
26a: 33% yield (started with 3.37 mmol of 16), amorphous; 1H-NMR (δ, CDCl3): 1.71 and 1.99 (each 9H, m, CH2), 3.63 and 3.98 (each 3H, m, OCH2), 3.91 (3H, s, OCH3), 5.47 (3H, s, OCH), 5.81 (1H, s, C=CH), 6.49, 6.50, 7.58 and 7.59 (each 1H, d, J = 15.8 Hz, CH=CH), 7.09 and 7.36 (each 1H, s, Ar-H), 7.137 and 7.144 (each 2H, d, J = 8.2 Hz, Ar-H), ESI-MS (positive, m/z): 607.30 [M+H]+.
Sodium hydride (60% dispersion, 3 equiv) was added to a solution of 18a or 26a at 0 °C in THF. The mixture was stirred at 0 °C for 30 min under N2 and then stirred at rt for 2 h under N2. Ethyl propionate (2 equiv) was added to the reaction mixture at rt, and the mixture was stirred at rt for 20 h. The mixture was diluted with EtOAc and then washed with water. The aqueous layer was extracted three times with EtOAc. The combined organic layer washed brine, and then dried over anhydrous Na2SO4. After removal of the solvent in vacuo, the product was purified by CombiFlash® column chromatography eluting with n-hexane–EtOAc gradient to give the desired 18b or 26b.
18b: 29% yield (started with 0.10 mmol of 18a), amorphous, 1H-NMR (δ, CDCl3): 1.34 (3H, t, J = 7.2 Hz, CH2CH3), 1.69 (8H, m, CH2), 1.93 (4H, m, CH2), 3.62 and 3.94 (each 2H, m, OCH2), 3.91 (6H, s, OCH3), 4.29 (2H, q, J = 7.2 Hz, CH2CH3), 5.49 (2H, t, J = 3.1 Hz, OCH), 5.96 and 7.89 (each 1H, d, J = 15.4 Hz, COCH=CH), 7.00 and 7.75 (each 2H, d, J = 15.8 Hz, CH=CH), 7.09 (2H, s, Ar-H), 7.18 (4H, s, Ar-H), ESI-MS (negative, m/z): 633.35 [M−H]−.
26b: 46% yield (started with 1.11 mmol of 26a), amorphous, 1H-NMR (δ, CDCl3): 1.35 (3H, t, J = 7.2 Hz, CH2CH3), 3.95 (3H, s, OCH3), 4.30 (2H, q, J = 7.2 Hz, CH2CH3), 5.95 and 7.88 (each 1H, d, J = 15.6 Hz, COCH=CH), 6.90 and 6.95 (each 1H, d, J = 8.2 Hz, Ar-H), 6.94, 6.96, 7.69 and 7.75 (each 1H, d, J = 15.6 Hz, CH=CH), 7.05 and 7.14 (each 1H, d, J = 1.8 Hz, Ar-H), 7.07 and 7.17 (each 1H, dd, J = 8.2, 1.8 Hz, Ar-H), ESI-MS (positive, m/z): 705.50 [M+H]+.
PPTS (1.2 equiv) was added to a solution of 18b or 26b at rt in EtOH, and the mixture was stirred at rt for 7 h under N2. After removal of the solvent in vacuo, the product was purified by CombiFlash® column chromatography eluting with n-hexane-EtOAc gradient to give the desired 18 or 26.
18: 66% yield (started with 0.03 mmol of 18b), amorphous, 1H-NMR (δ, CDCl3): 1.34 (3H, t, J = 7.2 Hz, CH2CH3), 3.95 (6H, s, OCH3), 4.29 (2H, q, J = 7.2 Hz, CH2CH3), 5.96 and 7.89 (each 1H, d, J = 15.6 Hz, COCH=CH), 6.95 (2H, d, J = 8.2 Hz, Ar-H), 6.97 and 7.75 (each 2H, d, J = 15.4 Hz, CH=CH), 7.05 (2H, d, J = 1.7 Hz, Ar-H), 7.17 (2H, dd, J = 8.2, 1.7 Hz, Ar-H), ESI-MS (positive, m/z): 467.15 [M+H]+.
