

Abstract
Crohn’s disease (CD) and ulcerative colitis (UC), collectively termed inflammatory bowel disease (IBD), are immunological disorders that represent the prototypes of chronic intestinal inflammation. Their pathogenesis involves the dysregulated interaction between the intestinal microbiota and the gut-associated mucosal immune system that takes place when genetically predisposed individuals are exposed to detrimental environmental triggers. In recent years, the therapeutic dogma in IBD has shifted away from the administration of non-specific immunosuppressives towards a pathway-based approach. In this review we will present an oulook of IBD treatment, based on this new conceptual approach. Firstly, we will provide an overview of the major aspects of IBD pathogenesis with emphasis on specific pathway-based defects. Subsequently, we will examine in detail the development of novel therapeutic approaches that can be used to target genetics, dysbiosis, the epithelial barrier, pro inflammatory cytokines, and leukocyte trafficking. Most of these strategies are still in the developmental phase, but promising approaches include: fecal microbiota transplantation as a means to correct IBD-related dysbiosis; administration of modified phosphatidylcoline (PC) to enhance the function of the intestinal mucous and tighten the defective epithelial barrier; the reduction of over-reactive pro-inflammatory pathways through the blockade of novel, non-TNF inflammatory mediators, via monoclonal antibodies against the common p40 chain of IL-12 and IL-23, JAK kinase inhibitors, or antisense oligonuclotides against inhibitors of the immunosuppressive cytokine TGF-β1; and, finally, inhibition of leukocyte trafficking to the gut via neutralization of the gut-specific integrin α4β7 integrin. Availability of such diverse treatment modalities with specific pathway-based targets will increase the therapeutic options for patients with IBD.
INTRODUCTION
IBD is a collective term for UC and CD. These clinical entities are the prototypes of chronic persistent inflammation of the intestines with a combined prevalence of more than 300/100.000 individuals in Western populations (1). UC and CD share several clinicopathological features, such as a fluctuating chronic pattern, preference for younger individuals, acute and chronic inflammatory infiltrates within the lamina propria, as well as common extra-intestinal manifestations. Nevertheless, they are also distinguished by certain separating features (2, 3). UC affects the colon, exclusively, with the inflammatory reaction being confined to the mucosa and spreading continuously from the anus and extends proximally. In contrast, CD may affect any region of the GI tract and any layer of the bowel wall, leading to the disease-specific phenotypes of fibrostenosis and/or fistula formation.
UC and CD may cause continuous clinical symptoms due to anatomic and functional damage of the GI tract. They are also often associated with specific complications that necessitate surgical intervention, which further impairs bowel function. A systemic inflammatory response may occasionally develop with serious and occasionally life-threatening consequences. Finally, patients with IBD frequently develop inflammation in extra-intestinal tissues, which add up to the overall disease burden and present unique therapeutic challenges. As a result, IBD is associated with serious compromise of the quality of life, loss of productivity, as well as frequent utilization of health care resources and considerable costs (4).
CD and UC are immune-mediated conditions; hence, their management has traditionally focused on anti-inflammatory treatments. In the last two decades, the therapeutic dogma has shifted away from general immunosuppressive treatment with corticosteroids and thiopurines towards a pathway-based approach. The latter initially implicates identification of specific immunomodulatory molecules that possess defined pathogenetic roles. Subsequently, these pathways are either neutralized via the administration of monoclonal antibodies or enhanced by using recombinant proteins. These new therapies aim not only to clinical improvement, but, most importantly, to prevention of long-term sequelae through the complete abrogation of inflammatory activity (the so-called deep remission) (5). Analyzing the effects of such treatments is necessary for their incorporation into updated therapeutic algorithms while at the same time, facilitate the elucidation of the precise roles of specific pathways for the initiation and perpetuation of intestinal inflammation. In the present review, we will discuss current and developing therapeutic approaches to IBD in relation to the increasing understanding of its pathogenesis.
OVERVIEW OF IBD PATHOGENESIS
The development of chronic inflammation in IBD signifies the miscommunication between the gut microbiota and the intestinal mucosal immune system, resulting in the failure of mucosal homeostasis (6). This dysregulated interaction critically depends upon the integrity of the epithelial barrier, is determined by genetic defects, and requires the presence of triggering environmental factors (Figure 1). Diverse lines of research have brought about the importance of each of the aforementioned factors.
Figure 1. Pathogenesis-driven therapies in IBD.
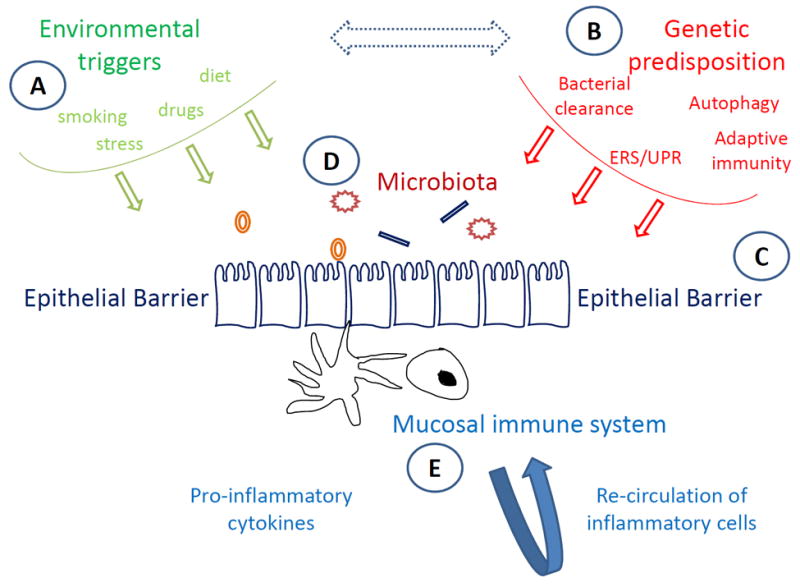
Homeostasis within the intestinal mucosa is maintained through a tightly regulated interaction between microbiota and the gut-associated immune system, which depends on the integrity of epithelial defense mechanisms. In patients with IBD, genetic and environmental pressures lead to failure of single or multiple components of mucosal homeostasis, resulting in dysregulated immune-bacterial interactions characterized by the persistence of pro-inflammatory pathways. Current therapeutic paradigms in IBD involve manipulation at various steps of the pathogenetic process. Environmental (A) and genetic (B) factors act at early, pre-clinical stages of IBD, and thus, can only be modified in selected subpopulations who suffer from single, well-characterized defects. Similarly, reinforcement of the epithelial barrier (C) can be achieved through the enhancement of mucus composition and function, whereas correction of dysbiosis may be reversed via flora manipulation (D). Nevertheless, the majority of current treatments aim at reducing the overactive pro-inflammatory pathways that take place within the inflamed mucosa (E). This can be accomplished either by blocking pro-inflammatory cytokines, primarily, but not exclusively, TNF-α, or by inhibiting the trafficking of inflammatory cells to the affected intestinal mucosa.
Application of high-throughput genomic analysis has resulted in the identification of more than 150 loci that are associated with modified risk for developing IBD (7), among which 110 are shared by both disease subtypes (UC and CD) (8). The majority of identified polymorphisms involve genes that encode for proteins with critical roles in immune responses against microorganisms. In fact, major pathways that have emerged from these genome-wide association studies include autophagy, intracellular recognition of pathogen-associated molecular patterns, and endoplasmic reticulum stress/unfolded protein responses (9). The sole existence of a genetic polymorphism is not sufficient to generate the inflammatory phenotype of IBD; nonetheless, it places upon the individual the risk for developing the disease when challenged by specific environmental pressures. Identification of the responsible environmental triggers has proven extremely difficult due to the significant overlap between diverse factors and the possible temporal dissociation between these factors and the initiation of disease (10). The strongest association has been established for smoking, which appears to be protective for UC and harmful for CD (11). Additional factors include medications (antibiotics in early life, estrogens and non-steroid anti-inflammatory drugs in adulthood), stress and dietary habits (12). It should also be noted that environmental factors may also influence the risk for developing IBD indirectly by inducing epigenetic changes to the genome or altering the composition of the microbiota (13).
