

Abstract
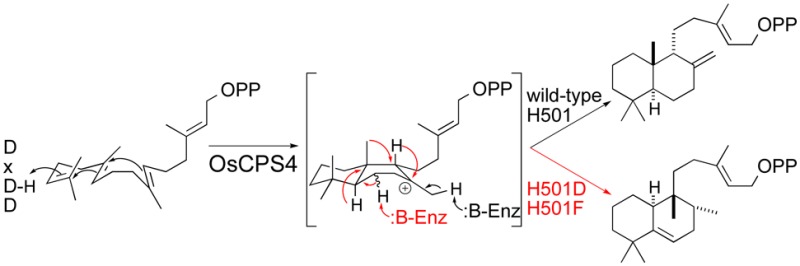
Through site-directed mutagenesis targeted at identification of the catalytic base in the rice (Oryza sativa) syn-copalyl diphosphate synthase OsCPS4, changes to a single residue (H501) were found to induce rearrangement rather than immediate deprotonation of the initially formed bicycle, leading to production of the novel compound syn-halimadienyl diphosphate. These mutational results are combined with quantum chemical calculations to provide insight into the underlying reaction mechanism.
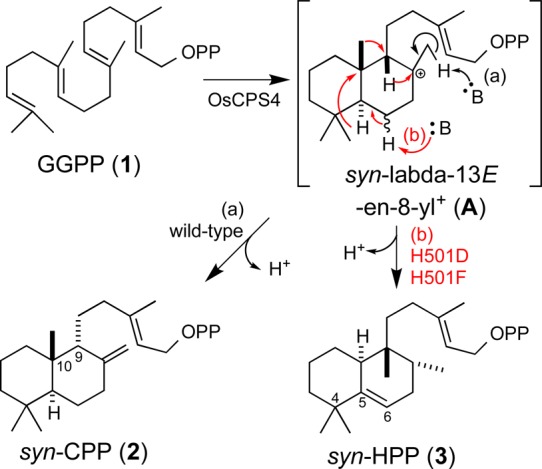
Labdane-related diterpenoids are a large and structurally varied set of natural products, and over 7000 of these compounds are known.1 These natural products are unified by their biosynthetic origins, particularly their characteristic trans-decalin core ring structure, which is formed by class II diterpene cyclases (DTCs). These enzymes catalyze protonation-initiated (bi)cyclization of the general diterpene precursor, (E,E,E)-geranylgeranyl diphosphate (GGPP, 1), with direct deprotonation of the resulting labda-13E-en-8-yl+ intermediate at the C8 methyl substituent yielding copalyl diphosphate (CPP). There are four stereoisomers of CPP depending on the prochiral conformation 1 is folded into before bicyclization occurs. One such stereoisomer is termed syn-CPP (2) (9R,10S), as it is formed from a pro-chair-boat configuration of 1 that leads to syn orientation of the C-9 hydrogen and C-10 methyl substituents (Scheme 1).1
Scheme 1. Cyclization of GGPP (1) to syn-CPP (2) or syn-HPP (3) by Wild-Type or H501 Mutants of OsCPS4, Respectively.
While all vascular plants contain a DTC to produce the ent-CPP (9R,10R) required for gibberellin phytohormone biosynthesis,2 the only plant CPP synthase (CPS) shown to catalyze formation of 2 is OsCPS4.3 This CPS is involved in the production of more specialized labdane-related diterpenoids (i.e., secondary metabolites), such as the allelochemical momilactones4,5 and antifungal phytoalexins.6 Nevertheless, OsCPS4 is phylogenetically related to the ent-CPP producing plant CPSs from in gibberellin biosynthesis.3 Thus, investigation of OsCPS4 represents an opportunity to investigate the enzymatic structure–function relationships underlying its conversion to production of the stereochemically differentiated syn-CPP (2), as well as DTC catalysis more generally.
The DTCs utilize a general acid–base catalytic mechanism, initiating bicyclization by protonating the terminal carbon–carbon double bond (C=C) of 1. This triggers a carbocation cascade, with sequential anti addition from the two internal C=C leading to the bicyclic intermediate labda-13E-en-8-yl+, which can be rearranged by sequential 1,2-shifts of hydride and methyl groups, with the C10 → C9 methyl migration forming the halimadane skeletal structure and subsequent C4 → C5 methyl migration forming the clerodane backbone (Scheme S1).1 The final carbocation seems to be invariably quenched by deprotonation, although this may be preceded by the addition of water, resulting in a hydroxylated product. Notably, while DTCs/CPSs are characterized by a highly conserved (D,E)xDD motif from which the middle aspartic acid acts as the catalytic acid,7 the catalytic base does not appear to be nearly as well conserved. This may be due in part to variation in positioning of the base, relative to the acid, underlying the production of different (stereo)-isomers of CPP, as well as the addition of water and/or rearrangement, by various DTCs.