26: 79% yield (started with 0.51 mmol of 26b), amorphous, 1H-NMR (δ, CDCl3): 1.35 (3H, t, J = 7.2 Hz, CH2CH3), 3.95 (3H, s, OCH3), 4.30 (2H, q, J = 7.2 Hz, CH2CH3), 5.95 and 7.88 (each 1H, d, J = 15.6 Hz, COCH=CH), 6.90 and 6.95 (each 1H, d, J = 8.2 Hz, Ar-H), 6.94, 6.96, 7.69 and 7.75 (each 1H, d, J = 15.6 Hz, CH=CH), 7.05 and 7.14 (each 1H, d, J = 1.8 Hz, Ar-H), 7.07 and 7.17 (each 1H, dd, J = 8.2, 1.8 Hz, Ar-H), ESI-MS (positive, m/z): 453.20 [M+H]+.
General procedure for synthesis of curcumin analogues (19, 24, 25, 27, 30, 32)
HCl (10%) was added to a solution of 3 or 17, 20, 22, 29, 31 in THF, and the mixture was stirred at 70 °C overnight. The mixture was extracted three times with EtOAc. The combined organic layers were washed with H2O and brine. After being dried over anhydrous MgSO4, the solvent was removed in vacuo. The crude products were recrystallized from MeOH or were purified by CombiFlash® column chromatography eluting with n-hexane–EtOAc.
19: 47% yield (started with 0.21 mmol of 3), amorphous, 1H-NMR (δ, CDCl3): 2.34 and 2.45 (each 1H, t, J = 7.0 Hz, CH2), 2.60 and 2.96 (each 1H, m, CH2), 3.92 and 3.95 (each 3H, s, OCH3), 6.69, 7.06, 7.65 and 7.72 (each 1H, d, J = 15.8 Hz, CH=CH), 6.91 and 6.94 (each 1H, d, J = 8.2 Hz, Ar-H), 7.04 and 7.08 (each 1H, d, J = 1.7 Hz, Ar-H), 7.11 and 7.17 (each 1H, dd, J = 8.2, 1.7 Hz, Ar-H), ESI-MS (positive, m/z): 441.20 [M+H]+.
24: 44% yield (started with 0.17 mmol of 22), amorphous, 1H-NMR (δ, acetone-d6): 2.21, 2.37, 2.43 and 3.00 (each 1H, t, J = 7.2 Hz, CH2), 6.79, 7.18, 7.60 and 7.63 (each 1H, d, J = 15.8 Hz, CH=CH), 6.87 and 6.88 (each 1H, d, J = 8.2 Hz, Ar-H), 7.07 and 7.09 (each 1H, dd, J = 8.2, 2.0 Hz, Ar-H), 7.21 and 7.33 (each 1H, d, J = 2.0 Hz, Ar-H), ESI-MS (positive, m/z): 413.00 [M+H]+.
25: 13% yield (started with 0.31 mmol of 17), amorphous, 1H-NMR (δ, acetone-d6+CD3OD): 2.20, 2.31, 2.48 and 2.97 (each 1H, t, J = 7.2 Hz, CH2), 3.89 (3H, s, OCH3), 6.78, 7.27, 7.62 and 7.66 (each 1H, d, J = 15.8 Hz, CH=CH), 6.83 and 6.87 (each 1H, d, J = 8.2 Hz, Ar-H), 7.03 and 7.05 (each 1H, dd, J = 8.2, 1.8 Hz, Ar-H), 7.33 and 7.39 (each 1H, d, J = 1.8 Hz, Ar-H), ESI-MS (negative, m/z): 425.10 [M−H]−.
27: quantitative yield (started with 0.70 mmol of 20), amorphous solid, 1H-NMR (δ, CD3OD): 3.89 (6H, s, OCH3), 4.70 (4H, s, OCH2COOH), 5.99 (1H, s, C=CH), 6.68 and 7.57 (each 2H, d, J = 15.8 Hz, CH=CH), 6.91 (2H, d, J = 8.4 Hz, Ar-H), 7.15 (2H, dd, J = 8.4, 1.9 Hz, Ar-H), 7.26 (2H, d, J = 1.9 Hz, Ar-H), ESI-MS (positive, m/z): 485.15 [M+H]+.