No matter what the genetic defects and environmental triggers may be, the final outcome is disruption of mucosal homeostasis. The latter is maintained by the healthy interaction between three participating parties, namely, the microbiota, the gut-associated immune system, and the epithelial barrier (14). In IBD, any of these parameters may be defective. The existence of IBD-specific alterations of the flora has been described and is collectively referred to as “dysbiosis” (15). Encompassed within this concept are a decreased diversity of microorganisms, changes in the relative proportions of the major bacterial phyla (i.e. Firmicutes vs. Bacteriodetes), as well as altered detection frequencies for certain microorganisms (16). Nevertheless, it is not clear whether such changes precede (and even cause) the development of IBD or whether they represent a by-product of mucosal inflammation. On the other hand, there is now ample evidence that immune cells within the lamina propria of patients with IBD exist in an activated state and demonstrate hyper-reactivity against bacterial antigens (17). There is still debate regarding the primary abnormality, whether it is loss of regulatory mechanisms or inherent over-reactivity of effector pathways. In any case, the end result is the amplification of proinflammatory responses within the affected mucosa. This is coupled by increased recruitment of leukocytes, mainly lymphocytes, via the combined action of chemokines and cell-adhesion molecules that is reflected in the robust inflammatory infiltrate seen in pathological specimens from IBD patients (18). Recruited immunocytes produce a variety of inflammatory mediators that further amplify immunological reactions and result in bowel damage. Pivotal among such factors are cytokines that are secreted by several cell-types and are further diversified into subgroups (19). Th1 and Th17 pathways appear to predominate in the inflamed mucosa of CD patients, whereas Th2 and Th17 factors are abundant in UC (20). It should be noted, however, that strict terminal polarization of effector responses does not seem to take place and significant overlap exists between the immunological pathways in UC and CD (21). One additional point that needs to be considered is that there is now increasing evidence that over-reactivity of adaptive effector responses may be preceded by an initial phase that is characterized by failure of innate immunity mechanisms (22). This “innate immunodeficiency” is translated into failure of effective clearance of bacteria and their products by phagocytic cells within the lamina propria (23). This is also supported by the association of IBD with polymorphisms in genes that encode for proteins that participate in the recognition and elimination of intruding microorganisms. Other polymorphisms affect components of the epithelial barrier itself (24). This is a complex structure that allows the regulated communication between intraluminal commensals and immunocytes at the lamina propria. It is formed by the single layer of epithelial cells and the dense network of interconnecting proteins of tight junctions, and further supported by the mucus layer, as well as the secretion of several natural antimicrobial peptides and repair factors (25). There are now several lines of evidence showing that failure of the integrity of the epithelial barrier may be an early event in the natural history of IBD, which allows for the uncontrolled influx of bacterial products into the lamina propria and the propagation of pro-inflammatory mucosal responses (26).
It is obvious that identification of defined defects in the factors that participate in mucosal homeostasis not only offers insights into the pathogenesis of IBD, but also establishes the basis for the development of targeted therapies. These will be discussed below in detail.
A “PATHOGENETIC” APPROACH TO IBD TREATMENT
Correcting the genetic defect
For the majority of IBD cases, the underlying genetic predisposition is not due to a single mutation that is inherited in a Mendelian manner. Instead, most IBD-associated polymorphisms have only a minor contribution to disease heritability (27). In fact, the 163 identified loci account for only 13.6% of CD and 7.5% of UC variance (8). Most probably, IBD patients may bear multiple minor traits, which add up and interact with environmental factors to result in the functional defect, and eventually to the clinical phenotype. This is not, however, the case for the rare patient who bears single mutations in critical genes that have immediate and direct results. In fact, it is now recognized that several monogenic diseases present with IBD-like chronic intestinal inflammation (28). Due to the definitive effect of these genetic defects on the functional level, a common clinical phenotype is the development of early-onset IBD.
A representative example is the occurrence of very-early, CD-like intestinal disease in individuals bearing mutations in the IL-10 or IL-10R genes (29, 30). Indeed, a recent study examined 66 patients with early-onset disease and found frequent mutations in IL-10RA (5 patients), IL-10RB (8 patients) or IL-10 (3 patients) genes (31). What is important from the therapeutic standpoint is the potential for correcting the genetic defect in such cases via allogeneic hematopoietic stem cell transplantation (HSCT). Along this line, this therapy was applied to 5 patients with mutations in IL-10R deficiency (31). The concept behind this approach is that HSCT would reconstitute IL-10R signaling in PBMCs and correct the functional defect; as a proof of principle, this was actually the case in all transplanted patients. HSCT induced a clinical remission in all patients that was sustained throughout a 2-year follow up period. These results are complementary to previous studies showing that bone marrow/HSCT is a valuable treatment option for other monogenic diseases that cause intestinal inflammation, such as immune dysregulation, polyendocrinopathy, enteropathy, X linked (IPEX) syndrome, chronic granulomatous disease (CGD), or X linked lymphoproliferative syndrome 2 (XIAP deficiency) (32).
Although anecdotal reports have reported induction of remission following allogeneic HSCT in patients with adult-onset, sporadic IBD (33, 34), the aforementioned results in specific inherited syndromes cannot be generalized (31). It should be noted that HSCT can only correct defects with functional consequences on hematopoietic cells. Furthermore, the underlying mutation should be clearly defined (if it is to be corrected with HSCT), which is not the case for the majority of UC or CD cases. Finally, the potential for adverse effects should not be underestimated, in particular when other therapeutic options exist.
Reversing dysbiosis
As previously mentioned, IBD-specific changes in the gut microbiota have been long recognized and are increasingly typified in recent years (16). Nevertheless, the majority of such changes refer to alterations of the relative proportions of the major subgroups of bacteria and not the involvement of single pathobionts. Consequently, correction of the observed abnormalities has been proven very difficult, if not impossible, to achieve. This is reflected in the failure of antibiotics or probiotics to provide permanent and long-lasting suppression of intestinal inflammation in IBD patients. Moreover, analysis of existing data is compromised by the effects of heterogeneity between studies that have to do with the type of antibiotic regimen or the strains of probiotic administered, the duration and dose of each specific treatment, and the concomitant therapies (35). Two exceptions, however, exist and may offer important pathogenetic insights. First, the multi-probiotic preparation VSL#3 is very effective in preventing the development or recurrence of pouchitis in patients with UC who have undergone ileal-pouch-anal anastomosis (IPAA) (36). Second, antibiotics prevent the short-term post-operative recurrence of CD in the neoterminal ileum following right hemicolectomy for L1-type disease (Montreal classification) (37). Therefore, it appears that antibiotics and probiotics are most effective when given in a preventive fashion; this, in turn, may imply that the role of the microbiota in IBD is more significant during the onset of intestinal inflammation, rather than after it is fully established. This concept is further supported by studies in SAMP1/YitFc mice (38). These mice spontaneously develop chronic intestinal inflammation that is fully established by 10 weeks of age and remains throughout the mouse lifespan (39). Disease in SAMP1/YitFc mice truly resembles CD, as it is localized at the terminal ileum, extends transmurally, and is associated with the development of fibrotic strictures and the occasional formation of granulomas. Ileitis in SAMP1/YitFc mice is significantly ameliorated by broad-spectrum antibiotic administration and completely abrogated by probiotics (40, 41). Nevertheless, this happens only when treatment is administered before the onset of ileitis, whereas no effect was observed when treatment was initiated during established inflammation.
A different approach, which has gained increasing attention in recent years, is fecal microbiota transplantation (FMT). This technique aims at replacing the “dysbiotic” flora of IBD patients with that of a healthy donor in hopes that this will re-establish mucosal homeostasis (42). The donor is tested for various communicable diseases and the feces are infused into the recipient’s GI tract via a variety of ways, such as through a nasogastric or nasojejunal tube, through upper or lower GI endoscopy, or via retention enemas. Although this approach has proven highly effective in patients with recurrent Cl. difficile colitis, results from IBD studies have not allowed for definitive conclusions (43). In either UC or CD, when FMT was applied in cases with concomitant Cl. difficile infection, the majority of patients reported resolution or reduction of symptoms (44). Results, however, were more ambiguous when patients with IBD and no Cl. difficile infection were studied. In a minority of studies, the effect of FMT on flora composition of recipients was also examined (45, 46). It is interesting that FMT increased the phylotype richness (which, as said, is decreased in UC) and resulted in similarity of flora of the recipient compared to that of the donor. Nevertheless, these modifications were transient in nature and not uniformly translated to clinical improvement. It should be noted that large heterogeneity between trials exists and this makes the interpretation of results, as a whole, unreliable. The resulting difficulties in clinical trial design and analysis are clearly depicted in the recent publication of two double-blind, randomized, controlled trials on FMT in patients with UC (47, 48). Importanty, both trials were prematurely terminated at interim analysis by the data safety and monitoring board because of futility. Both studies were seriously criticized for overestimating the effect of FMT, which led to the trials being underpowered. In one study, however, further analysis after inclusion of additional 22 subjects, who had already been enrolled, significance was accomplished for the primary end point of remission (47). This “partially positive” trial differed from the “negative” one in the route of fecal administration (enema vs. oral gavage), number of FMTs (six vs. two), and allowance for anti-TNF administration (vs. no allowance). Furthermore, a smaller number of fecal donors were included (six vs. fifteen). Interestingly, receiving feces from a particular donor may have been responsible for the positive result of the one trial. Another important signal was that the effect of FMT on the recipient microbiome appeared to significantly differ between clinical responders and non-responders. In all, these conflicting results further emphasize that better designed studies are needed to clarify the role, if any, of FMT in patients with IBD and no concurrent Cl. difficile infection.