The catalytic base for the plant CPSs involved in gibberellin phytohormone biosynthesis has been recently identified as dependent on a histidine-asparagine dyad that is specifically conserved in these ent-CPP producing DTCs.8 By contrast, the catalytic base in the DTC active site of the diterpene synthases involved in conifer resin acid biosynthesis has been identified as a very similarly positioned tyrosine-histidine dyad, which is specifically conserved in these normal CPP (9S,10S) producing enzymes.9,10 In both cases, alanine substitution for the histidines leads to the addition of water, with the mutant DTCs yielding hydroxylated derivatives of CPP.8,9
Consistent with its closer phylogenetic relationship to other CPSs, OsCPS4 contains the histidine (H251) of the CPS catalytic base dyad, although the other position is a cysteine (C310) instead of asparagine. However, alanine substitution for this histidine did not eliminate activity, nor alter product outcome, with expression of the OsCPS4:H251A mutant in E. coli engineered to produce 1 via a modular metabolic engineering system11 still leading to the exclusive production of 2 (Figure S1). Similarly, alanine substitution for the cysteine also does not significantly alter activity (i.e., OsCPS4:C310A also produces 2, Figure S1). In addition, while substitution of aspartate for the catalytic histidine in the DTC active site of the diterpene synthases involved in conifer resin acid biosynthesis also leads to incorporation of water,9 such substitution in OsCPS4 simply reduces catalytic activity (i.e., OsCPS4:H251D produces only trace amounts of 2, Figure S1). Accordingly, OsCPS4 appears to use a novel general base, consistent with the unique stereochemistry of its product.
The identity of the catalytic base in OsCPS4 was further investigated by protein structure modeling. A model was constructed on the basis of the most closely related known crystal structure, that of the CPS from Arabidopsis thaliana (AtCPS),12,13 using the SWISS-MODEL server.14−17 The previously identified catalytic base groups both include histidines, which were found across the active site from the DxDD motif. In addition to H251, two other similarly positioned histidines (H275 and H501) are present in the modeled OsCPS4 active site (Figure 1A).
Figure 1.
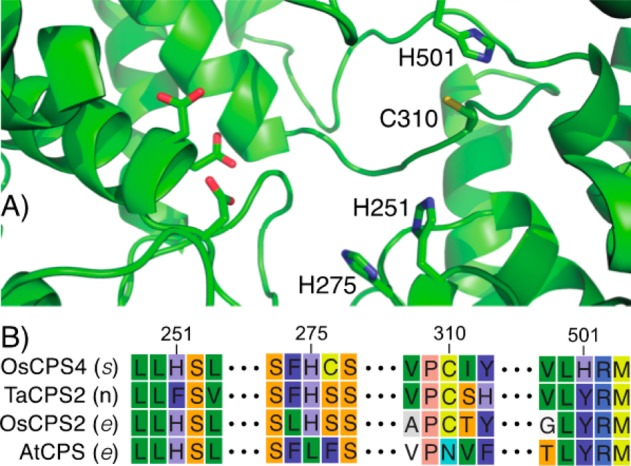
OsCPS4 active site. (A) From modeled structure, shown in cartoon format with stick representation of the side chains of the aspartate residues of the DxDD motif (on the left) and the targeted histidines and cysteine (on the right), created using PyMol.18 (B) Conservation of targeted residues in selected CPSs discussed in text, with stereochemistry of CPP products as indicated (s, syn; n, normal; e, ent), numbering as in panel A (i.e., from OsCPS4).
