

Abstract
Over the past few years, CuH-catalyzed hydroamination has been discovered and developed as a robust and conceptually novel approach for the synthesis of enantioenriched secondary and tertiary amines. The success in this area of research was made possible through the large body of precedent in copper(I) hydride catalysis and the well-explored use of hydroxylamine esters as electrophilic amine sources in related copper-catalyzed processes. This mini-review details the background, advances, and mechanistic investigations in CuH-catalyzed hydroamination.
Keywords: hydroamination, copper catalysis, copper(I) hydride, chiral amine synthesis, asymmetric catalysis
Graphical Abstract
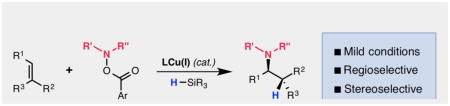
CuH-catalyzed hydroamination has recently been developed as a viable method for synthesizing a broad range of chiral aliphatic amines in excellent efficiencies and enantioselectivites. This minireview highlights advancements made in this area of catalysis along with the precedent that has led to these discoveries.
1. Introduction
Due to the ubiquitous and indispensible presence of aliphatic amines in biologically active molecules, the development of general methods for their preparation have been the target of considerable effort in the past few decades.[1] Nucleophilic substitution, reductive amination, and metal-catalyzed allylic substitution are general and widely used strategies for amine synthesis.[2] Hydroamination, the net addition of an amino group and hydrogen atom across a carbon–carbon double bond, represents a complementary and direct approach to access amines from readily available alkene and alkyne precursors. While the overall process is generally feasible from a thermodynamic perspective, the hydroamination of unactivated substrates is not kinetically viable in the absence of a catalyst. In addition, the use of a catalyst provides the opportunity for control over the chemo-, regio-, and stereoselectivity of the reaction. Consequently, efficient, general, and selective catalysis for hydroamination has been a much sought-after goal for synthetic chemists.
Hydroamination reactions can be broadly classified in terms of regiochemical outcome. Markovnikov hydroamination results in the installation of an amino group on the more substituted carbon of the alkene while an anti-Markovnikov process installs an amino group on the less substituted carbon. Markovnikov hydroamination commonly generates α-branched amine products of type 2 and introduces a stereocenter. Thus, enantioselective variants of this process are especially valuable for their potential applicability to the enantioselective preparation of medicinally relevant amines with the formation of a new stereogenic center. On the other hand, the anti-Markovnikov hydroamination of simple alkenes provides direct access to linear, aliphatic amines of type 1, a common structural motif found in pharmaceuticals, agrochemicals, and natural product isolates. The factors that govern regioselectivity for particular hydroamination processes will be discussed in more detail later in this mini-review.
A variety of approaches toward catalytic hydroamination have been advanced, including metal-catalyzed,[3] Brønsted acid-catalyzed,[4] radical-mediated,[5] and pericyclic processes.[6] In the vast majority of these, a primary or secondary amine is directly employed as a substrate, resulting in the formal addition of the N–H bond of the amine across the carbon–carbon π bond. These protocols are, therefore, ideal from the standpoint of atom economy, and the amine precursors are readily available. While these previously reported processes represent considerable advances in catalysis, and many of the reported methods are efficient and selective, the development of hydroamination methods that are reliably stereoselective, mild, and applicable to a wide range of both functionalized and unfunctionalized substrates, including unactivated olefins, remains an important objective of ongoing research.
In 2013, two research groups independently reported the use of ligated copper(I) hydride (CuH) species as key catalytic intermediates for enantio- and regioselective hydroamination reactions.[7] In these protocols, the amino group is derived from an electrophilic aminating reagent while the hydrogen atom is delivered from a hydridic source, the CuH species. Although this reversed polarity diminishes the atom economy of the hydroamination process, this strategy possesses several advantages over previously reported hydroamination approaches. For instance, the steric and electronic properties of the aminating reagent can be systematically adjusted, a property that was exploited in several of the recently reported hydroamination protocols. Furthermore, while CuH complexes are well known for reducing certain carbonyl groups,[8] these species are otherwise remarkably chemoselective toward olefin or alkyne insertion. Thus, sensitive functional groups are often tolerated, and the substrate scope is generally broad. In addition, the reactions proceed with excellent regio- and enantiocontrol for a wide range of substrates. Finally, the use of mild reaction conditions and an earth-abundant transition metal are additional attractive features of this approach.
2. Development and study of copper(I) hydride complexes, hydroxylamine esters and related reactions
The development of CuH-catalyzed hydroamination drew upon a large body of precedent pertaining to the use of copper(I) hydride (CuH) complexes for reductive transformations in organic synthesis, as well as the use of hydroxylamine esters as electrophilic aminating reagents for the Cu-catalyzed formation of C—N bonds. Although copper(I) hydride was first prepared as a polymeric solid in the 1840s,[9] well-defined, phosphine-ligated CuH complexes were not studied until the latter half of the 20th century.[10] In the late 1980s, Stryker popularized the use of a hexameric CuH complex, [(Ph3P)CuH]6, as a mild reagent for the 1,4-reduction of α, β-unsaturated ketones and esters.[11]
Shortly after the development of CuH complexes as stoichiometric reagents, efforts were made to utilize these complexes in a catalytic fashion. In an early report, Brunner demonstrated that Cu-phosphine complexes catalyzed the hydrosilylation of acetophenone with modest levels of enantioinduction.[12] Subsequently, high-pressure hydrogen gas was also found to regenerate the CuH species for the catalytic, conjugate reductions of α, β-unsaturated ketones.[13] Lipshutz later developed a more practical procedure for the use of [(Ph3P)CuH]6 under catalytic conditions by employing Bu3SnH or PhSiH3 as the stoichiometric hydride source.[14] The first highly enantioselective application of CuH chemistry was achieved by employing a chiral bisphosphine ligand (Tol-BINAP, L1) to effect the asymmetric 1,4-reduction of prochiral α, β-unsaturated esters.[15a] In this system, phosphine-ligated CuH species 3 is believed to undergo enantioselective addition into the C–C double bond of unsaturated ester 4, followed by transmetalation of resulting copper(I) enolate 5 with the hydride source, polymethylhydrosiloxane (PMHS), to regenerate CuH species 3 (Scheme 2). Several other classes of α, β-unsaturated compounds were subsequently demonstrated to be suitable substrates for this process.[15] Important developments by Lipshutz have led to the identification of several catalyst systems, including those based on the BIPHEP and SEGPHOS-ligated CuH species, that are exceptionally active and enantioselective for ketone and imine reduction.[16]
Scheme 2.
