

Abstract
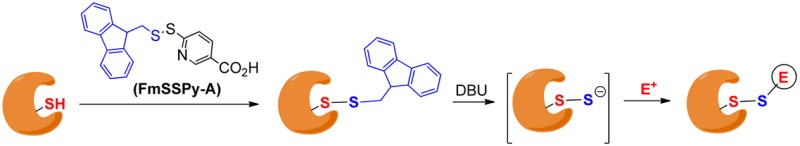
The development of a functional disulfide, FmSSPy-A (Fm = 9-fluorenylmethyl; Py = pyridinyl), is reported. It can effectively convert small molecule and protein thiols (−SH) to form −S-SFm adducts under mild conditions. This method allows for a H2S-free and biomimetic protocol to generate highly reactive persulfides (in their anionic forms). The high nucleophilicity of persulfides toward a number of thiol-blocking reagents is also demonstrated. The method holds promise for further understanding the chemical biology of persulfides and S-sulfhydration.
Hydrogen sulfide (H2S) is a biologically important signaling molecule that exerts diverse roles in various physiological and/or pathological processes.1 The functions of H2S are mediated by its many biological reactions. Among them protein S-sulfhydration is particularly attractive. This reaction has been recognized as an important oxidative posttranslational modification for cell signaling. It provides a possible mechanism by which H2S or H2S-derived reactive sulfur species alter the functions of a wide range of cellular proteins and enzymes.2 To date, the chemical foundation of S-sulfhydration is still unclear. The chemistry and chemical biology of the sulfhydration products, i.e. persulfides (RSSH), are still poorly understood.3 This is primarily due to the instability of RSSH. Moreover, the biological importance of RSSH was only recently appreciated due to their links to H2S signaling.4
Methods that allow easy and reliable access to RSSH (both in small molecules and proteins) are critical for the study of RSSH and S-sulfhydration. For biologically important RSSH (such as GSSH and protein-SSH), the common preparation method is the reaction between a disulfide (RSSR) and H2S (to form a RSSH and a RSH, Scheme 1a).5 However, this reaction requires high concentrations of the reactants, which is not biologically relevant. In addition the reaction is under equilibrium, thus H2S and RSH always remain in the solutions. This makes it very difficult (unless the remaining H2S and RSH can be completely removed) to analyze the property of RSSH exclusively (as H2S and thiol may have similar reactivity). For small molecule RSSH, currently the standard method is to convert thiols to acylated disulfides and then hydrolysis (by HCl/MeOH) to produce RSSH (Scheme 1b). In this method, the hydrolysis appears to be slow (usually needs >12 h). This long process of RSSH formation is not suitable for the study of RSSH. For example, although RSSH are believed to be stronger nucleophiles than their thiol analogues (RSH), the reactions between RSSH and thiol blocking reagents rarely give informative results.3,6
Scheme 1. Common Methods for Persulfide Formation.

Because of the aforementioned problems, it is necessary to develop novel methods for the generation of RSSH from the corresponding RSH. We expected methods that allow fast and H2S-free RSSH formation would be most useful. In addition, RSSH have a lower pKa (∼6) than RSH (∼8).3,7 Under physiological pH, RSSH are expected to exist mainly as RSS– and their nucleophilicity may dominate their biochemical reactions. As such, methods that can directly produce RSS– would be ideal for biomimic studies of RSSH. These considerations triggered our idea of developing base-promoted RSSH formation strategies. Herein we report a new method that can effectively convert RSH to anionic RSSH. The reactivity of the resultant RSSH toward commonly used thiol-blocking reagents is also reported.
Our idea is illustrated in Scheme 2. Briefly, we expected that base-sensitive disulfide substrates were suitable precursors of RSSH. 9-Fluorenylmethyl (Fm) based thioesters have been used as thiol precursors, and the mild deprotection conditions are attractive.8 Therefore, we selected Fm as the protecting group. To obtain Fm-based disulfides from the RSH starting materials, we designed an Fm-pyridinyl disulfide (1), which should effectively convert RSH to RSSFm. Since this method will also be used on protein substrates, another disulfide with a carboxylate (2) was proposed. It should have improved solubility in aqueous solutions. Both 1 and 2 were readily prepared from disulfide formation of the corresponding dithiopyridine derivatives and FmSH (see Supporting Information).
