

Abstract
Serous tubal intraepithelial carcinomas (STICs) have been proposed to be the most likely precursor of ovarian, tubal and ‘primary peritoneal’ (pelvic) high-grade serous carcinoma (HGSC). As somatic mutation of TP53 is the most common molecular genetic change of ovarian HGSC, occurring in more than 95% of cases, we undertook a mutational analysis of 29 pelvic HGSCs that had concurrent STICs to demonstrate the clonal relationship of STICs and HGSCs. In addition, we correlated the mutational data with p53 immunostaining to determine the role of p53 immunoreactivity as a surrogate for TP53 mutations in histological diagnosis. Somatic TP53 mutations were detected in all 29 HGSCs analysed and the identical mutations were detected in 27 of 29 pairs of STICs and concurrent HGSCs. Missense mutations were observed in 61% of STICs and frameshift/splicing junction/nonsense mutations in 39%. Interestingly, there were two HGSCs with two distinctly different TP53 mutations each, but only one of the mutations was detected in the concurrent STICs. Missense mutations were associated with intense and diffuse (≥ 60%) p53 nuclear immunoreactivity, while most of the null mutations were associated with complete loss of p53 staining (p < 0.0001). Overall, this p53 staining pattern yielded a sensitivity of 87% and a specificity of 100% in detecting TP53 missense mutations. In conclusion, the above findings support the clonal relationship of STIC and pelvic HGSC and demonstrate the utility of p53 immunostaining as a surrogate for TP53 mutation in the histological diagnosis of STIC. In this regard, it is important to appreciate the significance of different staining patterns. Specifically, strong diffuse staining correlates with a missense mutation, whereas complete absence of staining correlates with null mutations.
Keywords: ovarian cancer, tubal intraepithelial carcinoma, p53, serous, TP53 mutation
The proposal that serous tubal intraepithelial carcinoma (STIC) is the precursor of ovarian high-grade serous carcinoma (HGSC) is based on several lines of investigation [1,2]. First, STICs are found in approximately 10–15% of Fallopian tubes removed prophylactically from women at high risk of developing ovarian carcinoma because of a germline BRCA mutation. Second, STICs are detected in 50–60% of cases of sporadic (without germline mutations of BRCA) ovarian, tubal,and so-called primary peritoneal HGSCs [3,4]. Third, STICs frequently up-regulate oncogene products, such as cyclin E1, Rsf-1, and fatty acid synthase, that are also overexpressed in HGSC [5]. Fourth, STICs have relatively shorter telomeres compared with concurrent ovarian HGSC, as occurs with precursor lesions in other sites. Finally, in a small series of five cases, STICs and concurrent ovarian HGSCs, the same TP53mutations were detected in STICs and HGSCs [12], indicating a potential clonal relationship.
Besides exploiting the presence of TP53 mutations in STIC and HGSC as a method of showing a clonal relationship, detection of TP53 mutations in tissue specimens has utility in confirming the histological diagnosis of STIC since it has been reported that TP53 mutations occur in over 95% of ovarian HGSCs [6,7]. For histological diagnosis, however, detection of mutated TP53 is not practical and therefore immunohistochemical detection of p53 protein has been used as a surrogate marker. There have been only a few studies correlating p53 expression with TP53 mutation in ovarian HGSC [8–11] and none that we are aware of in STICs. Accordingly, we undertook the present study of STICs with concurrent pelvic HGSCs in order to (1) confirm a clonal relationship of STIC with HGSC in a relatively large series of cases, and (2) clarify the relationship of immunohistochemical expression of p53 protein with the mutational status of the TP53 gene.
Materials and methods
Case selection
A total of 29 pelvic (not uterine) HGSCs with concurrent STICs were obtained from the Johns Hopkins Hospital (Baltimore, MD), Memorial Sloan Kettering Cancer Center (New York, NY), and Legacy Health System (Portland, OR). Histological diagnosis of STICs was based on reported morphological criteria and confirmed by all the investigators [5]. Tissue collection conformed to the guidelines of the Institutional Research Board of all three participating institutions. All the tissue specimens that were evaluated were formalin-fixed and paraffin-embedded. Fallopian tubes were processed using the SEE-FIM protocol in 27 cases [12]. In two cases, only representative sections were obtained. HGSCs were classified as primary ovarian, tubal or peritoneal based on conventional criteria [4].