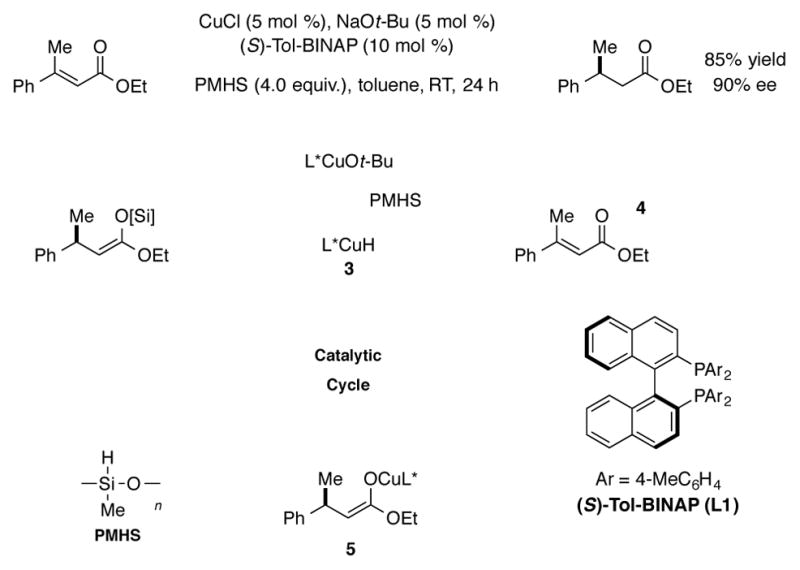
Enantioselective CuH-catalyzed reduction of prochiral enoates.
Although activated olefins, such as enoates, enones, and vinylnitriles, have been thoroughly investigated as substrates for CuH catalysis, other types of alkenes have only been explored recently. In 2009, Yun reported a Cu-catalyzed enantioselective hydroboration of simple styrenes in which the olefin substrate was proposed to undergo hydrocupration in the presence of a bisphosphine-ligated CuH species, providing an important precedent for the use of less activated alkenes in CuH-mediated processes.[17]
Prior to their use as aminating reagents for CuH-catalyzed hydroamination, hydroxylamine esters had already been widely employed as electrophilic sources of nitrogen in C—N bond forming reactions. Early on, Boche demonstrated that organolithium and Grignard reagents undergo uncatalyzed amination in the presence of N,N-dialkylhydroxylamine O-sulfonate esters to form tertiary amines.[18] Subsequent reports detailed the use of a copper catalyst to increase reaction rates and yields for the electrophilic amination of Grignard reagents.[19]
The use of readily prepared and stable N,N-dialkylhydroxylamine O-benzoates for Cu-catalyzed amination was first reported by Johnson in 2004. In the presence of catalytic CuOTf or CuCl2, these reagents react with diorganozinc compounds to afford the tertiary amine product with high synthetic efficiency under mild conditions.[20] A mechanistic study of this process was later reported in which the authors proposed that Zn to Cu transmetalation first occurs to give organocopper intermediate 7, which displaces the benzoate leaving group of hydroxylamine ester 6 in an SN2-like fashion to form transient Cu(III) intermediate 8 (Scheme 3). Tertiary amine product 9 and copper(I) benzoate species 10 are then generated upon rapid reductive elimination.[21] Subsequent work has also demonstrated that N-heterocyclic carbene (NHC) or phosphine ligated copper catalysts can mediate the coupling of aryl boronic esters and silyl ketene acetals with hydroxylamine O-benzoates to form the respective aminated products.[22]
Scheme 3.
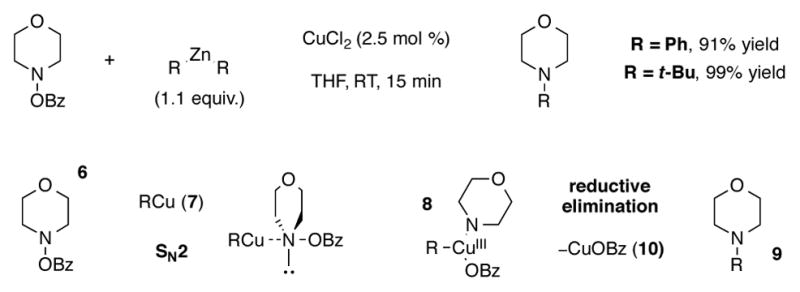
Copper-catalyzed electrophilic amination of organozinc reagents.