30: 42% yield (started with 0.34 mmol of 29), amorphous solid, 1H-NMR (δ, CD3OD): 2.06 (4H, quintet, J = 7.2 Hz, CH2CH2CH2), 2.49 (4H, t, J = 7.2 Hz, CH2CO), 3.87 (6H, s, OCH3), 4.07 (4H, t, J = 6.2 Hz, OCH2), 5.98 (1H, s, C=CH), 6.65 and 7.57 (each 2H, d, J = 15.8 Hz, CH=CH), 6.96 (2H, d, J = 8.4 Hz, Ar-H), 7.16 (2H, dd, J = 8.4, 2.0 Hz, Ar-H), 7.22 (2H, d, J = 2.0 Hz, Ar-H), ESI-MS (positive, m/z): 541.25 [M+H]+.
32: 65% yield (started with 0.24 mmol of 31), amorphous solid, 1H-NMR (δ, CD3OD): 2.05 (4H, quintet, J = 6.4 Hz, CH2CH2CH2), 2.19 (2H, t, J = 7.2 Hz, CH2CH2CO), 2.33 (2H, d, J = 7.0 Hz, CH2C=C), 2.47 (4H, t, J = 7.2 Hz, CH2CH2CH2CO), 3.83 and 3.89 (each 3H, s, OCH3), 4.13 (4H, t, J = 6.4 Hz, OCH2), 6.84 and 7.68 (each 2H, d, J = 16.0 Hz, CH=CH), 6.96 (2H, d, J = 8.2 Hz, Ar-H), 7.19 (2H, dd, J = 8.2, 2.0 Hz, Ar-H), 7.22 (2H, d, J = 2.0 Hz, Ar-H), ESI-MS (positive, m/z): 613.20 [M+H]+.
Preparation of vanillin derivatives (20a and 29a)
Methyl bromoacetate (3 equiv, for compound 20a) or ethyl 4-bromobutyrate (3 equiv, for compound 29a) and K2CO3 (1.5 equiv) were added to a solution vanillin in DMF, and the mixture was stirred at 80 °C overnight (for compound 20a) or 2 days (for compound 29a) under N2. The mixture was quenched with 0.1N HCl to pH 6 and extracted three times with EtOAc. The combined organic layers were washed with H2O and brine. After being dried over anhydrous MgSO4, the solvent was removed in vacuo. The crude products were purified by CombiFlash® column chromatography eluting with n-hexane–EtOAc.
20a: 93% yield (started with 6.6 mmol of vanillin), white solid, mp. 116 °C, 1H-NMR (δ, CDCl3): 3.82 and 3.96 (each 3H, s, OCH3), 4.79 (2H, s, CH2), 6.88 (1H, d, J = 8.2 Hz, Ar-H), 7.43 (1H, dd, J = 8.2, 1.9 Hz, Ar-H), 7.45 (1H, d, J = 1.9 Hz, Ar-H), 9.87 (1H, s, CHO), ESI-MS (positive, m/z): 247.05 [M+Na]+.
29a: 99% yield (started with 6.6 mmol of vanillin), white solid, mp. 75–76 °C, 1H-NMR (δ, CDCl3): 1.26 (3H, t, J = 7.2 Hz, OCH2CH3), 2.20 (2H, quintet, J = 7.2 Hz, OCH2CH2CH2CO), 2.55 (2H, t, J = 7.2 Hz, OCH2CH2CH2CO), 3.92 (3H, s, OCH3), 4.15 (2H, q, J = 7.2 Hz, OCH2CH3), 4.17 (2H, t, J = 7.2 Hz, OCH2CH2CH2CO), 6.99 (1H, d, J = 8.2 Hz, Ar-H), 7.41 (1H, d, J = 1.7 Hz, Ar-H), 7.44 (1H, dd, J = 8.2, 1.7 Hz, Ar-H), 9.85 (1H, s, CHO), ESI-MS (positive, m/z): 267.05 [M+H]+, 289.00 [M+Na]+.