Laser capture microdissection and DNA extraction
Serial 10 – μm-thick sections were mounted onto PALM membrane slides, stained with haematoxylin, laser-captured using the PALM laser capture microdissection microscope (Zeiss, Thornwood, NY, USA), and catapulted into a tube cap according to the manufacturer's instructions. A total of 500–1000 highly pure normal-appearing Fallopian tubal epithelial cells and STIC cells were obtained; an immediately adjacent haematoxylin and eosin slide was used as a guide to identify the areas of interest. Selected HGSC cells were manually microdissected using a 30-gauge needle under microscopic visualization. DNA extraction was performed on highly pure microdissected cell samples using a QIAamp DNA Micro Kit (Qiagen, Valencia, CA, USA) following the company's protocol.
PCR amplification and TP53 mutation analysis
Mutations were analysed from exons 2–9, which harbour more than 90% of the TP53 gene mutations reported in ovarian HGSC [6,7]. These exons of TP53 gene were polymerase chain reaction (PCR)-amplified using intron-based primers (Supporting information, Supplementary Table 1). All PCR amplification products were visualized with ethidium bromide under ultraviolet light by electrophoresis in 2.5% agarose gel. All amplified PCR products were sequenced at the Agencourt Biosciences (Beverly, MA, USA) and all sequence variants were confirmed by at least three independent rounds.
Immunohistochemistry (IHC)
IHC was performed using a p53 monoclonal mouse antibody (clone Bp53-11, cat # 760–2542; Ventana Medical Systems, Tucson, AZ, USA) that recognizes the linear epitope mapping to the human p53 protein at the transcription domain within the NH2 terminus. Antigen retrieval and IHC followed a previous report [8]. IHC for p53 was performed in all the cases with positive controls from patients with known p53 overexpression and negative controls by replacing the primary antibody with 10% fetal bovine serum in Trisbuffered saline with Tween 20. The immunostaining for p53 was scored as the percentage of strongly positive nuclei. If ≥ 60% of nuclei were positive, the stain was interpreted as positive. This cut-off was based on our experience and a previous study showing that HGSCs with intense p53 nuclear staining correlated with TP53 missense mutations [8].
Results
The results of TP53 sequence analysis and p53 immunohistochemistry in STIC and HGSC are summarized in Table 1. All 29 cases examined were informative for somatic TP53 mutations. Only mutations producing amino acid change (missense) and null mutations (frameshifts, splicing junction, and nonsense) were considered.
Table 1. p53 alteration in serous tubal intraepithelial carcinoma and high-grade serous carcinoma.