Tightening the epithelial barrier
The competence of the epithelial barrier is maintained through the structural and functional integrity of its several discrete, but interconnected components. Among those are the mucus layer, the epithelial monolayer with the various types of cells (enterocytes, goblet cells, Paneth cells), the intercellular connective structures and tight junctions, as well as the secreted natural antimicrobial peptides and trefoil factors. Defects in any of these integral parts may result in failure of the overall “barrier” function, causing inadequate control of the bacterial influx into the lamina propria. Such events are now considered among the earliest that take place in the natural history of IBD and precede the development of pro-inflammatory mucosal reaction. It follows that restitution of the defective barrier is a desired therapeutic aim in patients with UC and CD. Nevertheless, very few efforts have reached the clinical stage, thus far (49). It should be noted that a major mode of action of mesalazine may be the improvement of epithelial integrity through its function as an agonist for peroxisome proliferator-activated receptor-γ (PPAR-γ) (50). Similarly, hyperbaric oxygen treatment may augment barrier function by reducing the adverse effects of tissue hypoxia (51, 52).
Phosphatidylcholine (PC) is the major mucus phospholipid and is essential for the protective function of colonic mucus. PC was found to be substantially decreased in the mucus of patients with UC as compared to healthy individuals, but also to patients with CD (53). In addition to its role in mucus stabilization, PC has also been shown to exert intrinsic anti-inflammatory properties (54). These data led investigators to hypothesize that replacement of PC in patients with UC may restore barrier function and ameliorate intestinal inflammation. The hypothesis has been tested so far in a number of studies, all of which demonstrated positive results. Initial studies used a slow-release PC that was first tested against placebo in chronic active UC (55). PC administration induced a clinical favorable outcome (remission or >50% improvement) in 90% of patients as opposed to a 10% response in patients in the placebo. The same PC compound was subsequently tested in the steroid-refractory UC population; PC induced a response (CAI index of≤3 or CAI improvement of >50%) in 50% of the patients, whereas placebo was successful only in 10% of treated patients (56). Similar results were recently reported for the administration of LT-02 (0, 0.8, 1.6, or 3.2 g), a modified release PC-enriched formula in patients with clinically active UC (Simple Clinical Colitis Activity Index ≥5), despite appropriate treatment with mesalazine (57). Patients were treated for 12-wks and followed for an additional 8-wks. Patients that received the high LT-02 dose (3.2 g) had a significantly higher SCCAI drop (51.7 %) compared to placebo (33.3 %, P=0.03). In other analyses, patients on high-dose LT-02 also achieved higher rates for remission (31.4 % vs. 15 %), mucosal healing (47.4 % vs. 32.5 %), and histologic remission (40.5 % vs. 20 %). Furthermore, treatment with LT-02 was associated with earlier (>2-wks) first symptom resolution, whereas, at the same time, twice as many patients reached complete symptom resolution compared to placebo. Treatment with PC was not associated with serious adverse events in the aforementioned studies. These encouraging results have prompted the design of phase III trials, which are currently recruiting patients (ClinicalTrials.gov Identifiers: NCT02280629 and NCT02142725).
Ameliorating pro-inflammatory responses
Anti-cytokine therapeutics
The elucidation of immunological pathways that underlie the pathogenesis of clinical and experimental intestinal inflammation has resulted in an abundance of therapeutic targets, the majority of which are pro-inflammatory cytokines. The first neutralization attempt was directed against tumor necrosis factor (TNF)-α. The high success of this therapy has led to the development of several anti-TNF monoclonal antibodies that have been incorporated into current therapeutic algorithms for UC (infliximab, adalimumab, golimumab) and CD (infliximab, adalimumab, certolizumab pegol) (58). More importantly, it has offered proof of principle that blockade of a single molecule/pathway may be sufficient to induce complete and sustainable remission of chronic intestinal inflammation. At the same time, however, a significant proportion of patients do not improve on anti-TNF treatments; this has led to the conclusion that other immunological pathways may be dominant in these groups of non-responders. Consequently, during the last two decades, the quest for the next appropriate target has been continuous, and, although failures have been many more than successes, it has provided the pool for the next IBD biologics.
In recent years, converging lines of evidence have supported an important role for the IL-23/Th17 pathway in chronic intestinal inflammation (59). First, genetic polymorphisms of the IL-23R gene, but also for several other molecules related to this pathway, have been associated with IBD. Second, mRNA and protein expression of IL-23, IL-17A and other associated cytokines are highly upregulated in the inflamed mucosa of patients with CD, and to a lesser extent, UC. Finally, animal studies have shown that inhibition of the IL-23/Th17 pathway is effective in ameliorating experimental colitis. This background evidence has led to clinical trials testing the efficacy of anti-IL-23 therapies in CD.
Ustekinumab is a monoclonal IgG1 antibody directed against the p40 subunit of IL-23. It should be emphasized that p40 is also a subunit of IL-12, a prototype Th1 cytokine. Thus, ustekinumab has the ability to block both Th1 and Th17 pathways. As these are considered the major mucosal immunological pathways that take place in CD, ustekinumab was tested in this patient population. An initial study demonstrated that subcutaneous or intravenous ustekinumab effectively induced clinical response in the short (4 and 6 wks), but not medium (week 8), term in CD (60). This study showed that patients that had previously received anti-TNF biologics tended to respond better to ustekinumab. Indeed, in subanalysis of the results, the effect of ustekinumab was almost 60% in anti-TNF experienced patients (placebo response rate: 26%). Furthermore, response was higher in patients with higher CRP values, indicating that anti-inflammatory therapies work better in patients with significant inflammation, probably due to the high availability of molecular targets (61). The aforementioned observations prompted a phase IIb trial, which included only patients with moderate to severe, active CD that was refractory to anti-TNF therapy (62). The efficacy of ustekinumab as induction or maintenance therapy was tested in 526 patients. In the induction study, ustekinumab was administered as intravenous therapy with increasing doses (1, 3 or 6 mg/kg). At week 6, all three ustekinumab groups achieved significantly higher clinical response rates (≥100 points decrease in CDAI) (placebo: 23.5%, ustekinumab 1 mg/kg: 37%, P=0.02, ustekinumab 3 mg/kg 34%, P=0.06, ustekinumab 6 mg/kg 40%, P=0.005). There were no significant differences regarding the rates of clinical remission. In the follow-up maintenance study, responders at week 6 (n=146) received either placebo or subcutaneous ustekinumab (90 mg) at weeks 8 and 16, and evaluated at week 22. Analysis of the results detected a significant increase in the number of patients on ustekinumab regarding either clinical response (69% vs. 42.5%, P<0.001) or clinical remission (42% vs. 27%, P<0.03), in comparison to patients on placebo. Ustekinumab demonstrated a good safety profile. These studies set the basis for a phase III trial that is currently ongoing and have generated hopes that this anti-IL-12/anti-IL23 therapy will find application in CD, particular in the difficult to treat population with refractoriness to anti-TNF.
A different approach to block the pro-inflammatory pathways in chronic intestinal inflammation is through the use of inhibitors of JAK kinases. JAK kinases are a family of intracellular tyrosine kinases, consisting of JAK1, JAK2, JAK3 and tyrosine kinase 2 (63). These are associated with intracellular signaling that is initiated by the bindings of various cytokines to their cognate receptors. Therefore, by inhibiting JAK kinase activity, the effects of multiple cytokines are blocked. This is a theoretical advantage compared to the neutralization of a single cytokine by monoclonal antibodies. Tofacitinib is a small molecule that is administered orally and selectively inhibits JAK1 and JAK3. This leads to inhibition of signaling through the common γ-chain (γc or CD132). This is a cytokine receptor subunit common to the receptor complexes for several cytokines, including IL-2, IL-7, IL-9, IL-15, IL-21 (64). As these cytokines play pivotal roles in immune function, their blockade by tofacitinib leads to anti-inflammatory effects. Among those are inhibition of the differentiation of effector lymphocytes of the Th2 and Th17 types, suppression of innate immune responses induced by LPS, and attenuation of proinflammatory signaling by interleukin-6 and interferon-γ.