A two-step, formal anti-Markovnikov hydroamination procedure was reported by Lalic for the conversion of terminal alkenes to tertiary amines through the intermediacy of an alkyl boronate ester.[23] In this procedure, alkene 11 undergoes uncatalyzed hydroboration in the first step to afford alkyl boronate ester (Scheme 4). The second step is proposed to proceed through transmetalation to produce organocopper intermediate 12 which undergoes amination with hydroxylamine O-benzoate reagent 13 to give the linear amine 14. In support of the proposed catalytic cycle, an alkylcopper species (IMesCuEt) was prepared and shown to be viable in the electrophilic amination as a stoichiometric intermediate and catalyst.
Scheme 4.
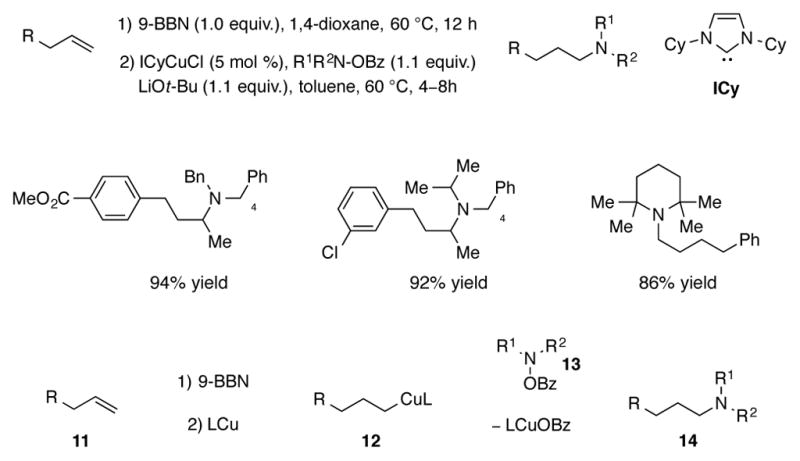
Copper-catalyzed two-step formal hydroamination of terminal alkenes.
Prior to the reports of Cu-catalyzed hydroamination, an analogous aminoboration process was reported in which a styrene was converted to the 1,2-aminoboronate in the presence of B2pin2 and a hydroxylamine O-benzoate reagent.[24] The process was proposed to proceed via borocupration, followed by interception of borylated alkylcopper species 15 by aminating reagent 13. The reaction proceeded in a stereospecific manner, and an enantioselective variant was also demonstrated using (S,S)-Me-DuPhos (L3) as the ligand for copper (Scheme 5). This transformation was later generalized to methylenecyclopropanes and bicyclic alkenes.[25] A recent report disclosed that terminal alkene substrates could be converted to either regioisomer of the 1,2-aminoboronate product with high selectivity, depending upon the boron source and catalyst system used.[26]
Scheme 5.
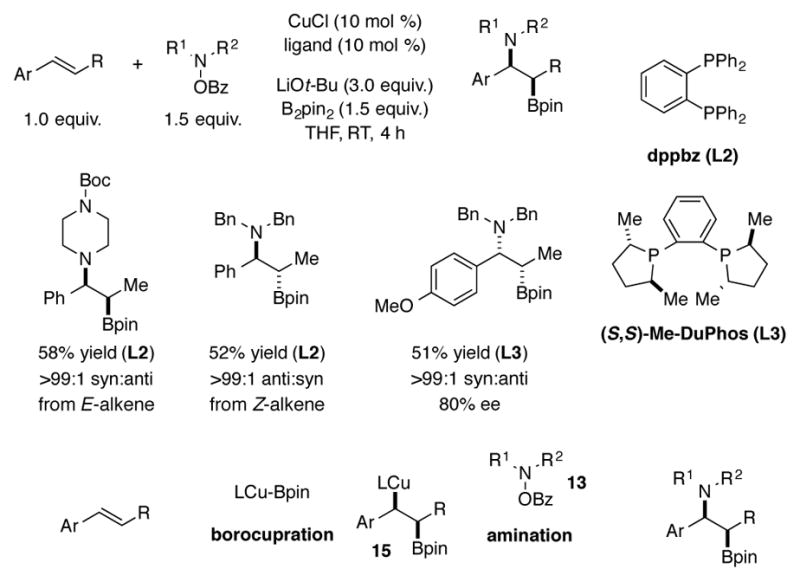
Copper-catalyzed aminoboration of styrenes.
3. Copper(I) hydride-catalyzed hydroamination – discovery, scope, and mechanistic insight
In 2013, two research groups independently reported the CuH-catalyzed hydroamination of alkenes with N,N-dialkylhydroxylamine O-benzoates to produce tertiary amine products. Miura and Hirano disclosed that styrenes react with hydroxylamine O-benzoate reagents to afford branched benzylic tertiary amines in the presence of a CuH catalyst and LiOt-Bu.[6a] Use of an electron poor 1,2-bis(diphenylphosphino)benzene derivative (CF3-dppbz, L4) as the ligand for copper afforded hydroamination product with high efficiency. By employing (S,S)-Me-DuPhos (L3) or (R,R)-Ph-BPE (L5) as the ligand, the reaction could be rendered enantioselective with enantiomeric excesses of up to 94% (Scheme 6). A variety of styrenes and β-substituted styrenes were found to be suitable substrates for this process.
Scheme 6.