| STIC-1 | STIC-2 | HGSC | |||||
|---|---|---|---|---|---|---|---|
|
|
|
|
|||||
| Case | TP53 mutation | IHC | TP53 mutation | IHC | TP53 mutation | IHC | Mutation type |
| 1 | E7 : 13380G>GA:248R>R/Q | 100 | E7 : 13380G>GA:248R>R/Q | 100 | E7 : 13380G>GA:248R>R/Q | 60 | Missense |
| 2 | E5 : 12457G>GT:157V>V/F | 100 | E5 : 12457G>GT:157V>V/F | 100 | E5 : 12457G>GT:157V>V/F | 80 | Missense |
| 3 | E8 : 13813C>CG:278P>P/R | 100 | NP | NP | E8 : 13813C>CG:278P>P/R | 100 | Missense |
| 4 | E7 : 13338A>AG,234Y>Y/C | 100 | E7 : 13338A>AG,234Y>Y/C | 100 | E7 : 13338A>AG,234Y>Y/C | 100 | Missense |
| 5 | E5 : 12478A>AG,164K>K/E | 100 | E5 : 12478A>AG,164K>K/E | 100 | E5 : 12478A>AG,164K>K/E | 100 | Missense |
| 6 | E4 : 11557T>TG,109F>F/C | 100 | E4 : 11557T>TG,109F>F/C | 100 | E4 : 11557T>TG,109F>F/C | 100 | Missense |
| 7 | E7 : 13349T>TC:238C>C/R | 100 | E7 : 13349T>TC:238C>C/R | 100 | E7 : 13349T>TC:238C>C/R | 100 | Missense |
| 8 | E8 :13772het_delA | 100 | NP | NP | E8 : 13772het_delA | 100 | Frameshift |
| 9 | E5 : 12512G>GA:175R>R/H | 100 | E5 : 12512G>GA:175R>R/H | 100 | E5 : 12512G>GA:175R>R/H | 100 | Missense |
| 10 | E7 : 13352A>AG:239N>N/D | 100 | NP | NP | E7 : 13352A>AG:239N>N/D | 100 | Missense |
| 11 | E7 : 13338A>AG:234Y>Y/C | 100 | E7 : 13338A>AG:234Y>Y/C | 100 | E7 : 13338A>AG:234Y>Y/C | 100 | Missense |
| 12 | WT (E5) | 100 | E5 : 12365A>AG:126Y>Y/C | 100 | E5 : 12365A>AG:126Y>Y/C | 100 | Missense |
| 13 | E5 : 12505G>GT:173V>V/L | 100 | E5 : 12505G>GT:173V>V/L | 100 | E5 : 12505G>GT:173V>V/L | 100 | Missense |
| 14 | E7 : 13350G>GA,238C>C/Y | 95 | E7 : 13350G>GA,238C>C/Y | 95 | E7 : 13350G>GA,238C>C/Y | 80 | Missense |
| 15 | E5 : 12458T>TG,157V>V/G | 70 | E5 : 12458T>TG,157V>V/G | 70 | E5 : 12458T>TG,157V>V/G | 90 | Missense |
| 16 | E5 : 12515G>AG:176C>Y/C | 60 | E5 : 12515G>AG:176C>Y/C | 90 | E5 : 12515G>AG:176C>Y/C | 75 | Missense |
| 17* | E8 : 13772het_delA | 60 | E8 : 13772het_delA | 100 | E8 : 13822A>T,281D>V | 80 | Missense |
| E8 : 13772het_delA | Frameshift | ||||||
| 18 | E8 : 13772hom_delA | 0 | E8 :13772hom_delA | 0 | E8 : 13772hom_delA | 0 | Frameshift |
| 19 | E7 : 13363hom_delC | 0 | E7 : 13363hom_delC | 0 | E7 : 13363hom_delC | 0 | Frameshift |
| 20 | E5 : 12418C>CT:144Q>Q/X | 0 | NP | NP | E5 : 12418C>CT:144Q>Q/X | 0 | Nonsense |
| 21 | I6 : 12742G>GT | 0 | NP | NP | I6 : 12742G>GT | 0 | Splice |
| 22 | WT | 0 | NP | NP | I6 : 13308A>AT | 0 | Splice |
| 23 | E7 : 13363het_delC | 0 | E7 : 13363het_delC | 0 | E7 : 13363het_delC | 0 | Frameshift |
| 24 | I4 : 11607G>GA | 0 | WT | 0 | I4 : 11607G>GA (3′ intron 1) | 0 | Splice |
| 25 | E5 : 12379_12380het_insTC | 0 | NP | NP | E5 : 12379_12380het_insTC | 0 | Frameshift |
| 26 | E7 : 13328_13329ins† | 0 | NP | NP | E7 : 13328_13329ins† | 0 | Frameshift |
| 27 | WT | 0 | NP | NP | E6 : 12661G>GT:198E>E/X | 0 | Nonsense |
| 28 | E7 : 13402_13403insA | 0 | NP | NP | E7 : 13402_13403insA | 0 | Frameshift |
| 29‡ | E7 : 13315het_delC | 0 | E7 : 13315het_delC | 0 | E6 : 12638C>CT:190P>P/L | 100 | Missense |
| E7 : 13315het_delC | Frameshift | ||||||
STIC = serous tubal intraepithelial carcinoma; HGSC = high-grade serous carcinoma; IHC = immunohistochemistry (% of tumour cells with intense nuclear staining); E = exon; WT = wild-type; NP = not present; I = intron.