The short-term effect of tofacitinib administration in patients with UC has been tested (65). Patients with moderate-to-severe, active UC were randomized to receive placebo or one of 4 doses of tofacitinib (0.5, 3, 19, 15 mg) twice daily for 8 weeks, and were followed for an additional 4 weeks. At 8-wks clinical response (primary endpoint) was seen in 42% of the placebo group and 32%, 48%, 61%, and 78% for the 0.5, 3, 10, and 15 mg, groups, respectively. The difference was significant for the comparison between placebo and the 15 mg group (P<0.001). Several secondary endpoints reached statistical significance for the high dose tofacitinib group, including clinical remission, endoscopic response, and endoscopic remission. There was also biological response (decrease in CRP, fecal calprotectin) and improvements in the Inflammatory Bowel Disease Questionnaire score. No major safety concerns were raised in these trials. It was noted, however, that, similarly to the trials in rheumatoid arthritis, tofacitinib therapy in UC was associated with a dose-dependent elevation of low-density and high-density lipoprotein cholesterol concentration. This, however, was reversed after treatment cessation. Moreover, 3 patients developed tofacitinib-induced leucopenia (absolute neutrophil count <1500). Taken together, tofacitinib appears to be a promising oral anti-inflammatory treatment for UC (66); nevertheless, its clinical application will largely depend upon the results of long-term, maintenance of response trials and the clarification of the aforementioned safety concerns. It should also be noted that administration of tofacitinib in patients with CD failed to induce a significant clinical benefit (67).
A final way to ameliorate pro-inflammatory mucosal responses is via the enhancement of immunosuppressive elements. This approach was taken in the recent clinical trial on the efficacy of Mongersen in CD (68). Morgensen is an orally administered anti-sense oligonucleotide that acts by inhibiting SMAD7 production. SMAD7 acts as an inhibitor of TGF-β1 and was found to be upregulated in CD. Thus, inactivation of SMAD7 by Morgensen restores TGF-β1 signaling, and, in turn, leads to suppression of inflammatory cytokine production. The efficacy of Mongersen (at increasing doses of 10, 40, or 160 mg per day for 2 weeks) in active CD was tested in a double-blind, placebo-controlled, phase 2 trial. The primary end-point (clinical remission at day 15) was reached by significantly more patients in the 40-mg and 160-mg Mongersen groups (55% and 65%, respectively), as compared to the placebo group (10%, P<0.001). Similar beneficial effects were seen for the secondary endpoint of clinical response which was reached by more patients receiving 10 mg (37%), 40 mg (58%), or 160 mg (72%) of Mongersen than those receiving placebo (17%). The majority of adverse events were related to complications and symptoms of CD. If these results are confirmed in larger trials, they will offer a novel and more convenient (due to the oral administration) therapeutic option for CD.
Anti-adhesion therapeutics
A major development in IBD therapeutics in recent years has been the emergence of treatments that target the trafficking of immunocytes to the inflamed bowel (69). This approach is based upon the fact that dysregulated leukocyte recruitment takes place in immune-mediated chronic inflammatory diseases, including CD and UC. In particular, lymphocytic trafficking has been explored in depth (70, 71). Naïve T-cells migrate to mesenteric lymph nodes, where they encounter antigens, become activated and acquire an effector phenotype. This transformation is associated not only with a specific set of cytokines, but also a distinct repertoire of adhesion molecule and chemokine receptors. These specific molecular signatures allow lymphocytes to recognize tissue-specific endothelial ligands on vascular structures and preferentially recirculate to the corresponding inflamed sites. This is critical for the maintenance of chronic inflammatory processes, including IBD. Thus, the molecules that mediate leukocyte traffic represent attractive therapeutic targets for chronic inflammatory conditions (72). The clinical application of such approaches has been aggressively sought for in recent years (73).
Integrins are cell-adhesion receptors, which are expressed on leukocytes as heterodimeric transmembrane glycoproteins consisting of the combination of one large (α) and one small (β) subunit (74). Integrins recognize and bind adhesion molecules on endothelial cells that belong to the immunoglobulin superfamily. Interactions between α4β7 integrin on lymphocytes and Mucosal Addressin Cell Adhesion Molecule-1 (MAdCAM-1) on endothelial cells are considered specific for recirculation of lymphocytes to the inflamed gut (75). Similarly, the expression of the chemokine CCL25/TECK is strictly restricted to the small intestine. Therefore, interactions between CCL25 and lymphocytes that bear its receptor, CCR9, may underlie the pathogenesis of CD with localization to the terminal ileum (76). This is supported by the fact that patients with small intestinal CD have increased numbers of CCR9+ T cells in the peripheral blood (77). The importance of tissue specificity for certain ligand/receptor combinations is obvious as blockade of these pathways will have effects that will be localized on the particular sites and not affect trafficking to other organs. This might have a significant impact from the safety standpoint as it may lead to decreased systemic toxicity. This was actually learned the hard way in the case of natalizumab. This is an IgG4 humanized monoclonal antibody against α4 integrin (78). It was found to be effective as an induction and maintenance therapy in CD (79). Nevertheless, its further clinical utilization was halted after the reports of progressive multifocal leukoencephalopathy (PML), due to JC virus reactivation in some cases (80). The biology of α4 integrin function explains this adverse outcome. Indeed, α4 is a component of the gut-specific α4β7 integrin (which recognizes MAdCAM-1), but also of the α4β1 integrin (74, 75). The latter binds to vascular cell adhesion molecule 1 (VCAM-1), and this interaction is critical for the prevention of JC virus infection of the brain. The latter is prohibited after blockade of α4 with natalizumab, therefore leading to PML. As a result, the use of natalizumab in CD has been practically abandoned and research has been directed to the blockade of gut-specific pathways. Two monoclonal antibodies that specifically target gut-specific trafficking of lymphocytes are vedolizumab and etrolizumab.
Vedolizumab is a humanized monoclonal antibody that has already been approved for use in both UC and CD (81). It specifically blocks α4β7/MAdCAM-1 binding; hence, it only exerts anti-inflammatory effects on the gut without affecting trafficking to other sites, including the brain. Its efficacy in UC was reported in the GEMINI I trial, which included an induction study (374 patients) and maintenance study in induction responders (373 patients) (82). The induction regimen consisted of two 300 mg intravenous injections at wk 0 and 2. Analysis showed that by wk 6, vedolizumab was significantly more effective that placebo at inducing clinical response (25.5% vs. 47%, P<0.001), clinical remission (5% vs. 17%, P=0.001), and mucosal healing (25% vs. 41%, P=0.001). Similarly, in the maintenance phase, continuous vedolizumab treatment every 4 or 8 wks was more effective than placebo at inducing clinical response (24% vs. 57% and 52%, both P<0.001), durable clinical remission (9% vs. 20.5%, P=0.008 and 24%, P=0.001), and mucosal healing (20% vs. 52% and 56%, both P<0.001) at week 52. The GEMINI II trial that tested the efficacy of vedolizumab in CD had the same design and included 368 patients in the induction study and 461 in the maintenance study (83). Results were not as good as in the UC trial; however, a clear superiority of vedolizumab over placebo was again shown. In the induction phase, by wk 6, more patients on vedolizumab achieved clinical remission (7% vs. 14.5%, P=0.02) and clinical response (26% vs. 31%, not statistically significant). In the maintenance study, significant differences between placebo and vedolizumab every 4 and 8 wks were seen at week 52 for clinical response (30% vs. 43.5%, P=0.01 and 45.5%, P=0.005) and clinical remission (22% vs. 39%, P<0.001 and 36%, P=0.004). Due to the aforementioned association of natalizumab with PML, safety reports from the vedolizumab studies were eagerly awaited. In all, among more than 3000 patients, no cases of PML were reported and no other safety alarms were raised.