General procedure for synthesis of symmetric curcumin analogues (20–23, 29, 31, 33–37)
In general, 2,4-pentanedione (0.5 equiv, for compounds 20, 23, 29, 36 and 37) or ethyl 4-acetyl-5-oxo-hexanoate (0.5 equiv, for compounds 21, 22, 31, 33, 34 and 35) and boric anhydride (0.35 equiv) dissolved in EtOAc were stirred for 1 h at 70 °C. The appropriate benzaldehyde (1 equiv) and tributylborate (1 equiv) dissolved in EtOAc were added, and the mixture was stirred for 30 min at 70 °C. n-Butylamine (1 equiv) dissolved in EtOAc was added dropwise over 15 min at 85 °C. Stirring was continued for 4 h at 100 °C. The mixture was then hydrolyzed by adding 1N HCl and heating at 50 °C for 45 min. The organic layer was separated, and the aqueous layer was extracted three times with EtOAc. The combined organic layers were washed with H2O and brine, and then dried over anhydrous Na2SO4, and the solvent was removed in vacuo. The crude products were purified by CombiFlash® column chromatography eluting with n-hexane–EtOAc.
20: 16% yield (started with 1.6 mmol of 20a), amorphous, 1H-NMR (δ, CDCl3): 3.81 and 3.94 (each 6H, s, OCH3), 4.74 (4H, s, CH2), 5.83 (1H, s, C=CH), 6.51 and 7.60 (each 2H, d, J = 15.8 Hz, CH=CH), 6.81 and 7.11 (each 2H, d, J = 8.7 Hz, Ar-H), 7.10 (2H, s, Ar-H), ESI-MS (negative, m/z): 511.15 [M−H]+.
21: 24% yield (started with 4.0 mmol of 20a), amorphous, 1H-NMR (δ, CDCl3): 1.25 (3H, t, J = 7.2 Hz, CH2CH3), 2.54 and 2.95 (each 2H, dd, J = 8.2, 7.2 Hz, CH2), 3.808, 3.811, 3.94 and 3.96 (each 3H, s, OCH3), 4.14 (2H, q, J = 7.2 Hz, CH2CH3), 4.74 and 4.75 (each 2H, s, OCH2), 6.51, 7.01, 7.59 and 7.71 (each 1H, d, J = 15.8 Hz, CH=CH), 6.81 and 6.82 (each 1H, d, J = 8.7 Hz, Ar-H), 7.10 and 7.15 (each 1H, d, J = 1.9 Hz, Ar-H), 7.11 and 7.16 (each 1H, dd, J = 8.7, 1.9 Hz, Ar-H), ESI-MS (negative, m/z): 611.40 [M−H]+.
22: 3% yield (started with 7.2 mmol of 3,4-dihydroxybenzaldehyde), amorphous, 1H-NMR (δ, CDCl3+CD3OD): 1.27 (3H, t, J = 7.0 Hz, CH2CH3), 2.28, 2.37, 2.54 and 2.91 (each 1H, m, CH2), 4.13 (2H, q, J = 7.0 Hz, CH2CH3), 6.64, 6.92, 7.59 and 7.64 (each 1H, d, J = 15.8 Hz, CH=CH), 6.82 and 6.85 (each 1H, d, J = 8.2 Hz, Ar-H), 7.02 and 7.08 (each 1H, dd, J = 8.2, 1.9 Hz, Ar-H), 7.08 and 7.13 (1H, d, J = 1.9 Hz, Ar-H), ESI-MS (positive, m/z): 441.05 [M+H]+.
23: 6% yield (started with 22 mmol of 3,4-dihydroxybenzaldehyde), amorphous solid; 1H-NMR (δ, acetone-d6): 5.94 (1H, s, C=CH), 6.56 and 7.53 (each 2H, d, J = 15.8 Hz, CH=CH), 6.88 (2H, d, J = 8.2 Hz, Ar-H), 7.04 (2H, d, J = 8.2 Hz, Ar-H), 7.17 (2H, s, Ar-H), ESI-MS (negative, m/z): 339.05 [M−H]−.
29: 9% yield (started with 12 mmol of 29a), amorphous solid; 1H-NMR (δ, CDCl3): 1.26 (6H, t, J = 7.0 Hz, OCH2CH3), 2.17 (4H, quintet, J = 7.2 Hz, CH2CH2CH2), 2.54 (4H, t, J = 7.2 Hz, CH2CO), 3.91 (6H, s, OCH3), 4.11 (4H, t, J = 7.2 Hz, OCH2), 4.15 (4H, q, J = 7.0 Hz, OCH2CH3), 5.82 (1H, s, C=CH), 6.49 and 7.60 (each 2H, d, J = 15.8 Hz, CH=CH), 6.89 (2H, d, J = 8.4 Hz, Ar-H), 7.07 (2H, d, J = 1.7 Hz, Ar-H), 7.12 (2H, dd, J = 8.4, 1.7 Hz, Ar-H), ESI-MS (positive, m/z): 597.25 [M+H]+.