Two different mutations are detected in the HGSC, probably due to intra-tumoural molecular heterogeneity.
20 base pair insertion.
Two clones of HGSC were present and were individually microdissected based on different IHC profiles.
TP53 mutations in STICs
Of the 48 STICs, 44 (92%) harboured a TP53 mutation and four (cases 12, 22, 24, and 27) contained wild-type TP53. Of the 44 mutations that were detected, 27 (61%) were missense mutations, while the remaining 17 (39%) were frameshift/splicing junction/nonsense mutations (Table 2). Specifically, there were 11 single base deletions, three insertions (one with single base insertion, one with two base insertion, and one with 20 base pair insertion), two with intronic point mutations that affect splicing junctions, and one nonsense mutation (Table 1 and Supporting information, Supplementary Table 2).
Table 2. Correlation between p53 immunostaining results and type of TP53 mutation in serous tubal epithelial carcinoma and high-grade serous carcinoma.
| p53 immunohistochemistry | |||
|---|---|---|---|
|
|
|||
| Lesion | TP53 mutation type | Positive n (%) | Negative n (%) |
| STIC | |||
| Missense | 27 (100.0) | 0 (0.0) | |
| Not missense | 3 (17.6) | 14 (82.4) | |
| HGSC | |||
| Missense | 17 (100.0) | 0 (0.0) | |
| Not missense | 2 (14.3) | 12 (85.7) | |
Fisher's exact test p value < 0.0001. n = number; STIC = serous tubal intraepithelial carcinoma; HGSC = high-grade serous carcinoma. p53 IHC positive: ≥ 60% strongly positive cells; p53 IHC negative: 0% positive cells.
TP53 mutations in concurrent pelvic HGSCs
TP53 mutations were detected in all 29 HGSCs. STICs and associated HGSCs had identical TP53 mutations in 27 (93%) of 29 cases. In two patients (7%), the TP53 mutation identified in the HGSC could not be detected in the concurrent STIC. There were four STICs with wild-type TP53 and two of them (cases 12 and 24) had another STIC present in the same tube and contained the same TP53 mutation as that in the associated HGSC. The two other STICs (cases 22 and 27) with wild-type TP53 were associated with an HGSC containing a TP53 mutation. In addition, there were two HGSCs (cases 17 and 29) with two distinctly different TP53 mutations but only one of the mutations was detected in the concurrent STIC. Missense mutations were observed in 17 (59%) HGSCs and null mutations in 14 (48%). TP53 mutations were not detected in the adjacent normal-appearing tubal epithelium in any of the cases, confirming that they were somatic mutations. All exons 4–8 were mutated but the most frequently mutated was exon 7, which occurred in 12 (39%) of the 31 total TP53 mutations in the HGSCs. There were nine (29%) TP53 mutations in exon 5, five mutations (16%) in exon 8, three (10%) in exon 6, and two (7%) in exon 4. Among all mutations, three different types of mutations were recorded in multiple specimens (Supporting information, Supplementary Table 2).
p53 immunohistochemical findings
In general, normal Fallopian tube epithelium either showed complete negativity or contained scattered nuclei that were weakly positive for p53, a finding consistent with functional p53 protein and wild-type TP53 (Figures 1A and 1B). All the cases but one had concordant p53 immunostaining profiles in the STIC and the associated HGSC, namely either both positive in 17 (59%) cases or both completely negative in 11 (38%) cases (Figures 1C and 1D). Interestingly, single STICs were usually p53 staining negative, whereas multiple STICs were generally positive (p = 0.02) but the number of cases was too limited for a firm conclusion. In one case (case 29), which contained two STICs, p53 immunoreactivity was absent in both STICs but the HGSC showed two discrete areas with different p53 staining (one diffusely and strongly positive, whereas the other was completely negative), but with indistinguishable morphology (Figure 2).