Etrolizumab is a fully humanized monoclonal antibody that also demonstrates gut selectivity, attributed to its specificity for the β7 subunit that is shared by integrins α4β7 and αEβ7 (84). The chief ligand for αEβ7 is the epithelial-specific adhesion molecule, E-cadherin. αEβ7/E-cadherin interaction is considered important for T-cell homing to the intestine (75). This adds an important advantage to etrolizumab over other anti-adhesion medications; that is, the reduction of intraepithelial leucocytes in the gut. Recently, etrolizumab was tested in a phase II, randomized controlled trial in moderate-to-severe active UC (85). In this study, 124 patients were randomized to receive as induction therapy, either placebo or one of two regimens of etrolizumab (100 mg or 300 mg plus loading dose). Analysis was performed at wk 10 and showed that the primary endpoint of clinical remission was achieved in significantly higher proportions of patients receiving etrolizumab than placebo [placebo 0/41, etrolizumab 100 mg 8/39 (21%), etrolizumab 300 mg plus loading 4/39 (10%)]. Treatment with etrolizumab was well tolerated. An important aspect of this study was that molecular markers of drug efficacy were studied. In particular, clinical remission rates were highly affected by the baseline mucosal mRNA expression of αE, as well as the numbers of αE+ cells in the colonic biopsy samples. Patients with the highest mucosal expression of the integrin αE were those who responded better. Furthermore, the occupancy of β7 receptors by etrolizumab was quantified on circulating CD4+ and CD8+β7+ T lymphocytes. These findings are complementary to a recent study that reported that patients with the highest expression of mucosal TNF-α were those who responded better to the anti-TNF agent, adalimumab (86). Taken together, these studies are gradually establishing the basis for a personalized medicine-approach to IBD. As a result, currently, several additional anti-adhesion and anti-chemokine medications are in various stages of development and/or clinical testing.
WHAT DO FAILURES TEACH US?
Although several biological agents have reached the “bedside” in recent years, there have been many more that were highly successful at the “bench,” but the subsequent clinical trials failed to show a clear beneficial effect for IBD patients. Although such negative results always lead to disappointment, there are, at the same time, important lessons that can be learned from these failures. These data need to be considered in the future development of novel therapeutic approaches.
First, two decades ago, a simplified model for IBD pathogenesis known as the “CD/Th1 vs. UC/Th2” model was proposed and dictated scientific thinking in regards to therapeutic design. It is now accepted that the inflammatory pathways that take place during IBD are much more complex and that the mucosal milieu found in UC and CD consists of multiple, diverse and highly overlapping pathways (21). The existence of 110 common genetic loci between the two diseases also highlights their similarities, rather than their differences. These concepts are reflected in the failure of the neutralization of lineage-specific cytokines to induce disease remission. Fontolizumab is a monoclonal antibody against the prototypic Th1 molecule, IFN-γ. Fontolizumab was tested in patients with CD, but, so far has failed to induce a meaningful therapeutic effect (87). Similarly, the main Th2 effector cytokine, IL-13 was the target of anrukinzumab, a humanized antibody that inhibits human IL-13. This treatment was tested in UC, as this condition is considered a typical Th2 response, highly dependent upon the actions of IL-13. Nevertheless, a recent randomized, double-blind, placebo-controlled study failed to demonstrate a statistically significant therapeutic effect of (88).
Second, inflammatory mediators exert a variety of immunological functions, which are oftentimes dichotomous and depend on the specific clinical or experimental setting. Such diversity may have important therapeutic implications. This was the case with abatacept, a soluble recombinant fusion protein containing cytotoxic T-lymphocyte-associated antigen 4 and IgG1. This molecule inhibits full activation of T cells after antigen presentation by blocking co-stimulatory signals induced by CD28/CD80 and CD28/CD86 interactions. Although this treatment was effective in rheumatoid arthritis, it failed to produce clinical benefit in either CD or UC (89). The reason for such failure is not clear. However, one possible explanation is that it may be the result of concomitant inhibition of the activation of regulatory T-cells (Tregs). This, in turn, may neutralize any effect on pathogenic T cell development and lead to treatment failure. Therefore, blockade of pathways specific for T-effector cell development may be required for a therapeutic effect.
Third, safety issues, including unexpected ones, should always be kept in mind. The primary function of the immune system is the fight against infectious agents and its blockade may lead to uncontrollable local growth, or even systemic dissemination, of microorganisms. In addition, manipulation of the immune system may have paradoxical effects due to the redundant functions of diverse pathways. Such was the case with visilizumab, a monoclonal antibody that induces apoptosis in T-cells by blocking the CD3 chain of the T-cell receptor (90). This biological agent was found, in initial trials, to be effective in inducing remission in steroid-resistant UC (91, 92). The drug was also tested in CD. Nevertheless, any efficacy of this medication was hampered by the induction of a cytokine release syndrome and infectious complications (93). Therefore, T-cell apoptosis was associated not only with disease remission, but also with deleterious adverse events. Similarly, blockade of IL-17A was considered a very promising therapy for CD, given the pivotal importance that has been attributed to the IL-23/Th17 pathway in IBD. In a recent study, secukinumab, a human anti-IL-17A monoclonal antibody, was administered in patients with moderate to severe CD (94). Quite unexpectedly, this treatment was associated with worsening of disease and serious adverse effects, which led to the premature discontinuation of the trial. Although these results have not been explained yet, it should be kept in mind that IL-17A is a major regulator of intestinal mucosal homeostasis and contributor to defense against certain microorganisms, including fungi. Failure of these mucosal defenses in the advent of anti-IL-17A blockade may explain the failure of such treatments.
Finally, it has been increasingly recognized that, instead of representing a single entity with two subcategories, IBD represents a broader clinical syndrome, with chronic intestinal inflammation being the predominant clinical phenotype. At the same time, the immunopathogenesis may differ between separate cases and distinct groups are recognized with unique signatures regarding environmental triggers, alterations of the microbiome, genetic polymorphisms, and immunological pathways. It is therefore expected that reversal of chronic intestinal inflammation will depend on the correction of the responsible abnormalities and may differ between patient subgroups. Such differences may also explain why in clinical trials with biological some patients respond dramatically, whereas, in others, the severity of disease is not substantially affected. In a characteristic scenario, several patients do not respond to anti-TNF blockade, although drug trough levels are well within the adequate therapeutic range.
CONCLUSIONS