Notably, while substitution of either alanine or aspartate for H275 did not alter product outcome, and only slightly reduced overall yield (Figure S2), such substitutions for H501 had a clear effect (Figure 2A–C). The OsCPS4:H501A mutant was found to exhibit both reduced yield and a mixture of 2 with an unidentified product whose mass spectra indicated that it was not a hydroxylated version of 2 (i.e., from incorporation of water), as the molecular weight of the dephosphorylated derivative appeared to be 290 (equivalent to that for 2; Figure 2D). With the OsCPS4:H501D mutant, although 2 is still observed and the overall yield lags that observed with the wild-type enzyme (i.e., substantial amounts of 1 are still present), this compound was produced in much higher amounts, enabling purification and structural analysis of the dephosphorylated derivative by NMR (Table S1, Figures S3–S5). The compound was found to be a halima-5,13E-dien-15-ol whose stereochemical configuration indicates its derivation from rearrangement of syn-labda-13E-en-8-yl+ (A, for structure see Scheme 1). Accordingly, the enzymatic product is termed here syn-halima-5,13E-dienyl diphosphate (syn-HPP, 3). Given that substitution of phenylalanine and, especially, tyrosine for the catalytic histidine in AtCPS has recently been shown to result in product rearrangement,19 analogous mutants were constructed for OsCPS4. Strikingly, these had contrasting effects, with phenylalanine substitution exhibiting the expected production of 3, with improved yield relative to the aspartate mutant (both overall and as an increased proportion of the product mix), while tyrosine substitution does not alter the production of 2, which is observed in good yield, with very little 1 left (Figure 2E and F).
Figure 2.

Effect of various substitutions for H501 on OsCPS4 product outcome. GC-MS chromatograms of the dephosphorylated products extracted from OsCPS4 (wild-type and indicated mutants) in E. coli engineered to produce 1, along with mass spectra for the observed novel product (numbering as in text, with prime ′ notation used to indicate that these are dephosphorylated derivatives).
Interestingly, H501 occupies a position that is otherwise invariably conserved as a tyrosine in plant DTCs, highlighting its functional role in the stereochemically unique reaction catalyzed by OsCPS4. By contrast, the position occupied by H275 is more variable. While this residue is generally an (iso)leucine in most plant DTCs, histidine can be found at this position in other closely related CPSs that produce different stereoisomers of CPP (Figure 1B), both the ent-CPP producing OsCPS2 from rice20 and the normal CPP producing TaCPS2 from wheat (Triticum aestivum).21 However, although it is possible that H501 acts as the catalytic base, the observed mixture of effects on product outcome from various substitutions differs from those previously observed for the catalytic bases in other DTCs,8,9,19 which seems to preclude clear assignment of the role H501 plays in shaping product outcome.
It is, further, not immediately evident why 3 is the observed alternative product. This is formed via rearrangement of A to a halimadienyl+ intermediate, with no C4 → C5 methyl migration to the clerodane backbone observed. Production of 3 contrasts with the effect of aromatic substitution for the catalytic histidine in AtCPS, where such further rearrangement is observed, with removal of the same proton originally added to initiate the reaction, presumably by returning to the same aspartate that acted as the catalytic general acid.19 However, quantum chemical calculations indicated that, at least with the relevant ent-labda-13E-en-8-yl+ intermediate, this last 1,2-methyl shift is much more energetically difficult than the preceding 1,2-shifts. Both the transition state structures and resulting tertiary carbocation intermediates for these 1,2-shifts are within ∼7 kcal mol–1 of the initial ent-labda-13E-en-8-yl+ intermediate, while the final C4 → C5 methyl shift has an energetic barrier of ∼14 kcal mol–1 and the resulting ent-cleroda-13E-en-4-yl+ intermediate is significantly higher in energy than the other intermediates.19 Moreover, the introduced aromatic group is relatively distant and, thus, seems unlikely to drive this last methyl shift, such that production of the clerodane skeletal structure by these AtCPS mutants is then likely driven by the lack of suitable general bases for deprotonation of any of the other, relatively more stable preceding intermediates.