Figure 1.
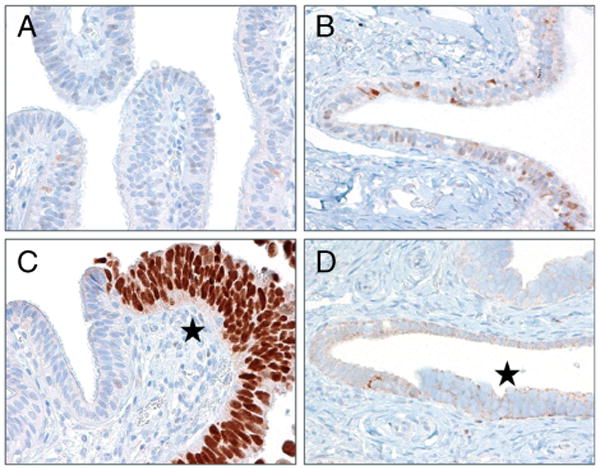
p53 immunoreactivity in representative normal Fallopian tubes and serous tubal intraepithelial carcinomas (STICs). (A) Normal Fallopian tube epithelium is negative for p53. (B) Normal Fallopian tube epithelium shows only single nuclei weakly positive for p53. This pattern is related to normal functional activation of wild-type p53 following a cellular stress. (C) An example of a STIC with a missense mutation of TP53 demonstrates intense and diffuse p53 positivity, while the adjacent normal-appearing Fallopian tube is negative for p53 staining (*, STIC cells). (D) Another STIC with a TP53 null mutation shows lack of nuclear p53 immunoreactivity (*, STIC cells).
Figure 2.
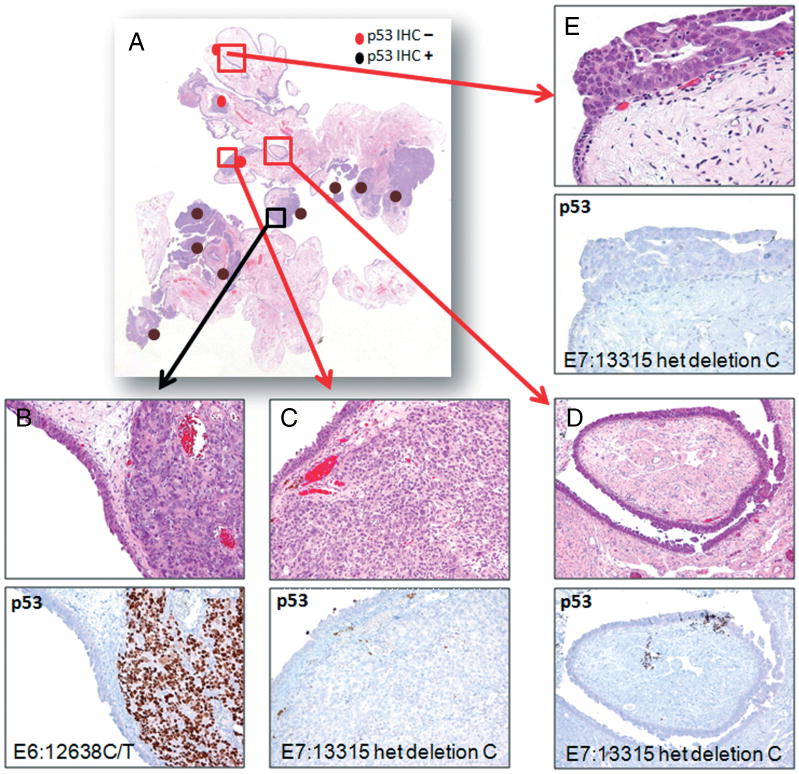
Histological appearance, p53 staining, and TP53 mutation status in lesions of case 29. (A) Low magnification of a haematoxylin and eosin-stained section shows a segment of Fallopian tube and paratubal soft tissue containing multiple foci of high-grade serous carcinoma and serous tubal intraepithelial carcinomas (STICs). (B) The high-grade serous carcinoma is diffusely and intensely positive for p53. (C) High-grade serous carcinoma from another region is completely negative for p53. (D, E) Two discrete STICs are negative for p53.