As the pathogenesis of IBD is gradually revealed and the underlying immunological pathways delineated, an abundance of therapeutic targets will continue to emerge. Although the majority of such drugs will never reach the clinical application stage, they will continue to offer important insights into disease pathogenesis and treatment. In parallel, the identification of environmental, microbiomic, molecular, and immunological signatures will allow the recognition of subgroups of patients with defined characteristics. As the pathogenetic mechanisms will be narrowed down in such cases, this will allow for the application of more targeted therapies that will be more effective and demonstrate a safer profile.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med. 2009;361:2066–2078. doi: 10.1056/NEJMra0804647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ordas I, Eckmann L, Talamini M, Baumgart DC, Sandborn WJ. Ulcerative colitis. Lancet. 2012;380:1606–1619. doi: 10.1016/S0140-6736(12)60150-0. [DOI] [PubMed] [Google Scholar]
- 3.Baumgart DC, Sandborn WJ. Crohn’s disease. Lancet. 2012;380:1590–1605. doi: 10.1016/S0140-6736(12)60026-9. [DOI] [PubMed] [Google Scholar]
- 4.Burisch J, Jess T, Martinato M, Lakatos PL. The burden of inflammatory bowel disease in Europe. J Crohns Colitis. 2013;7:322–337. doi: 10.1016/j.crohns.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 5.Zallot C, Peyrin-Biroulet L. Deep remission in inflammatory bowel disease: looking beyond symptoms. Curr Gastroenterol Rep. 2013;15:315. doi: 10.1007/s11894-013-0315-7. [DOI] [PubMed] [Google Scholar]
- 6.Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol. 2010;28:573–621. doi: 10.1146/annurev-immunol-030409-101225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Graham DB, Xavier RJ. From genetics of inflammatory bowel disease towards mechanistic insights. Trends Immunol. 2013;34:371–378. doi: 10.1016/j.it.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Limbergen J, Radford-Smith G, Satsangi J. Advances in IBD genetics. Nat Rev Gastroenterol Hepatol. 2014;11:372–385. doi: 10.1038/nrgastro.2014.27. [DOI] [PubMed] [Google Scholar]
- 10.O’Toole A, Korzenik J. Environmental triggers for IBD. Curr Gastroenterol Rep. 2014;16:396. doi: 10.1007/s11894-014-0396-y. [DOI] [PubMed] [Google Scholar]
- 11.Parkes GC, Whelan K, Lindsay JO. Smoking in inflammatory bowel disease: impact on disease course and insights into the aetiology of its effect. J Crohns Colitis. 2014;8:717–725. doi: 10.1016/j.crohns.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 12.Ananthakrishnan AN. Epidemiology and risk factors for IBD. Nat Rev Gastroenterol Hepatol. 2015 doi: 10.1038/nrgastro.2015.34. [DOI] [PubMed] [Google Scholar]
- 13.Karatzas PS, Gazouli M, Safioleas M, Mantzaris GJ. DNA methylation changes in inflammatory bowel disease. Ann Gastroenterol. 2014;27:125–132. [PMC free article] [PubMed] [Google Scholar]
- 14.Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature. 2011;474:298–306. doi: 10.1038/nature10208. [DOI] [PubMed] [Google Scholar]
- 15.Huttenhower C, Kostic AD, Xavier RJ. Inflammatory bowel disease as a model for translating the microbiome. Immunity. 2014;40:843–854. doi: 10.1016/j.immuni.2014.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peterson CT, Sharma V, Elmen L, Peterson SN. Immune homeostasis, dysbiosis and therapeutic modulation of the gut microbiota. Clin Exp Immunol. 2015;179:363–377. doi: 10.1111/cei.12474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cader MZ, Kaser A. Recent advances in inflammatory bowel disease: mucosal immune cells in intestinal inflammation. Gut. 2013;62:1653–1664. doi: 10.1136/gutjnl-2012-303955. [DOI] [PubMed] [Google Scholar]
- 18.Hart AL, Ng SC, Mann E, Al-Hassi HO, Bernardo D, Knight SC. Homing of immune cells: role in homeostasis and intestinal inflammation. Inflamm Bowel Dis. 2010;16:1969–1977. doi: 10.1002/ibd.21304. [DOI] [PubMed] [Google Scholar]
- 19.Bamias G, Arseneau KO, Cominelli F. Cytokines and mucosal immunity. Curr Opin Gastroenterol. 2014;30:547–552. doi: 10.1097/MOG.0000000000000118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Strober W, Fuss IJ. Proinflammatory cytokines in the pathogenesis of inflammatory bowel diseases. Gastroenterology. 2011;140:1756–1767. doi: 10.1053/j.gastro.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bamias G, Nyce MR, De La Rue SA, Cominelli F. New concepts in the pathophysiology of inflammatory bowel disease. Ann Intern Med. 2005;143:895–904. doi: 10.7326/0003-4819-143-12-200512200-00007. [DOI] [PubMed] [Google Scholar]
- 22.Bamias G, Corridoni D, Pizarro TT, Cominelli F. New insights into the dichotomous role of innate cytokines in gut homeostasis and inflammation. Cytokine. 2012;59:451–459. doi: 10.1016/j.cyto.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marks DJ, Harbord MW, MacAllister R, Rahman FZ, Young J, Al-Lazikani B, Lees W, Novelli M, Bloom S, Segal AW. Defective acute inflammation in Crohn’s disease: a clinical investigation. Lancet. 2006;367:668–678. doi: 10.1016/S0140-6736(06)68265-2. [DOI] [PubMed] [Google Scholar]
- 24.Mokry M, Middendorp S, Wiegerinck CL, Witte M, Teunissen H, Meddens CA, Cuppen E, Clevers H, Nieuwenhuis EE. Many inflammatory bowel disease risk loci include regions that regulate gene expression in immune cells and the intestinal epithelium. Gastroenterology. 2014;146:1040–1047. doi: 10.1053/j.gastro.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 25.Gersemann M, Wehkamp J, Stange EF. Innate immune dysfunction in inflammatory bowel disease. J Intern Med. 2012;271:421–428. doi: 10.1111/j.1365-2796.2012.02515.x. [DOI] [PubMed] [Google Scholar]
- 26.Pastorelli L, De Salvo C, Mercado JR, Vecchi M, Pizarro TT. Central role of the gut epithelial barrier in the pathogenesis of chronic intestinal inflammation: lessons learned from animal models and human genetics. Front Immunol. 2013;4:280. doi: 10.3389/fimmu.2013.00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cho JH, Brant SR. Recent insights into the genetics of inflammatory bowel disease. Gastroenterology. 2011;140:1704–1712. doi: 10.1053/j.gastro.2011.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Uhlig HH. Monogenic diseases associated with intestinal inflammation: implications for the understanding of inflammatory bowel disease. Gut. 2013;62:1795–1805. doi: 10.1136/gutjnl-2012-303956. [DOI] [PubMed] [Google Scholar]
- 29.Shah N, Kammermeier J, Elawad M, Glocker EO. Interleukin-10 and interleukin-10-receptor defects in inflammatory bowel disease. Curr Allergy Asthma Rep. 2012;12:373–379. doi: 10.1007/s11882-012-0286-z. [DOI] [PubMed] [Google Scholar]
- 30.Glocker EO, Kotlarz D, Boztug K, Gertz EM, Schaffer AA, Noyan F, Perro M, Diestelhorst J, Allroth A, Murugan D, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. 2009;361:2033–2045. doi: 10.1056/NEJMoa0907206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kotlarz D, Beier R, Murugan D, Diestelhorst J, Jensen O, Boztug K, Pfeifer D, Kreipe H, Pfister ED, Baumann U, et al. Loss of interleukin-10 signaling and infantile inflammatory bowel disease: implications for diagnosis and therapy. Gastroenterology. 2012;143:347–355. doi: 10.1053/j.gastro.2012.04.045. [DOI] [PubMed] [Google Scholar]
- 32.Uhlig HH, Schwerd T, Koletzko S, Shah N, Kammermeier J, Elkadri A, Ouahed J, Wilson DC, Travis SP, Turner D, et al. The diagnostic approach to monogenic very early onset inflammatory bowel disease. Gastroenterology. 2014;147:990–1007. e1003. doi: 10.1053/j.gastro.2014.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oyama Y, Craig RM, Traynor AE, Quigley K, Statkute L, Halverson A, Brush M, Verda L, Kowalska B, Krosnjar N, et al. Autologous hematopoietic stem cell transplantation in patients with refractory Crohn’s disease. Gastroenterology. 2005;128:552–563. doi: 10.1053/j.gastro.2004.11.051. [DOI] [PubMed] [Google Scholar]
- 34.Cassinotti A, Annaloro C, Ardizzone S, Onida F, Della Volpe A, Clerici M, Usardi P, Greco S, Maconi G, Porro GB, et al. Autologous haematopoietic stem cell transplantation without CD34+ cell selection in refractory Crohn’s disease. Gut. 2008;57:211–217. doi: 10.1136/gut.2007.128694. [DOI] [PubMed] [Google Scholar]