Scheme 2. Design of Functional Disulfides.

With these functionalized disulfides in hand, we tested their reactions with thiols to form the corresponding Fm-disulfides. A cysteine derivative 3a was used as the first model. The reaction between 1 and 3a gave the desired product 4a in an excellent yield (Table 1, entry 1). However, the reaction was found to be slow in organic solvents (such as DCM, THF, and DMF) even in the presence of triethylamine (TEA) at room temperature. As such, the reaction needed 24 h to go completion. Interestingly, 2 showed much better reactivity than 1 under the same conditions. The reaction between 2 and 3a provided 4a in a similar yield in less than 30 min. The enhanced reactivity of 2 can be explained by the introduction of the electron-withdrawing carboxylate group, which should increase the reactivity of the disulfide bond. The generality of using 2 to introduce the Fm-disulfides was then tested with other thiol substrates, including a sterically hindered tertiary thiol 3b and two peptide substrates 3c and 3d. In all cases, almost quantitative yields were obtained. In addition, this reaction worked nicely in many different solvents, such as DMF, MeCN, DMSO, THF, and aqueous solutions (PBS buffers).
Table 1. Conversion of Thiols (RSH) to RSSFm by 1 and 2.
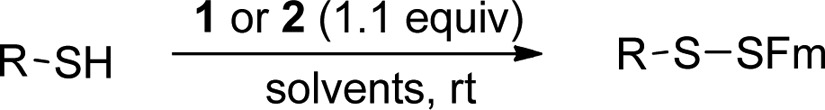
Next we tested the generation of RSSH from the RSSFm precursors. RSSH are unstable species. Derivation and the characterization of their derivatives would provide conclusive evidence of RSSH formation. RSSH are thought to be more nucleophilic than their RSH analogs, due to the α effect. The work by Carroll clearly demonstrated the nucleophilicity of protein-SSH to certain -SH blocking reagents (with undefined conversion yields).5c However, previous studies did not provide much information about the reactivity of small molecule RSSH toward thiol-specific electrophiles. We expected base-promoted RSSH formation from RSSFm should provide RSS– as the reactive species. We also expected RSS– could effectively react with thiol-blocking reagents. To verify this hypothesis, RSSFm 4a was treated with different bases including DBU, piperidine, morpholine, and NaOH. We found the best conditions to be the use of 2 equiv of DBU, which completely removed the Fm in 20 min. The isolated product was a polysulfide (or disulfide (RSSR) and elemental sulfur (S8)) which is the typical decomposition product of persulfides. We also tried to capture the persulfide with iodoacetamide (IAM), a standard thiol-alkylating reagent. As expected, the desired adduct 6a was obtained in a high yield (Scheme 3). The optimized reaction conditions were the following: RSSFm and 2 equiv of IAM were dissolved in a mixture of THF/DCM at room temperature. To this mixture 2 equiv of DBU were added. The product was obtained in 30 min. This reaction also worked nicely with the tertiary persulfide substrate 4b. It should be noted that the RSS– intermediates generated in this method were found to be unstable. Attempts to produce them first (only treating 4a or 4b with DBU) and then trap RSSH with IAM failed. RSSH decomposed tri- and tetrasulfides were found to be the products. These demonstrated the instability of small molecule RSSH.
Scheme 3. Generation of Persulfides and Tagging with IAM.