31: 8% yield (started with 3.8 mmol of 29a), amorphous solid; 1H-NMR (δ, CDCl3): 1.26 (9H, t, J = 7.0 Hz, OCH2CH3), 2.18 (4H, quintet, J = 7.2 Hz, CH2CH2CH2), 2.54 (6H, t, J = 7.2 Hz, CH2CO), 2.95 (2H, dd, J = 8.6, 7.0 Hz, CH2C=C), 3.93 (6H, s, OCH3), 4.13 (4H, t, J = 7.2 Hz, OCH2), 4.15 (6H, q, J = 7.0 Hz, OCH2CH3), 6.90 (2H, d, J = 8.4 Hz, Ar-H), 6.99 and 7.72 (each 2H, d, J = 15.2 Hz, CH=CH), 7.13 (2H, d, J = 1.7 Hz, Ar-H), 7.17 (2H, dd, J = 8.4, 1.7 Hz, Ar-H). ESI-MS (positive, m/z): 697.30 [M+H]+.
33: 5% yield (started with 3.0 mmol of 3,4-Dimethoxybenzaldehyde), amorphous, 1H-NMR (δ, CDCl3): 1.25 (3H, t, J = 6.9 Hz, OCH2CH3), 2.48 (2H, dd, J = 8.3, 7.5 Hz, CH2CO), 2.78 (2H, dd, J = 8.3, 7.5 Hz, CH2C=C), 3.92 and 3.93 (each 6H, s, OCH3), 4.13 (2H, q, J = 6.9 Hz, OCH2CH3), 6.69 and 7.64 (each 2H, d, J = 15.8 Hz, CH=CH), 6.89 (2H, d, J = 8.3 Hz, Ar-H), 7.08 (2H, d, J = 2.0 Hz, Ar-H), 7.17 (2H, dd, J = 8.3, 2.0 Hz, Ar-H), EI-MS (m/z): 496 [M]+.
34: 15% yield (started with 2.8 mmol of 5-hydroxy-3,4-Dimethoxybenzaldehyde), amorphous, 1H-NMR (δ, CDCl3): 1.25 (3H, t, J = 7.0 Hz, OCH2CH3), 2.53 (2H, dd, J = 8.0, 7.5 Hz, CH2C=C), 2.93 (2H, dd, J = 8.0, 7.5 Hz, CH2CO), 3.92 and 3.95 (each 6H, s, OCH3), 4.15 (2H, q, J = 7.0 Hz, OCH2CH3), 6.70 and 6.90 (each 2H, d, J = 1.9 Hz, Ar-H), 7.00 and 7.65 (each 2H, d, J = 15.3 Hz, CH=CH), EI-MS (m/z): 528 [M]+.
35: 15% yield (started with 2.7 mmol of 3,5-Dimethoxy-4-hydroxybenzaldehyde), amorphous, 1H-NMR (δ, CDCl3): 1.23 (3H, t, J = 7.0 Hz, OCH2CH3), 2.56 (2H, t, J = 7.3 Hz, CH2C=C), 2.96 (2H, t, J = 7.3 Hz, CH2CO), 3.96 (12H, s, OCH3), 4.13 (2H, q, J = 7.0 Hz, OCH2CH3), 6.80 and 6.86 (each 2H, s, Ar-H), 6.99 and 7.70 (each 2H, d, J = 15.3 Hz, CH=CH), EI-MS (m/z): 528 [M]+.
36: 3% yield (started with 2.7 mmol of 2-fluoro-4,5-dimethoxybenzaldehyde), amorphous solid, 1H-NMR (δ, CDCl3): 3.91 (12H, s, OCH3), 5.87 (1H, s, C=CH), 6.59 and 7.73 (each 2H, d, J = 16.1 Hz, CH=CH), 6.66 (2H, d, J = 11.7 Hz, Ar-H), 6.98 (2H, d, J = 6.8 Hz, Ar-H), EI-MS (m/z): 431 [M−H]+.