Correlation of p53 pattern of expression (IHC) with type of mutation
There was a significant correlation between TP53 mutation type and the p53 immunoreactivity pattern (Table 2). Missense mutations were associated with intense and diffuse p53 nuclear immunoreactivity, while frameshift mutations due to either deletion or insertions, nonsense mutations, and splicing junction mutations were associated with a complete loss of p53 staining (p < 0.0001, two-tailed Fisher's exact test) (Table 2). Overall, the intense and diffuse (≥ 60%) p53 staining yielded a sensitivity of 87% and a specificity of 100% in detecting TP53 missense mutations.
The HGSC was classified as ovarian in 19 (65.5%) cases, peritoneal in six (20.7%) cases, and tubal in four (13.8%). The correlation between the p53 immunohistochemical pattern, TP53 mutation type, and the presence of single or multiple STICs is summarized in Table 3. No statistical significance was detected.
Table 3. Correlation between primary site of high-grade serous carcinoma and p53 immunostaining results, type of TP53 mutation, and single and multiple serous tubal intraepithelial carcinomas.
| STIC characteristics | Total n (%) | Ovary n (%) | Peritoneum n (%) | Tube n (%) | p value |
|---|---|---|---|---|---|
| p53 IHC positive | 18 (60.7) | 12 (66.7) | 4 (22.2) | 2 (11.1) | 0.08 |
| p53 IHC negative | 11 (39.3) | 7 (63.6) | 2 (18.2) | 2 (18.2) | |
| Missense mutation | 17 (57.1) | 11 (64.7) | 4 (23.5) | 2 (11.8) | 0.08 |
| Not missense mutation | 12 (42.9) | 8 (66.6) | 2 (16.7) | 2 (16.7) | |
| Single STIC | 10 (35.7) | 7 (70.0) | 1 (10.0) | 2 (20.0) | 0.09 |
| Multiple STICs | 19 (64.3) | 12 (63.2) | 5 (26.3) | 2 (10.5) |
STIC = serous tubal intraepithelial carcinoma; IHC = immunohistochemistry. p53 IHC positive: ≥ 60% strongly positive cells; p53 IHC negative: 0% positive cells.
Discussion
The findings in this study confirm that STICs and associated HGSCs are clonally related, as the same TP53 mutation was detected in both STIC and HGSC in all 27 informative cases in which a mutation was present. In two cases, a TP53 mutation was detected in the HGSC but not in the associated STIC which contained wild-type TP53 . This raises two questions. First, why does the STIC not harbour the same mutation as the HGSC and second, which is even more puzzling, can a lesion that morphologically is an unequivocal STIC contain wild-type TP53 ? One possible explanation to account for an HGSC harbouring a TP53 mutation in the absence of a TP53 mutation in a concurrent STIC is that despite thorough sampling using the SEE-FIM protocol, an occult STIC with the same mutation as HGSC was missed. We have previously reported that in some cases in which a STIC was not initially found, an occult STIC was subsequently detected when the paraffin block was levelled [4]. Furthermore, in the present study two discrete STICs in the same tube were identified in 19 cases, supporting the possibility that another occult STIC may have been present in these cases but was not sampled. The question of why a morphologically unequivocal STIC has wild-type TP53 is more difficult to answer. One possible explanation is that the TP53 mutation was in an exon not evaluated. More likely, despite careful microdissection, the presence of wild-type TP53 in four STICs may have been due to contamination of the STIC with normal cells, which obscured detection of a mutation.