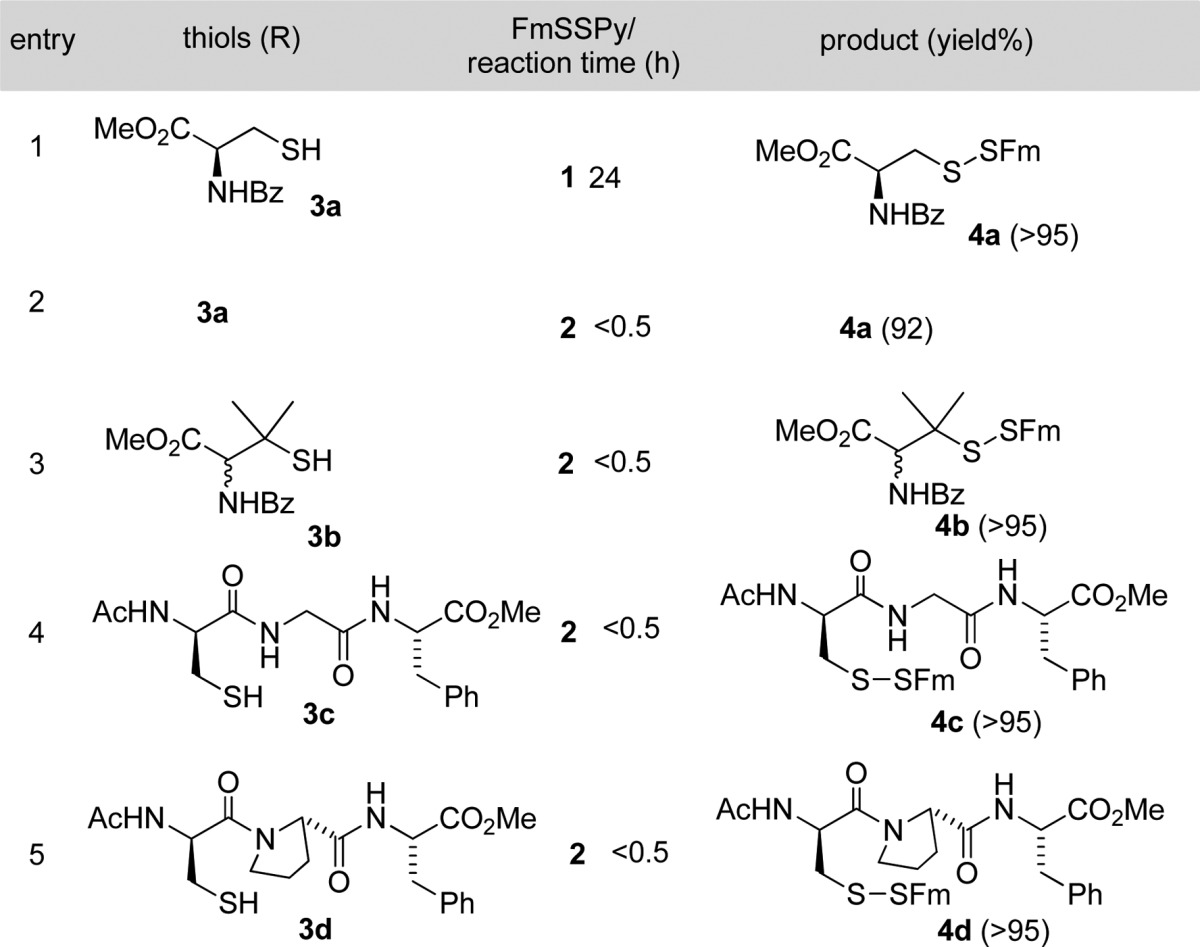
We then tested the reactions of the persulfide precursors 4a and 4b with a series of other commonly used thiol-blocking reagents including methyl acrylate, monobromobimane (mBB), benzyl bromide, and methylmethanethiosulfonate (MMTS). As shown in Table 2, the desired products were obtained in good to excellent yields. The main byproducts formed in the reactions were the decomposition products of RSSH, i.e. the corresponding tri- or tetrasulfides. Peptide substrates 4c and 4d were also used in these studies and proved to be suitable substrates. To the best of our knowledge, these are the first examples to clearly show persulfides (in the form of RSS-) can be derivatized by thiol-blocking reagents.
Table 2. Trapping Persulfides by Various Electrophiles.

Methyl sulfonyl benzothiazol (MSBT) is another very effective thiol-blocking reagent discovered by our laboratory,9 and it was also tested in this reaction. Interestingly, the reaction of MSBT 8 gave a homotrisulfide adduct 10 instead of the desired disulfide 9 (Scheme 4). Benzothiazolethione 11 was also isolated as the product. Based on these products, we proposed that the reaction indeed proceeded to form 9. However, BT-linked disulfide was quite sensitive to −SH based nucleophiles. So the presence of persulfide 7 could rapidly react with 9 to give 10 and 11. To verify this mechanism, we carried out a crossover experiment by adding a BT-disulfide 12 in the reaction of 4b. The generation of 4b-derived persulfide (by DBU) led to a clean crossover reaction with 12. Trisulfide product 13 was obtained in a high yield.