37: 8% yield (started with 2.7 mmol of 5-hydroxy-3,4-dimethoxybenzaldehyde), amorphous solid, 1H-NMR (δ, CDCl3): 3.88 and 3.94 (each 6H, s, OCH3), 5.79 (1H, s, C=CH), 6.48 and 7.51 (each 2H, d, J = 16.1 Hz, CH=CH), 6.63 and 6.83 (each 2H, d, J = 1.9 Hz, Ar-H), EI-MS (m/z): 427 [M−H]+.
Synthesis of compound (28)
Aniline (0.04 mL, 0.44 mmol), EDCI (45 mg, 0.24 mmol) and DMAP (29 mg, 0.23 mmol) were added to a solution of 27 (57 mg, 0.12 mmol) in CH2Cl2 (5 mL), and the mixture was stirred at 40 °C for 22 h under N2. The mixture was diluted with CH2Cl2 and then washed with sat. NH4Cl (aq.), sat. NaHCO3 (aq.), and brine, then dried over anhydrous Na2SO4. After removal of the solvent in vacuo, the product was purified by CombiFlash® column chromatography eluting with n-hexane–EtOAc gradient to give the desired 28 (26 mg, 35%).
28: amorphous solid, 1H-NMR (δ, CDCl3): 4.00 (6H, s, OCH3), 4.68 (4H, s, OCH2CO), 5.84 (1H, s, C=CH), 6.53 and 7.61 (each 2H, d, J = 15.8 Hz, CH=CH), 6.98 (2H, d, J = 8.2 Hz, Ar-H), 7.13 (2H, d, J = 1.9 Hz, Ar-H), 7.16 (2H, t, J = 8.4 Hz, Ar-H), 7.17 (2H, dd, J = 8.2, 1.9 Hz, Ar-H), 7.36 (4H, t, J = 8.4 Hz, Ar-H), 7.60 (4H, d, J = 8.4 Hz, Ar-H), 8.76 (2H, s, NH), ESI-MS (positive, m/z): 635.20 [M+H]+.
5.2. Biology
5.2.1. Cell lines
The human lung adenocarcinoma cell line CL1-5 was established previously.16 H1975 was obtained from American Type Culture Collection (ATCC) (Manassas, VA). These cells were maintained in RPMI-1640 medium containing 10% FBS (Life Technologies) with penicillin and streptomycin (100 mg/mL) at 37 °C in a humidified atmosphere of 5% CO2. The IEC-18 rat intestinal epithelial cell line was obtained from Bioresource Collection and Research Center (BCRC, Taiwan) and maintained in DMEM containing 10% FBS (Life Technologies) with penicillin and streptomycin (100 mg/mL).
5.2.2. Western blot analysis
Human tumor cells were plated in 10-cm dish at a density of 1 × 106 cells/well and incubated overnight in culture medium containing 10% FBS, and then cultured for 24 h in serum-free medium. Cells were treated with 10 μM curcumin analogs for 1 h in a serum-free condition followed by stimulation with 20 ng/mL EGF (Sigma, MO) for 20 min. Whole cell lysates were prepared by protein extraction reagent for mammalian cells (Pierce, Rockford) in the presence of protease inhibitors and phosphatase inhibitors (Sigma, MO). Solubilized protein concentration was determined after clarified by centrifugation using BCA method (Pierce, Rockford). Proteins were separated by 10% SDS-polyacrylamide gel electrophoresis (SAD-PAGE) (40 μg/lane), and then proteins were transferred to the PVDF membrane. Antibodies to pan-EGFR and phosphorylated (Tyr1068) EGFR were purchased from Santa Cruz Biotechnology (Santa Cruz, CA) and Cell Signaling Technology (Beverly, MA), respectively. Bound primary antibodies were detected using Enhanced Chemiluminescence System (Santa Cruz, CA), and chemiluminescent signals were captured by LAS 3000 system (Fujifilm, Tokyo, Japan).4
5.2.3. Proliferation assay
Stock solutions of curcumin analogs and gefitinib were prepared in DMSO and stored at −20 °C. The compounds were diluted in fresh media before each experiment, and the final DMSO concentration was lower than 0.1%. Antiproliferative activities of compounds were determined by MTT [3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide] (Sigma, St Louis, MO) assay. IEC-18 cells were plated in 96-well plates at a density of 5 × 103 cells/well. The IEC-18 cells were treated with curcumin analogs and gefitinib for 24 h followed by addition of 0.5 mg/mL MTT solution into the culture medium. After a further 1.5 h of incubation with MTT, the medium was removed and DMSO was added to the plates. The color intensity was measured at 570 nm using a multi-label plate reader (Vector3; Perkin-Elmer, USA). Assays were performed in triplicate and calculated IC50 shown as an average with standard deviation (SD).