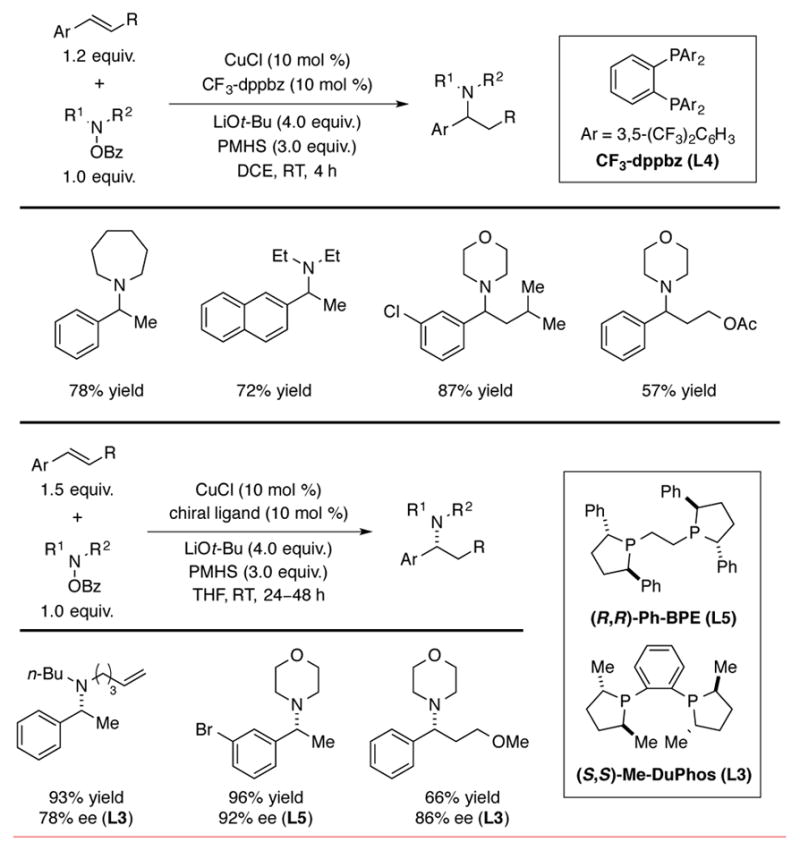
CuH-catalyzed hydroamination of styrenes.
The proposed mechanism for this transformation is shown in Scheme 7. Initially, a copper(I) alkoxide species (LCuOt-Bu, 16) is generated under reaction conditions and undergoes transmetalation with the hydrosilane to form CuH complex 17. Styrene 18 reacts with the CuH complex 17 to form chiral benzylcopper species 19, which then intercepts hydroxylamine O-benzoate reagent 20 to generate enantioenriched benzylic amine 21 and phosphine-bound copper(I) benzoate 22. Copper(I) benzoate 22 then undergoes transmetalation with LiOt-Bu 23 to regenerate copper(I) alkoxide 16 to complete the catalytic cycle.
Scheme 7.
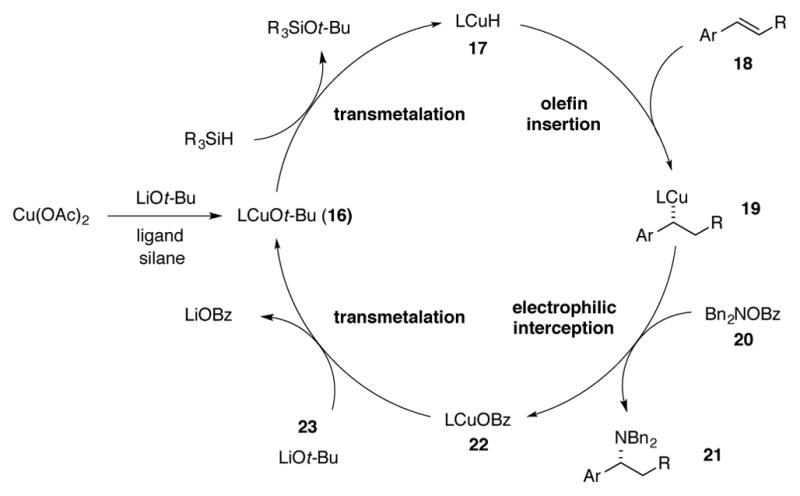
Proposed mechanism for CuH-catalyzed hydroamination of styrenes.
In an independent and contemporaneous study reported by Buchwald, a DTBM-SEGPHOS (L6) based copper catalyst was reported to effect the enantioselective hydroamination of arylalkenes in excellent yield and enantioselectivity (Scheme 8).[6b] This CuH-catalyzed process was found to be suitable for a range of arylalkenes, including β-substituted and β,β-disubstituted styrenes. A reaction was conducted on a 10 mmol scale using β-substituted styrene 24 with 1 mol % catalyst loading to demonstrate the scalability of the procedure. In contrast to the requirement for LiOt-Bu under Miura and Hirano’s protocol (Scheme 7), the addition of exogenous base was unnecessary for this system, and transmetalation of the copper(I) benzoate species with hydrosilane was proposed to take place directly. In both reports, complete Markovnikov regioselectivity was observed, which was explained by the electronic stabilization of the benzylcopper species relative to the isomeric primary alkylcopper.[27]
Scheme 8.