Scheme 4. (A) Reaction between Persulfides and MSBT; (B) Crossover Reaction To Prove the Formation of Persulfide Intermediate.

Having demonstrated the method in small molecule substrates, we explored whether it could be used for protein substrates. Bovine serum albumin (BSA) was selected as the model, as its persulfide adduct was previously prepared using a H2S dependent method.9b Freshly reduced (by DTT) BSA was first treated with FmSSPy-A 2 to test if this reagent could effectively label all free −SH residues. After treatment, the concentration of remaining −SH on BSA was determined spectrophotometrically with 5,5-dithiobis(2-nitrobenzoic acid) (DTNB). This assay did not result in any significant signals (Figure S1 in Supporting Information), demonstrating all free thiols were blocked by 2. The modified protein was also subjected to DBU treatment in the presence of mBB. The labeled protein samples were characterized by trypsin digestion and LC-MS/MS analysis. The formation of −SFm and – S–S-bimane fragments were clearly observed (Figure S2). The conversion yields varied from 4.2% to 71.4% (Table S1).
We then tested if this method could be used to form persulfide adducts on specific cysteines in proteins. It is known that nonreduced BSA has one free −SH (Cys34). Therefore, nonreduced BSA was treated with 2 and followed by DBU/IAM. The resultant protein intermediate and final product were subjected to tryptic digestion and LC-MS/MS. Control samples (without the treatment of 2) were also obtained and compared. As shown in Figure 1, the formation of BSA-S-SFm on Cys34 was clearly identified by MS analysis (Figure 1B). Moreover, an IAM-modified fragment on Cys34 was also observed (Figure 1C).
Figure 1.

Reaction schematics and extracted ion chromatograms for LC-MS/MS analysis of Cys34-labeled and unlabeled tryptic peptides from BSA. (A) Tryptic peptide 14 (GLVLIAFSQYLQQ34CPFDEHVK) containing unlabeled Cys34. (B) Tryptic peptide 16 (GLVLIAFSQYLQQ34C[SFm]PFDEHVK) with Cys34 labeled with FmSSPy-A 2. (C) Tryptic peptide 19 (GLVLIAFSQYLQQ34C[SSCH2CONH2]PFDEHVK) where BSA-SSH was trapped with iodoacetamide (IAM), leading to the labeling of Cys34-SSH with IAM. The MS/MS spectra for peptides 14, 16, and 19 are shown in Figure S3.
In summary, here we reported a novel functional disulfide (i.e., compound 2) which could effectively convert small molecule and protein thiols (−SH) to form −S-SFm adducts under mild conditions. This allows for a H2S-free protocol (by using DBU) to generate highly reactive persulfides in their anionic forms. We also demonstrated the high nucleophilicity of persulfides toward a number of thiol-blocking reagents. The use of DBU in this process may, under some circumstances, impact the reactivity studies. Nevertheless, this biomimetic persulfide formation strategy should be useful in further understanding the chemical biology of persulfides.
Acknowledgments
This work was supported by the NIH (R01HL116571 and P41 GM103493). Part of the mass spectromtery analyses was performed in the Environmental Molecular Sciences Laboratory, a DOE national scientific user facility located at Pacific Northwest National Laboratory, which is operated by Battelle Memorial Institute for the DOE under Contract DE-AC05-76RL01830.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.orglett.5b03557.