Previous semiempirical quantum chemical calculations suggested that the various tertiary carbocation intermediates in rearrangement of syn-labda-13E-en-8-yl+ (A) are all of similar energy (spanning a range of only 5 kcal mol–1). Specifically, the 9-yl+B (see Figure 3) formed by initial 1,2-hydride migration is predicted to be <1 kcal mol–1 lower in energy than A, and the syn-halima-13E-en-10-yl+C formed by subsequent 1,2-methyl migration is predicted to be 4 kcal mol–1 lower in energy than A and essentially isoenergetic with the syn-halimad-13E-en-5-yl+D formed by subsequent 1,2-hydride migration, while the syn-cleroda-13E-en-4-yl+E formed by the final 1,2-methyl migration is predicted to be only 1 kcal mol–1 higher in energy than A.22 Here quantum chemical calculations, using density functional theory (DFT) methods previously applied to characterization of terpene carbocation rearrangements (see Supporting Information for details)23,24 and with proven utility,25 were used to investigate not only these intermediates but also the corresponding transition state structures for their interconversion (Figure 3, numbers in normal text and brackets; see also Figure S6). This analysis was broadly supportive of the previously reported results, although the relative stability of A appears to have been overestimated by the utilized semiempirical method (Figure 3). In addition, the barrier for the final 1,2-methyl shift is predicted to be smaller than that found previously for rearrangement of ent-labda-13E-en-8-yl+,19 both the barrier from D (lower by ∼4 kcal mol–1) and the overall barrier from A (lower by ∼12 kcal mol–1).26 Accordingly, there is not a large energetic barrier to this final 1,2-methyl shift, indicating that there is a suitably positioned general base in the OsCPS4 active site for deprotonation of D (note that this would not interfere with initial bicyclization, as this does not proceed via a corresponding 5-yl+ intermediate), which alleviates any need to undergo full rearrangement to E to allow for removal of the initiating proton, unlike what is observed upon aromatic replacement of the catalytic histidine in AtCPS.19 Identification of this otherwise “silent” general base, as well as elucidation of the exact role of H501, will be the subject of future investigation and may require determination of a high-resolution structure for OsCPS4.
Figure 3.

Energies of carbocation minima and transition-state structures involved in hydride and methyl shifts en route to 3. Relative energies (B3LYP/6-31+G(d,p) in normal text, mPW1PW91/6-31+G(d,p)//B3lYP/31+G(d,p) [in brackets], and PM3 from ref (22) underlined) are shown in kcal mol–1; in this model, R = (CH2)2C(Me)=CHCH2OPO3H2.
Regardless of the exact mechanism underlying this observed change in product outcome, it is notable that these OsCPS4:H501 mutants appear to represent the first identified syn-HPP synthase, which then offers access to derived natural products. For example, 3 is likely to be the precursor to various labdane-related diterpenoids isolated from Vitex agnus and Vitex rotundifolia, which are Chinese medicinal plants that have been widely used in Korea, China, and Japan for the treatment of inflammation, headache, migraine, chronic bronchitis, eye pain, and gastrointestinal infections.27,28 Finally, these studies further illustrate the plasticity of DTCs, which underlies the expansive evolution of the labdane-related diterpenoid superfamily of natural products.
Acknowledgments
This work was supported by grants from the NIH (GM076324 to R.J.P.) and NSF (CHE-1361807 to D.J.T.).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.orglett.6b00181.
Description of experimental methods; GC-MS chromatograms for other OsCPS4 mutants described; NMR chemical shift data and spectra for 3′; Details of quantum chemical calculations (PDF)
Author Present Address
§ Department of Biology, University of North Carolina, Chapel Hill, North Carolina 27599, United States.
The authors declare no competing financial interest.
Supplementary Material
References
- Peters R. J. Nat. Prod. Rep. 2010, 27, 1521–1530. 10.1039/c0np00019a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zi J.; Mafu S.; Peters R. J. Annu. Rev. Plant Biol. 2014, 65, 259–286. 10.1146/annurev-arplant-050213-035705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M.; Hillwig M. L.; Prisic S.; Coates R. M.; Peters R. J. Plant J. 2004, 39, 309–318. 10.1111/j.1365-313X.2004.02137.x. [DOI] [PubMed] [Google Scholar]