- 35.Hansen JJ, Sartor RB. Therapeutic Manipulation of the Microbiome in IBD: Current Results and Future Approaches. Curr Treat Options Gastroenterol. 2015;13:105–120. doi: 10.1007/s11938-014-0042-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holubar SD, Cima RR, Sandborn WJ, Pardi DS. Treatment and prevention of pouchitis after ileal pouch-anal anastomosis for chronic ulcerative colitis. Cochrane Database Syst Rev. 2010 doi: 10.1002/14651858.CD001176.pub2. CD001176. [DOI] [PubMed] [Google Scholar]
- 37.Doherty GA, Bennett GC, Cheifetz AS, Moss AC. Meta-analysis: targeting the intestinal microbiota in prophylaxis for post-operative Crohn’s disease. Aliment Pharmacol Ther. 2010;31:802–809. doi: 10.1111/j.1365-2036.2010.04231.x. [DOI] [PubMed] [Google Scholar]
- 38.Pizarro TT, Pastorelli L, Bamias G, Garg RR, Reuter BK, Mercado JR, Chieppa M, Arseneau KO, Ley K, Cominelli F. SAMP1/YitFc mouse strain: a spontaneous model of Crohn’s disease-like ileitis. Inflamm Bowel Dis. 2011;17:2566–2584. doi: 10.1002/ibd.21638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rivera-Nieves J, Bamias G, Vidrich A, Marini M, Pizarro TT, McDuffie MJ, Moskaluk CA, Cohn SM, Cominelli F. Emergence of perianal fistulizing disease in the SAMP1/YitFc mouse, a spontaneous model of chronic ileitis. Gastroenterology. 2003;124:972–982. doi: 10.1053/gast.2003.50148. [DOI] [PubMed] [Google Scholar]
- 40.Bamias G, Marini M, Moskaluk CA, Odashima M, Ross WG, Rivera-Nieves J, Cominelli F. Down-regulation of intestinal lymphocyte activation and Th1 cytokine production by antibiotic therapy in a murine model of Crohn’s disease. J Immunol. 2002;169:5308–5314. doi: 10.4049/jimmunol.169.9.5308. [DOI] [PubMed] [Google Scholar]
- 41.Pagnini C, Saeed R, Bamias G, Arseneau KO, Pizarro TT, Cominelli F. Probiotics promote gut health through stimulation of epithelial innate immunity. Proc Natl Acad Sci U S A. 2010;107:454–459. doi: 10.1073/pnas.0910307107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smits LP, Bouter KE, de Vos WM, Borody TJ, Nieuwdorp M. Therapeutic potential of fecal microbiota transplantation. Gastroenterology. 2013;145:946–953. doi: 10.1053/j.gastro.2013.08.058. [DOI] [PubMed] [Google Scholar]
- 43.Anderson JL, Edney RJ, Whelan K. Systematic review: faecal microbiota transplantation in the management of inflammatory bowel disease. Aliment Pharmacol Ther. 2012;36:503–516. doi: 10.1111/j.1365-2036.2012.05220.x. [DOI] [PubMed] [Google Scholar]
- 44.Cammarota G, Ianiro G, Cianci R, Bibbo S, Gasbarrini A, Curro D. The involvement of gut microbiota in inflammatory bowel disease pathogenesis: Potential for therapy. Pharmacol Ther. 2015 doi: 10.1016/j.pharmthera.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 45.Angelberger S, Reinisch W, Makristathis A, Lichtenberger C, Dejaco C, Papay P, Novacek G, Trauner M, Loy A, Berry D. Temporal bacterial community dynamics vary among ulcerative colitis patients after fecal microbiota transplantation. Am J Gastroenterol. 2013;108:1620–1630. doi: 10.1038/ajg.2013.257. [DOI] [PubMed] [Google Scholar]
- 46.Kump PK, Grochenig HP, Lackner S, Trajanoski S, Reicht G, Hoffmann KM, Deutschmann A, Wenzl HH, Petritsch W, Krejs GJ, et al. Alteration of intestinal dysbiosis by fecal microbiota transplantation does not induce remission in patients with chronic active ulcerative colitis. Inflamm Bowel Dis. 2013;19:2155–2165. doi: 10.1097/MIB.0b013e31829ea325. [DOI] [PubMed] [Google Scholar]
- 47.Moayyedi P, Surette MG, Kim PT, Libertucci J, Wolfe M, Onischi C, Armstrong D, Marshall JK, Kassam Z, Reinisch W, et al. Fecal Microbiota Transplantation Induces Remission in Patients With Active Ulcerative Colitis in a Randomized Controlled Trial. Gastroenterology. 2015;149:102–109. e106. doi: 10.1053/j.gastro.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 48.Rossen NG, Fuentes S, van der Spek MJ, Tijssen JG, Hartman JH, Duflou A, Lowenberg M, van den Brink GR, Mathus-Vliegen EM, de Vos WM, et al. Findings From a Randomized Controlled Trial of Fecal Transplantation for Patients With Ulcerative Colitis. Gastroenterology. 2015;149:110–118. e114. doi: 10.1053/j.gastro.2015.03.045. [DOI] [PubMed] [Google Scholar]
- 49.Torres J, Danese S, Colombel JF. New therapeutic avenues in ulcerative colitis: thinking out of the box. Gut. 2013;62:1642–1652. doi: 10.1136/gutjnl-2012-303959. [DOI] [PubMed] [Google Scholar]
- 50.Rousseaux C, Lefebvre B, Dubuquoy L, Lefebvre P, Romano O, Auwerx J, Metzger D, Wahli W, Desvergne B, Naccari GC, et al. Intestinal antiinflammatory effect of 5-aminosalicylic acid is dependent on peroxisome proliferator-activated receptor-gamma. J Exp Med. 2005;201:1205–1215. doi: 10.1084/jem.20041948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Buchman AL, Fife C, Torres C, Smith L, Aristizibal J. Hyperbaric oxygen therapy for severe ulcerative colitis. J Clin Gastroenterol. 2001;33:337–339. doi: 10.1097/00004836-200110000-00018. [DOI] [PubMed] [Google Scholar]
- 52.Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med. 2011;364:656–665. doi: 10.1056/NEJMra0910283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ehehalt R, Wagenblast J, Erben G, Lehmann WD, Hinz U, Merle U, Stremmel W. Phosphatidylcholine and lysophosphatidylcholine in intestinal mucus of ulcerative colitis patients. A quantitative approach by nanoElectrospray-tandem mass spectrometry. Scand J Gastroenterol. 2004;39:737–742. doi: 10.1080/00365520410006233. [DOI] [PubMed] [Google Scholar]
- 54.Treede I, Braun A, Sparla R, Kuhnel M, Giese T, Turner JR, Anes E, Kulaksiz H, Fullekrug J, Stremmel W, et al. Anti-inflammatory effects of phosphatidylcholine. J Biol Chem. 2007;282:27155–27164. doi: 10.1074/jbc.M704408200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stremmel W, Merle U, Zahn A, Autschbach F, Hinz U, Ehehalt R. Retarded release phosphatidylcholine benefits patients with chronic active ulcerative colitis. Gut. 2005;54:966–971. doi: 10.1136/gut.2004.052316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stremmel W, Ehehalt R, Autschbach F, Karner M. Phosphatidylcholine for steroid-refractory chronic ulcerative colitis: a randomized trial. Ann Intern Med. 2007;147:603–610. doi: 10.7326/0003-4819-147-9-200711060-00004. [DOI] [PubMed] [Google Scholar]
- 57.Karner M, Kocjan A, Stein J, Schreiber S, von Boyen G, Uebel P, Schmidt C, Kupcinskas L, Dina I, Zuelch F, et al. First multicenter study of modified release phosphatidylcholine “LT-02” in ulcerative colitis: a randomized, placebo-controlled trial in mesalazine-refractory courses. Am J Gastroenterol. 2014;109:1041–1051. doi: 10.1038/ajg.2014.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Billiet T, Rutgeerts P, Ferrante M, Van Assche G, Vermeire S. Targeting TNFalpha for the treatment of inflammatory bowel disease. Expert Opin Biol Ther. 2014;14:75–101. doi: 10.1517/14712598.2014.858695. [DOI] [PubMed] [Google Scholar]
- 59.Siakavellas SI, Bamias G. Role of the IL-23/IL-17 axis in Crohn’s disease. Discov Med. 2012;14:253–262. [PubMed] [Google Scholar]
- 60.Sandborn WJ, Feagan BG, Fedorak RN, Scherl E, Fleisher MR, Katz S, Johanns J, Blank M, Rutgeerts P. A randomized trial of Ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with moderate-to-severe Crohn’s disease. Gastroenterology. 2008;135:1130–1141. doi: 10.1053/j.gastro.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 61.Toedter GP, Blank M, Lang Y, Chen D, Sandborn WJ, de Villiers WJ. Relationship of C-reactive protein with clinical response after therapy with ustekinumab in Crohn’s disease. Am J Gastroenterol. 2009;104:2768–2773. doi: 10.1038/ajg.2009.454. [DOI] [PubMed] [Google Scholar]
- 62.Sandborn WJ, Gasink C, Gao LL, Blank MA, Johanns J, Guzzo C, Sands BE, Hanauer SB, Targan S, Rutgeerts P, et al. Ustekinumab induction and maintenance therapy in refractory Crohn’s disease. N Engl J Med. 2012;367:1519–1528. doi: 10.1056/NEJMoa1203572. [DOI] [PubMed] [Google Scholar]
- 63.Coskun M, Salem M, Pedersen J, Nielsen OH. Involvement of JAK/STAT signaling in the pathogenesis of inflammatory bowel disease. Pharmacol Res. 2013;76:1–8. doi: 10.1016/j.phrs.2013.06.007. [DOI] [PubMed] [Google Scholar]
- 64.Vuitton L, Koch S, Peyrin-Biroulet L. Janus kinase inhibition with tofacitinib: changing the face of inflammatory bowel disease treatment. Curr Drug Targets. 2013;14:1385–1391. doi: 10.2174/13894501113149990160. [DOI] [PubMed] [Google Scholar]