Experimental procedures, characterization data of all compounds, MS/MS data for peptides 14, 16, and 19 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Wang R. Physiol. Rev. 2012, 92, 791. 10.1152/physrev.00017.2011. [DOI] [PubMed] [Google Scholar]; b Li L.; Rose P.; Moore P. K. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 169. 10.1146/annurev-pharmtox-010510-100505. [DOI] [PubMed] [Google Scholar]; c Olson K. R.; Donald J. A.; Dombkowski R. A.; Perry S. F. Respir. Physiol. Neurobiol. 2012, 184, 117. 10.1016/j.resp.2012.04.004. [DOI] [PubMed] [Google Scholar]; d Kashfi K.; Olson K. R. Biochem. Pharmacol. 2013, 85, 689. 10.1016/j.bcp.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Fukuto J. M.; Carrington S. J.; Tantillo D. J.; Harrison J. G.; Ignarro L. J.; Freeman B. A.; Chen A.; Wink D. A. Chem. Res. Toxicol. 2012, 25, 769. 10.1021/tx2005234. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Lavu M.; Bhushan S.; Lefer D. J. Clin. Sci. 2011, 120, 219. 10.1042/CS20100462. [DOI] [PubMed] [Google Scholar]; g Predmore B. L.; Lefer D. J.; Gojon G. Antioxid. Redox Signaling 2012, 17, 119. 10.1089/ars.2012.4612. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Olson K. R. Antioxid. Redox Signaling 2012, 17, 32. 10.1089/ars.2011.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Kimura H.; Shibuya N.; Kimura Y. Antioxid. Redox Signaling 2012, 17, 45. 10.1089/ars.2011.4345. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Kimura H. Amino Acids 2011, 41, 113. 10.1007/s00726-010-0510-x. [DOI] [PubMed] [Google Scholar]
- a Mustafa A. K.; Gadalla M. M.; Sen N.; Kim S.; Mu W.; Gazi S. K.; Barrow R. K.; Yang G.; Wang R.; Snyder S. H. Sci. Signaling 2009, 2, ra72. 10.1126/scisignal.2000464. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sen N.; Paul B. D.; Gadalla M. M.; Mustafa A. K.; Sen T.; Xu R.; Kim S.; Snyder S. H. Mol. Cell 2012, 45, 13. 10.1016/j.molcel.2011.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Yang G.; Zhao K.; Ju Y.; Mani S.; Cao Q.; Puukila S.; Khaper N.; Wu L.; Wang R. Antioxid. Redox Signaling 2013, 18, 1906. 10.1089/ars.2012.4645. [DOI] [PubMed] [Google Scholar]; d Krishnan N.; Fu C.; Pappin D. J.; Tonks N. K. Sci. Signaling 2011, 4, ra86. 10.1126/scisignal.2002329. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Gadalla M. M.; Snyder S. H. J. Neurochem. 2010, 113, 14. 10.1111/j.1471-4159.2010.06580.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C.-M.; Weerasinghe L.; Day J. J.; Fukuto J. M.; Xian M. Mol. BioSyst. 2015, 11, 1775. 10.1039/C5MB00216H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ida T.; Sawa T.; Ihara H.; Tsuchiya Y.; Watanabe Y.; Kumagai Y.; Suematsu M.; Motohashi H.; Fujii S.; Matsunaga T.; Yamamoto M.; Ono K.; Devarie-Baez N. O.; Xian M.; Fukuto J. M.; Akaike T. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 7606. 10.1073/pnas.1321232111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Artaud I.; Galardon E. ChemBioChem 2014, 15, 2361. 10.1002/cbic.201402312. [DOI] [PubMed] [Google Scholar]; b Francoleon N. E.; Carrington S. J.; Fukuto J. M. Arch. Biochem. Biophys. 2011, 516, 146. 10.1016/j.abb.2011.09.015. [DOI] [PubMed] [Google Scholar]; c Pan J.; Carroll K. S. ACS Chem. Biol. 2013, 8, 1110. 10.1021/cb4001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey T. S.; Zakharov L. N.; Pluth M. D. J. Am. Chem. Soc. 2014, 136, 10573. 10.1021/ja505371z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono K.; Akaike T.; Sawa T.; Kumagai Y.; Wink D. A.; Tantillo D. J.; Hobbs A. J.; Nagy P.; Xian M.; Lin J.; Fukuto J. M. Free Radical Biol. Med. 2014, 77, 82. 10.1016/j.freeradbiomed.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crich D.; Sana K.; Guo S. Org. Lett. 2007, 9, 4423. 10.1021/ol701583t. [DOI] [PubMed] [Google Scholar]
- a Zhang D.; Devarie-Baez N. O.; Li Q.; Lancaster J. R. Jr.; Xian M. Org. Lett. 2012, 14, 3396. 10.1021/ol301370s. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhang D.; Macinkovic I.; Devarie-Baez N. O.; Pan J.; Park C.-M.; Carroll K. S.; Filipovic M. R.; Xian M. Angew. Chem., Int. Ed. 2014, 53, 575. 10.1002/anie.201305876. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.