Moreover, we detected in two HGSCs two different TP53 mutations. There are two possible explanations to account for the presence of more than one TP53 mutation in two HGSCs. One possibility is that different initiating transformed clones arose independently from multiple STICs with different mutations leading to the development of a multiclonal HGSC. Alternatively, intra-tumoural heterogeneity is due to acquisition of an additional mutation from a common precursor cell during tumour progression. If the latter hypothesis is valid, this observation suggests tumour progression from the STIC to an HGSC, providing further evidence that the STIC is a precursor rather than a metastasis.
The reproducibility of the diagnosis of STIC based on morphological features alone is moderate at best [13], even among expert gynaecological pathologists; therefore, the need for additional immunohistochemical support for the diagnosis is critical. We have recently reported that the use of an algorithm incorporating morphology along with immunohistochemical staining for p53 and Ki-67 greatly improves reproducibility of the diagnosis [14]. Accordingly, correlation of the immunohistochemical findings with the mutational data has important practical implications. Diffuse, intense positivity for p53, defined as ≥ 60% positive cells, corresponded to a missense mutation, while complete loss of p53 staining corresponded to null mutations (due to frameshift, splicing junction, and nonsense mutation). On the other hand, weak and patchy staining generally corresponded to wild-type TP53. Accordingly, an immunohistochemical stain forp53 that is completely devoid of staining, assuming that proper controls have been performed, should be interpreted as consistent with a TP53 mutation.
Given the critical role of TP53 mutations in human cancer development [15,16], the results from this study provide new insight into the pathogenesis of pelvic HGSC. TP53 mutation likely represents one of the earliest events in initiating pelvic HGSC, as the mutations were recorded in the majority of STICs [17]. The dysregulated p53 compromises its role as a gatekeeper that prevents cellular transformation by tightly regulating cellular response to a variety of stresses including oncogene-induced stress, aberrant transcription, and chromatin remodelling [15,18–21]. In the presence of mutant TP53, epithelial cells replicate despite genotoxic events and as a result, the epithelial cells sequentially display genomic instability, which might contribute to the accumulation of molecular genetic changes and ultimately tumour development of pelvic HGSC [22].
In conclusion, the direct evidence showing STIC as the precursor of HGSC is still tantalizing and apparently future molecular genetic studies are required to address this important question. Nevertheless, the results from this study provide a step further to support the view that both HGSC and STIC are clonally related, as identical TP53 mutations occur in both STICs and concurrent HGSCs in the majority of cases. Finally, immunohistochemical staining for p53 can serve as a useful surrogate for a TP53 mutation but it is important to appreciate the significance of different staining patterns. Specifically, strong diffuse staining correlates with a missense mutation, whereas complete absence of staining correlates with null mutations. Accordingly, the latter pattern should be interpreted as compatible with a TP53 mutation.
Supplementary Material
Acknowledgments
This study was supported by a CDMRP grant (No OC100517) from the US Department of Defense.
Footnotes
Supporting Information on The Internet: The following supporting information may be found in the online version of this article.
Table S1. TP53 primer sequences.
Table S2. Details of the TP53 mutations identified.
No conflicts of interest were declared.
Author contribution statement: EK, RJK, TLW, and IMS conceived the study. RJK, RV, ASS, GH, and RS provided study material. EK performed the experiments. EK, RJK, TLW, and IMS analysed the data. EK, RJK, and IMS wrote the manuscript. All the authors were involved in revising the manuscript and approved the final version of the manuscript.