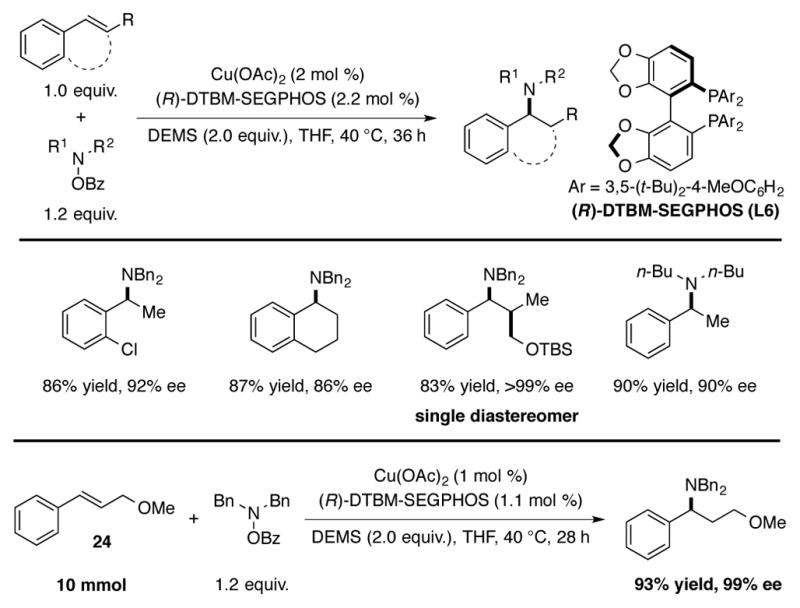
CuH-catalyzed hydroamination of styrenes with DTBM-SEGPHOS as the ligand.
The same DTBM-SEGPHOS-based CuH catalyst was also reported to effect the anti-Markovnikov hydroamination of terminal alkenes to generate linear aliphatic amines (Scheme 9).[6b] This protocol was found to be effective for a wide variety of terminal alkenes and amine electrophiles. Due to the mild reaction conditions, substrates containing functional groups such as a terminal epoxide and a terminal alkyl bromide were efficiently transformed to product. Anti-Markovnikov regioselectivity was rationalized by selective olefin insertion into the CuH species to form the sterically less-encumbered alkylcopper intermediate.
Scheme 9.
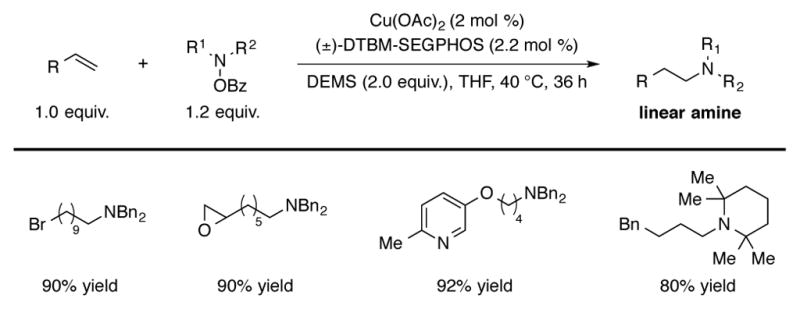
CuH-catalyzed Anti-Markovnikov hydroamination of terminal olefins.
The CuH-based hydroamination technology has also been extended to oxa- and azabicyclic alkenes (Scheme 10) by Miura and Hirano.[28] In the reported process, the optimal catalyst was found to be a combination of CuCl and (R,R)-Ph-BPE (L5). The enantioselectivities obtained ranged from moderate to excellent and a wide range of amine electrophiles, including one derived from the hindered 2,2,6,6-tetramethylpiperidine, could be successfully employed.
Scheme 10.
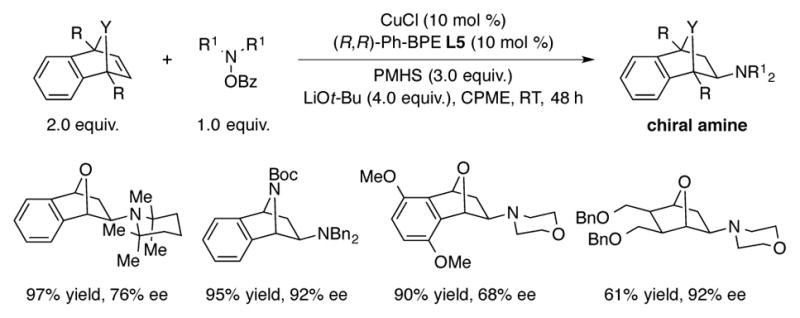
CuH-catalyzed hydroamination of oxa- and azabicyclic alkenes.
A CuH-catalyzed enantioselective synthesis of α-aminosilanes has been reported by Buchwald (Scheme 11).[29] Due to their potential application as amino acid bioisosteres in medicinal chemistry, there is considerable interest in the enantioselective preparation of these compounds.[30] Using the previously reported DTBM-SEGPHOS-based CuH catalyst system, high enantioselectivities and yields were observed for a series of vinylsilanes and hydroxylamine O-benzoate electrophiles. In this report, it was noted that (E)- and (Z)-vinylsilanes both reacted to provide the same enantiomer, but the (E)-vinylsilane isomers reacted faster and with a generally higher level of enantioselectivity. Hydroamination occurred with exclusive Markovnikov regioselectivity, which was rationalized by the hyperconjugative stabilization of the proposed alkylcopper intermediate by the adjacent silyl group.[31]
Scheme 11.
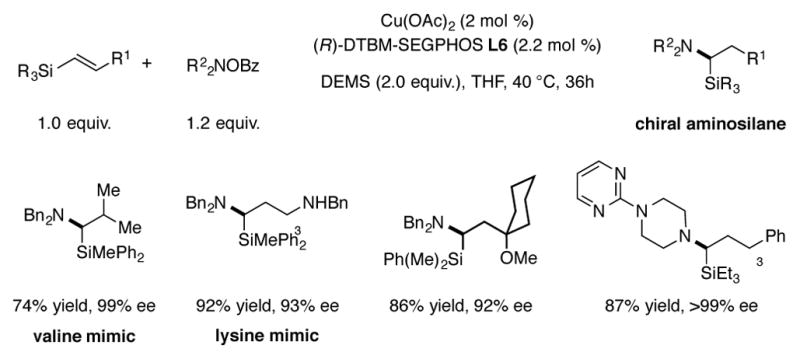
CuH-catalyzed enantioselective synthesis of α-aminosilanes.