Acknowledgements
This work was supported by Taiwan Ministry of Science and Technology (NSC 102-2325-B-039-005) awarded to Sheng-Chu Kuo, and by National Cancer Institute Grant CA177584 (USA) awarded to K. H. Lee. Authors are thankful to Dr. Susan Morris-Natschke (UNC) for the critical editing of this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Gupta SC, Prasad S, Kim JH, Patchva S, Webb LJ, Priyadarsini IK, Aggarwal BB. Nat. Prod. Rep. 2011;28:1937. doi: 10.1039/c1np00051a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen HW, Huang HC. Br. J. Pharmacol. 1998;124:1029. doi: 10.1038/sj.bjp.0701914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen HW, Lee JY, Huang JY, Wang CC, Chen WJ, Su SF, Huang CW, Ho CC, Chen JJ, Tsai MF, Yu SL, Yang PC. Cancer Res. 2008;68:7428. doi: 10.1158/0008-5472.CAN-07-6734. [DOI] [PubMed] [Google Scholar]
- 4.Chen HW, Yu SL, Chen JJ, Li HN, Lin YC, Yao PL, Chou HY, Chien CT, Chen WJ, Lee YT, Yang PC. Mol. Pharmacol. 2004;65:99. doi: 10.1124/mol.65.1.99. [DOI] [PubMed] [Google Scholar]
- 5.Lee JY, Lee YM, Chang GC, Yu SL, Hsieh WY, Chen JJ, Chen HW, Yang PC. PloS One. 2011;6:e23756. doi: 10.1371/journal.pone.0023756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perez-Soler R. The Oncologist. 2004;9:58. doi: 10.1634/theoncologist.9-1-58. [DOI] [PubMed] [Google Scholar]
- 7.Kari C, Chan TO, Rocha de Quadros M, Rodeck U. Cancer Res. 2003;63:1. [PubMed] [Google Scholar]
- 8.Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. New Engl. J. Med. 2005;352:786. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 9.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Masuda T, Matsumura H, Oyama Y, Takeda Y, Jitoe A, Kida A, Hidaka K. J. Nat. Prod. 1998;61:609. doi: 10.1021/np970555g. [DOI] [PubMed] [Google Scholar]
- 11.Miyashita N, Yoshikoshi A, Grieco PA. J. Org. Chem. 1977;42:3. doi: 10.1021/jo00423a022. [DOI] [PubMed] [Google Scholar]
- 12.Pedersen U, Rasmussen PB, Lawesson SO. Liebigs Ann. Chem. 1985:1557. [Google Scholar]
- 13.Lin L, Shi Q, Su CY, Shih CC, Lee KH. Bioorg. Med. Chem. 2006;14:2527. doi: 10.1016/j.bmc.2005.11.034. [DOI] [PubMed] [Google Scholar]
- 14.Lin L, Shi Q, Nyarko AK, Bastow KF, Wu CC, Su CY, Shih CC, Lee KH. J. Med. Chem. 2006;49:3963. doi: 10.1021/jm051043z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ishida J, Ohtsu H, Tachibana Y, Nakanishi Y, Bastow KF, Nagai M, Wang HK, Itokawa H, Lee KH. Bioorg. Med. Chem. 2002;10:3481. doi: 10.1016/s0968-0896(02)00249-3. [DOI] [PubMed] [Google Scholar]
- 16.Chu YW, Yang PC, Yang SC, Shyu YC, Hendrix MJ, Wu R, Wu CW. Am. J. Respir. Cell Mol. Biol. 1997;17:353. doi: 10.1165/ajrcmb.17.3.2837. [DOI] [PubMed] [Google Scholar]