- 65.Sandborn WJ, Ghosh S, Panes J, Vranic I, Su C, Rousell S, Niezychowski W. Tofacitinib, an oral Janus kinase inhibitor, in active ulcerative colitis. N Engl J Med. 2012;367:616–624. doi: 10.1056/NEJMoa1112168. [DOI] [PubMed] [Google Scholar]
- 66.Panes J, Su C, Bushmakin AG, Cappelleri JC, Mamolo C, Healey P. Randomized trial of tofacitinib in active ulcerative colitis: analysis of efficacy based on patientreported outcomes. BMC Gastroenterol. 2015;15:14. doi: 10.1186/s12876-015-0239-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sandborn WJ, Ghosh S, Panes J, Vranic I, Wang W, Niezychowski W. A phase 2 study of tofacitinib, an oral Janus kinase inhibitor, in patients with Crohn’s disease. Clin Gastroenterol Hepatol. 2014;12:1485–1493. e1482. doi: 10.1016/j.cgh.2014.01.029. [DOI] [PubMed] [Google Scholar]
- 68.Monteleone G, Neurath MF, Ardizzone S, Di Sabatino A, Fantini MC, Castiglione F, Scribano ML, Armuzzi A, Caprioli F, Sturniolo GC, et al. Mongersen, an oral SMAD7 antisense oligonucleotide, and Crohn’s disease. N Engl J Med. 2015;372:1104–1113. doi: 10.1056/NEJMoa1407250. [DOI] [PubMed] [Google Scholar]
- 69.Lobaton T, Vermeire S, Van Assche G, Rutgeerts P. Review article: anti-adhesion therapies for inflammatory bowel disease. Aliment Pharmacol Ther. 2014;39:579–594. doi: 10.1111/apt.12639. [DOI] [PubMed] [Google Scholar]
- 70.Springer TA. Traffic signals on endothelium for lymphocyte recirculation and leukocyte emigration. Annu Rev Physiol. 1995;57:827–872. doi: 10.1146/annurev.ph.57.030195.004143. [DOI] [PubMed] [Google Scholar]
- 71.von Andrian UH, Mackay CR. T-cell function and migration. Two sides of the same coin. N Engl J Med. 2000;343:1020–1034. doi: 10.1056/NEJM200010053431407. [DOI] [PubMed] [Google Scholar]
- 72.Arseneau K, Cominelli F. Targeting leukocyte trafficking for the treatment of inflammatory bowel disease. Clin Pharmacol Ther. 2015;97:22–28. doi: 10.1002/cpt.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bamias G, Clark DJ, Rivera-Nieves J. Leukocyte traffic blockade as a therapeutic strategy in inflammatory bowel disease. Curr Drug Targets. 2013;14:1490–1500. doi: 10.2174/13894501113149990158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 75.Gorfu G, Rivera-Nieves J, Ley K. Role of beta7 integrins in intestinal lymphocyte homing and retention. Curr Mol Med. 2009;9:836–850. doi: 10.2174/156652409789105525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kunkel EJ, Butcher EC. Chemokines and the tissue-specific migration of lymphocytes. Immunity. 2002;16:1–4. doi: 10.1016/s1074-7613(01)00261-8. [DOI] [PubMed] [Google Scholar]
- 77.Papadakis KA, Prehn J, Moreno ST, Cheng L, Kouroumalis EA, Deem R, Breaverman T, Ponath PD, Andrew DP, Green PH, et al. CCR9-positive lymphocytes and thymus-expressed chemokine distinguish small bowel from colonic Crohn’s disease. Gastroenterology. 2001;121:246–254. doi: 10.1053/gast.2001.27154. [DOI] [PubMed] [Google Scholar]
- 78.Yu Y, Schurpf T, Springer TA. How natalizumab binds and antagonizes alpha4 integrins. J Biol Chem. 2013;288:32314–32325. doi: 10.1074/jbc.M113.501668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ghosh S, Goldin E, Gordon FH, Malchow HA, Rask-Madsen J, Rutgeerts P, Vyhnalek P, Zadorova Z, Palmer T, Donoghue S. Natalizumab for active Crohn’s disease. N Engl J Med. 2003;348:24–32. doi: 10.1056/NEJMoa020732. [DOI] [PubMed] [Google Scholar]
- 80.Van Assche G, Van Ranst M, Sciot R, Dubois B, Vermeire S, Noman M, Verbeeck J, Geboes K, Robberecht W, Rutgeerts P. Progressive multifocal leukoencephalopathy after natalizumab therapy for Crohn’s disease. N Engl J Med. 2005;353:362–368. doi: 10.1056/NEJMoa051586. [DOI] [PubMed] [Google Scholar]
- 81.Raine T. Vedolizumab for inflammatory bowel disease: Changing the game, or more of the same? United European Gastroenterol J. 2014;2:333–344. doi: 10.1177/2050640614550672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Feagan BG, Rutgeerts P, Sands BE, Hanauer S, Colombel JF, Sandborn WJ, Van Assche G, Axler J, Kim HJ, Danese S, et al. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2013;369:699–710. doi: 10.1056/NEJMoa1215734. [DOI] [PubMed] [Google Scholar]
- 83.Sandborn WJ, Feagan BG, Rutgeerts P, Hanauer S, Colombel JF, Sands BE, Lukas M, Fedorak RN, Lee S, Bressler B, et al. Vedolizumab as induction and maintenance therapy for Crohn’s disease. N Engl J Med. 2013;369:711–721. doi: 10.1056/NEJMoa1215739. [DOI] [PubMed] [Google Scholar]
- 84.Stefanich EG, Danilenko DM, Wang H, O’Byrne S, Erickson R, Gelzleichter T, Hiraragi H, Chiu H, Ivelja S, Jeet S, et al. A humanized monoclonal antibody targeting the beta7 integrin selectively blocks intestinal homing of T lymphocytes. Br J Pharmacol. 2011;162:1855–1870. doi: 10.1111/j.1476-5381.2011.01205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vermeire S, O’Byrne S, Keir M, Williams M, Lu TT, Mansfield JC, Lamb CA, Feagan BG, Panes J, Salas A, et al. Etrolizumab as induction therapy for ulcerative colitis: a randomised, controlled, phase 2 trial. Lancet. 2014;384:309–318. doi: 10.1016/S0140-6736(14)60661-9. [DOI] [PubMed] [Google Scholar]
- 86.Atreya R, Neumann H, Neufert C, Waldner MJ, Billmeier U, Zopf Y, Willma M, App C, Munster T, Kessler H, et al. In vivo imaging using fluorescent antibodies to tumor necrosis factor predicts therapeutic response in Crohn’s disease. Nat Med. 2014;20:313–318. doi: 10.1038/nm.3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Reinisch W, de Villiers W, Bene L, Simon L, Racz I, Katz S, Altorjay I, Feagan B, Riff D, Bernstein CN, et al. Fontolizumab in moderate to severe Crohn’s disease: a phase 2, randomized, double-blind, placebo-controlled, multiple-dose study. Inflamm Bowel Dis. 2010;16:233–242. doi: 10.1002/ibd.21038. [DOI] [PubMed] [Google Scholar]
- 88.Reinisch W, Panes J, Khurana S, Toth G, Hua F, Comer GM, Hinz M, Page K, O’Toole M, McDonnell Moorehead T, et al. Anrukinzumab, an anti-interleukin 13 monoclonal antibody, in active UC: efficacy and safety from a phase IIa randomised multicentre study. Gut. 2015 doi: 10.1136/gutjnl-2014-308337. [DOI] [PubMed] [Google Scholar]
- 89.Sandborn WJ, Colombel JF, Sands BE, Rutgeerts P, Targan SR, Panaccione R, Bressler B, Geboes K, Schreiber S, Aranda R, et al. Abatacept for Crohn’s disease and ulcerative colitis. Gastroenterology. 2012;143:62–69. e64. doi: 10.1053/j.gastro.2012.04.010. [DOI] [PubMed] [Google Scholar]
- 90.Yu QT, Saruta M, Papadakis KA. Visilizumab induces apoptosis of mucosal T lymphocytes in ulcerative colitis through activation of caspase 3 and 8 dependent pathways. Clin Immunol. 2008;127:322–329. doi: 10.1016/j.clim.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 91.Baumgart DC, Targan SR, Dignass AU, Mayer L, van Assche G, Hommes DW, Hanauer SB, Mahadevan U, Reinisch W, Plevy SE, et al. Prospective randomized open-label multicenter phase I/II dose escalation trial of visilizumab (HuM291) in severe steroid-refractory ulcerative colitis. Inflamm Bowel Dis. 2010;16:620–629. doi: 10.1002/ibd.21084. [DOI] [PubMed] [Google Scholar]
- 92.Plevy S, Salzberg B, Van Assche G, Regueiro M, Hommes D, Sandborn W, Hanauer S, Targan S, Mayer L, Mahadevan U, et al. A phase I study of visilizumab, a humanized anti-CD3 monoclonal antibody, in severe steroid-refractory ulcerative colitis. Gastroenterology. 2007;133:1414–1422. doi: 10.1053/j.gastro.2007.08.035. [DOI] [PubMed] [Google Scholar]
- 93.Baumgart DC, Lowder JN, Targan SR, Sandborn WJ, Frankel MB. Transient cytokine-induced liver injury following administration of the humanized anti-CD3 antibody visilizumab (HuM291) in Crohn’s disease. Am J Gastroenterol. 2009;104:868–876. doi: 10.1038/ajg.2008.138. [DOI] [PubMed] [Google Scholar]
- 94.Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, Higgins PD, Wehkamp J, Feagan BG, Yao MD, Karczewski M, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. 2012;61:1693–1700. doi: 10.1136/gutjnl-2011-301668. [DOI] [PMC free article] [PubMed] [Google Scholar]