Compounds containing an amine group with a stereocenter positioned β to it are found in a number of pharmaceuticals and natural product isolates. The enantioselective anti-Markovnikov hydroamination of 1,1-disubstituted alkenes represents an attractive route for the synthesis of this type of amine, but a process to effect this transformation remained elusive due to the low reactivity of 1,1-disubstituted alkenes towards many transition metal catalyzed processes, as well as the relatively undifferentiated nature of the enantiotopic faces of these substrates.[32] In 2014, Buchwald reported that the DTBM-SEGPHOS-based CuH catalyst system was able to facilitate this transformation.[33] Excellent levels of enantioselectivity were observed when there was a significant difference in size between the two substituents of the 1,1-disubstituted alkene (25–27, Scheme 12). In some cases, steric differences relatively far from the site of reactivity also resulted in synthetically useful levels of enantioinduction (28, Scheme 12). Other substrates, including 1,1-disubstituted vinylsilanes, limonene, deuterated alkenes, and estrone-derived olefins, all proved suitable for this transformation. Lastly, a hydroamination reaction with (R)-limonene was conducted on a 10 mmol scale to demonstrate the robustness of this methodology. As discovered by Lipshutz, triphenylphosphine could be used as a secondary ligand to enable low catalyst loadings (0.4 mol%) to be used in this system.[15b]
Scheme 12.

CuH-catalyzed enantioselective anti-Markovnikov hydroamination of 1,1-disubstituted alkenes.
CuH-catalyzed hydroamination has also been successfully applied to alkynes, a readily accessible class of substrates.[34] Under different sets of conditions, both enamines and alkylamines could be selectively prepared from the same starting materials. In the absence of a proton source, a series of arylalkynes were efficiently converted into the respective enamines (Scheme 13). Enamine products were formed with excellent regio- and diastereoselectivities (>20:1 α:β amination, >20:1 E/Z). The regiochemical outcome of the transformation was rationalized by stabilization of the vinylcopper species by the neighboring aryl group. When a protic additive (EtOH or i-PrOH) was included, arylalkynes and alkylacetylenes were converted to α-chiral branched aliphatic amines and linear alkylamines, respectively. Reports by Tsuji[35a] and Lalic[35b] on the CuH-catalyzed semireduction of alkynes to cis-alkenes in the presence of a proton source provided precedent for this transformation. The regioselectivity and enantioselectivity (for branched amines) of the process were again found to be uniformly high. This hydroamination technology was applied to the asymmetric synthesis of rivastigmine, a drug used to treat dementia, and the formal syntheses of duloxetine, atomoxetine, fluoxetine, and tolterodine, all pharmaceutical agents used to treat a variety of ailments.
Scheme 13.

CuH-catalyzed reductive hydroamination of alkynes for the synthesis of enamines, α-chiral amines, and linear aliphatic amines.
The proposed mechanisms for the two alkyne hydroamination processes, enamine formation and alkylamine formation, are displayed in Scheme 15. Alkyne insertion into copper(I) hydride species 29 forms vinylcopper 30, which could then branch into two separate pathways. If there is no proton source available, then alkenylcopper intermediate 30 intercepts hydroxylamine O-benzoate 31 to generate enamine 32 (Scheme 14, “direct hydroamination”). In the presence of an alcohol additive, protonation occurs to generate copper(I) alkoxide 33 and cis-alkene 34. Regeneration of the copper(I) hydride species 29 followed by olefin insertion regenerates aliphatic copper(I) intermediate 35. Electrophilic amination yields the enantioenriched aliphatic amine 36 and copper(I) benzoate 37 (Scheme 14, “reductive hydroamination”). Transmetalation of copper(I) benzoate 37 with silane 38 regenerates copper hydride species 29.
Scheme 15.
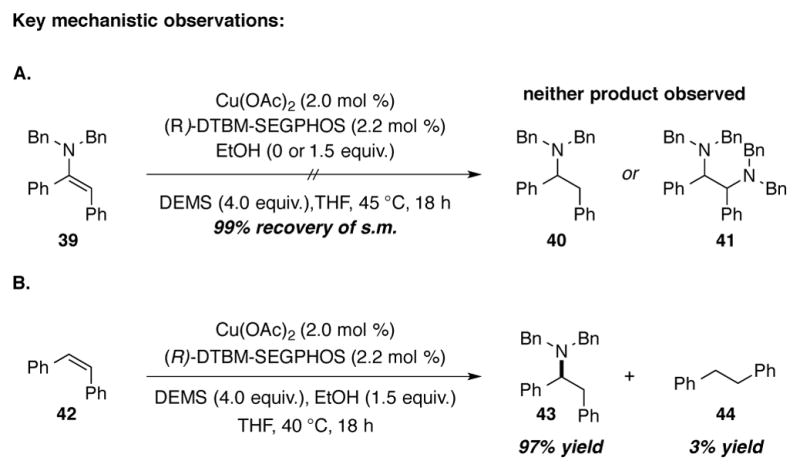
Experiments to determine the mechanism oft he hydroamination of alkynes.
Scheme 14.