- Xu M.; Galhano R.; Wiemann P.; Bueno E.; Tiernan M.; Wu W.; Chung I. M.; Gershenzon J.; Tudzynski B.; Sesma A.; Peters R. J. New Phytol. 2012, 193, 570–5. 10.1111/j.1469-8137.2011.04005.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato-Noguchi H.; Peters R. J. J. Chem. Ecol. 2013, 39, 175–185. 10.1007/s10886-013-0236-9. [DOI] [PubMed] [Google Scholar]
- Schmelz E. A.; Huffaker A.; Sims J. W.; Christensen S. A.; Lu X.; Okada K.; Peters R. J. Plant J. 2014, 79, 659–678. 10.1111/tpj.12436. [DOI] [PubMed] [Google Scholar]
- Prisic S.; Xu J.; Coates R. M.; Peters R. J. ChemBioChem 2007, 8, 869–874. 10.1002/cbic.200700045. [DOI] [PubMed] [Google Scholar]
- Potter K.; Criswell J.; Peters R. J. Angew. Chem., Int. Ed. 2014, 53, 7198–7202. 10.1002/anie.201402911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criswell J.; Potter K.; Shephard F.; Beale M. B.; Peters R. J. Org. Lett. 2012, 14, 5828–5831. 10.1021/ol3026022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mafu S.; Potter K. C.; Hillwig M. L.; Schulte S.; Criswell J.; Peters R. J. Chem. Commun. (Cambridge, U. K.) 2015, 51, 13485–7. 10.1039/C5CC05754J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyr A.; Wilderman P. R.; Determan M.; Peters R. J. J. Am. Chem. Soc. 2007, 129, 6684–6685. 10.1021/ja071158n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köksal M.; Hu H.; Coates R. M.; Peters R. J.; Christianson D. W. Nat. Chem. Biol. 2011, 7, 431–3. 10.1038/nchembio.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köksal M.; Potter K.; Peters R. J.; Christianson D. W. Biochim. Biophys. Acta, Gen. Subj. 2014, 1840, 184–90. 10.1016/j.bbagen.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold K.; Bordoli L.; Kopp J.; Schwede T. Bioinformatics 2006, 22, 195–201. 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- Biasini M.; Bienert S.; Waterhouse A.; Arnold K.; Studer G.; Schmidt T.; Kiefer F.; Cassarino T. G.; Bertoni M.; Bordoli L.; Schwede T. Nucleic Acids Res. 2014, 42, W252–8. 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guex N.; Peitsch M. C.; Schwede T. Electrophoresis 2009, 30 (Suppl. 1), S162–73. 10.1002/elps.200900140. [DOI] [PubMed] [Google Scholar]
- Kiefer F.; Arnold K.; Künzli M.; Bordoli L.; Schwede T. Nucleic Acids Res. 2009, 37, D387–92. 10.1093/nar/gkn750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stofer Vogel B.; Wildung M. R.; Vogel G.; Croteau R. J. Biol. Chem. 1996, 271, 23262–23268. 10.1074/jbc.271.38.23262. [DOI] [PubMed] [Google Scholar]
- Potter K. C.; Zi J.; Hong Y. J.; Schulte S.; Malchow B.; Tantillo D. J.; Peters R. J. Angew. Chem., Int. Ed. 2016, 55, 634–638. 10.1002/anie.201509060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prisic S.; Xu M.; Wilderman P. R.; Peters R. J. Plant Physiol. 2004, 136, 4228–4236. 10.1104/pp.104.050567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.; Zhou K.; Toyomasu T.; Sugawara C.; Oku M.; Abe S.; Usui M.; Mitsuhashi W.; Chono M.; Chandler P. M.; Peters R. J. Phytochemistry 2012, 84, 40–46. 10.1016/j.phytochem.2012.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi T.; Dekishima Y.; Hamano Y.; Dairi T.; Seto H.; Kakinuma K. J. Org. Chem. 2003, 68, 5433–8. 10.1021/jo026728a. [DOI] [PubMed] [Google Scholar]
- Tantillo D. J. Nat. Prod. Rep. 2011, 28, 1035–53. 10.1039/c1np00006c. [DOI] [PubMed] [Google Scholar]
- This is part 12 in our series on quantum calculations on diterpene-forming carbocation rearrangements; for part 11, see ref (19).
- Zu L.; Xu M.; Lodewyk M. W.; Cane D. E.; Peters R. J.; Tantillo D. J. J. Am. Chem. Soc. 2012, 134, 11369–71. 10.1021/ja3043245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conversion to an alternative conformer of D leads to a barrier that is lower still (by an additional 1.2 kcal mol–1), but whether the necessary conformational change could occur in the active site is not clear. See Supporting Information for details.
- Lee C.; Lee J. W.; Jin Q.; Lee H. J.; Lee S. J.; Lee D.; Lee M. K.; Lee C. K.; Hong J. T.; Hwang B. Y. Bioorg. Med. Chem. Lett. 2013, 23, 6010–4. 10.1016/j.bmcl.2013.08.004. [DOI] [PubMed] [Google Scholar]
- Ono M.; Yamasaki T.; Konoshita M.; Ikeda T.; Okawa M.; Kinjo J.; Yoshimitsu H.; Nohara T. Chem. Pharm. Bull. 2008, 56, 1621–4. 10.1248/cpb.56.1621. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.