References
- 1.Kurman RJ, Shih IeM. Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer—shifting the paradigm. Hum Pathol. 2011;42:918–931. doi: 10.1016/j.humpath.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gross AL, Kurman RJ, Vang R, et al. Precursor lesions of high-grade serous ovarian carcinoma: morphological and molecular characteristics. J Oncol. 2010 doi: 10.1155/2010/126295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Callahan MJ, Crum CP, Medeiros F, et al. Primary Fallopian tube malignancies in BRCA-positive women undergoing surgery for ovarian cancer risk reduction. J Clin Oncol. 2007;25:3985–3990. doi: 10.1200/JCO.2007.12.2622. [DOI] [PubMed] [Google Scholar]
- 4.Przybycin CG, Kurman RJ, Ronnett BM, et al. Are all pelvic (nonuterine) serous carcinomas of tubal origin? Am J Surg Pathol. 2010;34:1407–1416. doi: 10.1097/PAS.0b013e3181ef7b16. [DOI] [PubMed] [Google Scholar]
- 5.Kuhn E, Meeker A, Wang TL, et al. Shortened telomeres in serous tubal intraepithelial carcinoma: an early event in ovarian high-grade serous carcinogenesis. Am J Surg Pathol. 2010;34:829–836. doi: 10.1097/PAS.0b013e3181dcede7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.TCGA Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahmed AA, Etemadmoghadam D, Temple J, et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J Pathol. 2010;221:49–56. doi: 10.1002/path.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yemelyanova A, Vang R, Kshirsagar M, et al. Immunohistochemical staining patterns of p53 can serve as a surrogate marker for TP53 mutations in ovarian carcinoma: an immunohistochemical and nucleotide sequencing analysis. Mod Pathol. 2011;24:1248–1253. doi: 10.1038/modpathol.2011.85. [DOI] [PubMed] [Google Scholar]
- 9.Reles A, Wen WH, Schmider A, et al. Correlation of p53 mutations with resistance to platinum-based chemotherapy and shortened survival in ovarian cancer. Clin Cancer Res. 2001;7:2984–2997. [PubMed] [Google Scholar]
- 10.DiCioccio RA, Werness BA, Peng R, et al. Correlation of TP53 mutations and p53 expression in ovarian tumors. Cancer Genet Cytogenet. 1998;105:93–102. doi: 10.1016/s0165-4608(98)00011-9. [DOI] [PubMed] [Google Scholar]
- 11.Kobel M, Reuss A, Bois A, et al. The biological and clinical value of p53 expression in pelvic high-grade serous carcinomas. J Pathol. 2010;222:191–198. doi: 10.1002/path.2744. [DOI] [PubMed] [Google Scholar]
- 12.Lee Y, Miron A, Drapkin R, et al. A candidate precursor to serous carcinoma that originates in the distal Fallopian tube. J Pathol. 2007;211:26–35. doi: 10.1002/path.2091. [DOI] [PubMed] [Google Scholar]
- 13.Carlson JW, Jarboe EA, Kindelberger D, et al. Serous tubal intraepithelial carcinoma: diagnostic reproducibility and its implications. Int J Gynecol Pathol. 2010;29:310–314. doi: 10.1097/PGP.0b013e3181c713a8. [DOI] [PubMed] [Google Scholar]
- 14.Visvanathan K, Vang R, Shaw P, et al. Diagnosis of serous tubal intraepithelial carcinoma (STIC) based on morphologic and immunohistochemical features—a reproducibility study. Am J Surg Pathol. 2011;35:1766–1775. doi: 10.1097/PAS.0b013e31822f58bc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nature Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 16.Goh AM, Coffill CR, Lane DP. The role of mutant p53 in human cancer. J Pathol. 2011;223:116–126. doi: 10.1002/path.2784. [DOI] [PubMed] [Google Scholar]
- 17.Bowtell DD. The genesis and evolution of high-grade serous ovarian cancer. Nature Rev Cancer. 2010;10:803–808. doi: 10.1038/nrc2946. [DOI] [PubMed] [Google Scholar]
- 18.Bartkova J, Rezaei N, Liontos M, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–637. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- 19.Gorgoulis VG, Vassiliou LV, Karakaidos P, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 20.Bartkova J, Horejsi Z, Koed K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 21.Haigis KM, Sweet-Cordero A. New insights into oncogenic stress. Nature Genet. 2011;43:177–178. doi: 10.1038/ng0311-177. [DOI] [PubMed] [Google Scholar]
- 22.Sheu JJ, Guan B, Choi JH, et al. Rsf-1, a chromatin remodeling protein, induces DNA damage and promotes genomic instability. J Biol Chem. 2010;285:38260–38269. doi: 10.1074/jbc.M110.138735. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.