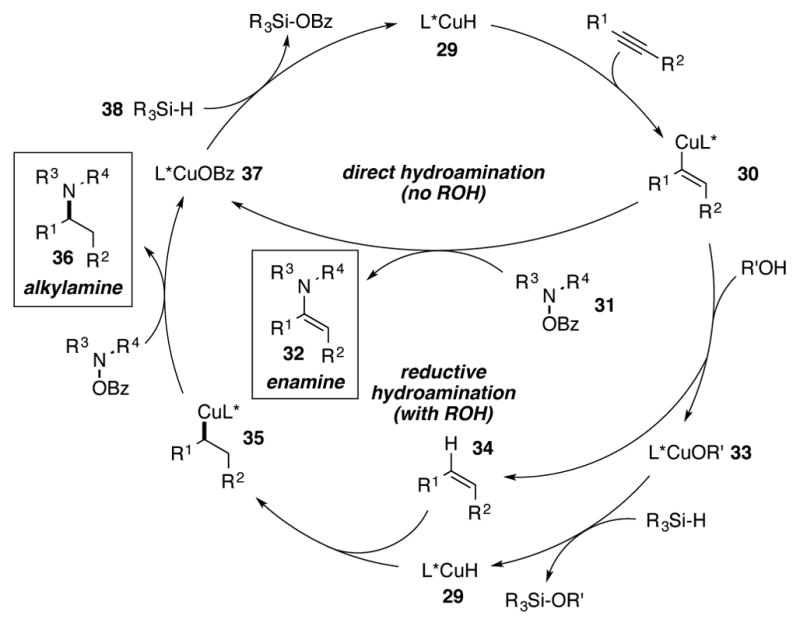
Proposed mechanism fort he direct and reductive hydroamination of alkynes.
Hydroamination of the alkyne substrate followed by reduction of the resultant enamine was considered as an alternative mechanism for the formation of aliphatic amine 36. To test this possibility, enamine 39 was subjected to standard hydroamination conditions with or without protic additives. In either case, starting material was recovered nearly quantitatively. Neither benzylic amine 40 nor diamination product 41 was observed (Scheme 15A). On the other hand, hydroamination of cis-stilbene (42) in the presence of ethanol as the proton source provided the reduced chiral amine product 43 in 97% yield, with only a trace of alkane 44 also observed (Scheme 15B). These experiments provide strong support for semireduction followed by alkene hydroamination as the operative mechanism for the formation of aliphatic amine 36.
In 2015, Buchwald extended the CuH-catalyzed hydroamination strategy to the direct enantioselective synthesis of secondary chiral amines (Scheme 16).[36] In initial experiments, it was found that N-benzylhydroxylamine O-benzoate 45 could be successfully employed as a monoalkylamine transfer agent for the hydroamination of styrene to afford the secondary amine in 60% yield and 96% ee. Low yields, however, were observed when this amine transfer agent was used for the hydroamination of ortho- or β-substituted styrenes. To improve the efficacy of the aminating reagent, various N-benzylhydroxylamine O-carboxylate esters were synthesized and tested in this hydroamination reaction. It was found that the monoalkylamine transfer agent containing a 4-(dimethylamino)benzoate group (46) provided the highest yields. This modified amine transfer reagent was shown to be generally useful for the installation of monoalkylamino groups on a range of functionalized and substituted arylalkenes. To highlight the potential applications of this chemistry, cinacalcet, a treatment for hyperparathyroidism, was synthesized with this protocol. Several pharmaceuticals, such as chlorpromazine, loratadine, and tolfenamic acid, were also successfully coupled with these modified amine transfer agents, illustrating the potential use of this methodology in late stage derivatization of clinically relevant precursors.
Scheme 16.

CuH-catalyzed synthesis of chiral secondary amines.
NMR studies demonstrated that in the presence of copper catalyst and hydrosilane, N-benzylhydroxylamine O-benzoate ester 45 is consumed much more rapidly, presumably through CuH-catalyzed reduction, than the analogous dialkylamine transfer agent 20 (Scheme 17). Competition experiments between hydroxylamine O-benzoate 45 and 4-(dimethylamino)benzoate 46 also show that the more electron-rich benzoate 46 is reduced by the CuH catalyst at a much slower rate than the parent benzoate 45. The slower unproductive consumption of the electron rich reagent by CuH was believed to account for the improved yields observed.
Scheme 17.
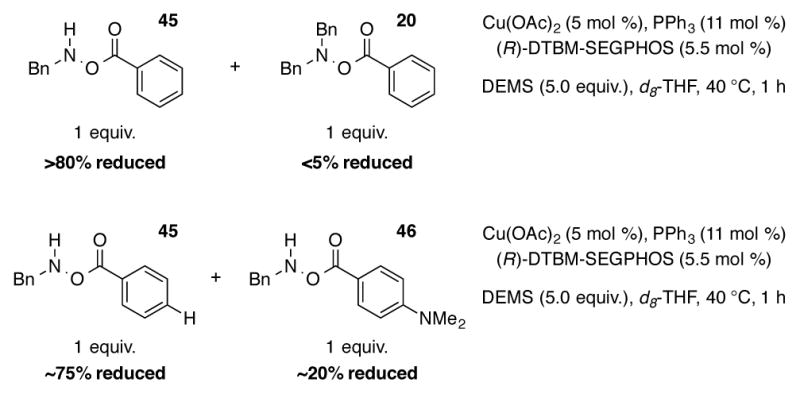
NMR competition experiments with various amine oxidants.
Unactivated internal olefins are a common yet rarely exploited class of feedstock chemicals in organic synthesis. In particular, there are few asymmetric transition metal-catalyzed hydrofunctionalization reactions that utilize unactivated internal olefins as substrates.[37] The scarcity of such transformations may be ascribed to several factors, including the low binding affinity of the alkene to the metal center, slow migratory insertion, and the possibility of undesired chain-walking isomerization processes.[38] Nevertheless, their use in enantioselective hydroamination is potentially valuable, since the resultant chiral α-branched amines possess stereocenters with minimally differentiated alkyl substituents. Such amines are often only accessed with difficulty.[39]
In 2015, Buchwald reported a CuH-catalyzed procedure for the hydroamination of unactivated internal olefins.[40] Notably, the protocol was suitable for the enantioselective synthesis of amines bearing methyl-ethyl stereocenters using trans-2-butene, a bulk commodity chemical, as the olefin substrate (Scheme 18). As in the case of the monoalkylamino aminating reagents discussed earlier, use of the more electron-rich 4-(diethylamino)benzoate amine electrophile was crucial for attaining high yields in this challenging transformation. A series of internal olefins and amine electrophiles were amenable to this chemistry. Chiral α-branched olefins were prepared with uniformly high (≥96% ee) levels of enantioselectivity. In cases where an unsymmetrical alkene was used, steric differentiation could be used to generate products in a moderately regioselective manner with each regioisomer generated in nearly optically pure form. Two pharmaceutical agents, cinacalcet and paroxetine, were readily diversified with this chemistry in a catalyst-controlled manner.
Scheme 18.
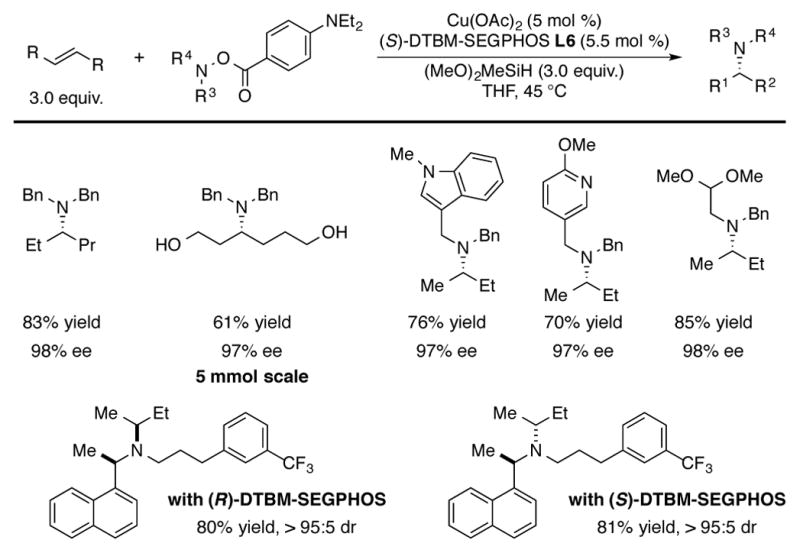
CuH-catalyzed hydroamination of unactivated internal olefins.
Computational studies on the hydrocupration step were presented in this study. The free energy barriers for the hydrocupration of styrenes, unactivated terminal olefins, and unactivated internal olefins were computed. The results suggest that unactivated internal olefins undergo hydrocupration more slowly than terminal alkenes or styrenes by several orders of magnitude (Scheme 19). Hydrocupration was tentatively proposed to be enantiodetermining, and calculations suggest that the high levels of enantioselectivity observed for this transformation originate from the spatial arrangement of the aryl groups on the phosphorus atom during the hydrocupration step. As is commonly observed for transition metal catalyzed reactions employing C2-symmetric ligands,[41] the aryl groups of the ligand were computed to adopt pseudoaxial and pseudoequatorial orientations in the transition state, wherein the equatorial aryl substituents closely approach the olefin. These steric interactions were proposed to play a dominant role in stereoinduction.
Scheme 19.
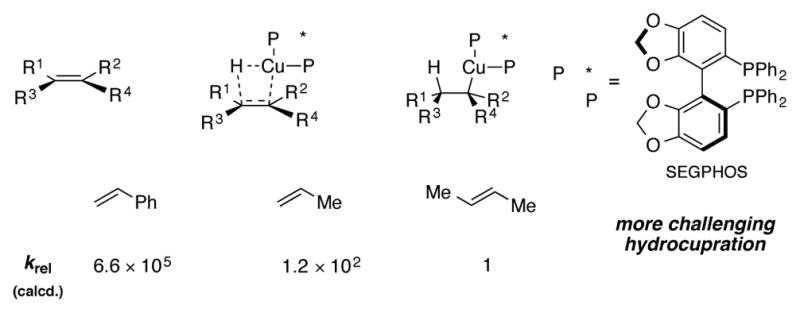
Calculations for the hydrocupration of olefins with a SEGPHOS-ligated copper(I) hydride complex.
4. Outlook and conclusions
Drawing on important precedent in copper hydride chemistry and electrophilic amination using hydroxylamine ester reagents, CuH-catalyzed hydroamination has recently been developed as a general and broadly applicable strategy for the regio- and stereoselective synthesis of amines. This methodology has rapidly evolved to encompass the use of a wide range of amine electrophiles and olefins, providing a strategy for the synthesis of secondary and tertiary amines, including several examples for which stereoselective strategies were previously unavailable.[42] Mechanistic and computational studies have provided insight into several aspects of this chemistry, including the basis for enantioinduction, the role of the amine electrophile, and the viability of copper-catalyzed isomerization processes.
There are still a number of features or versions of this chemistry that are not optimal. For example, amines derived from tri- and tetrasubstituted alkenes are currently not accessible, presumably due to the challenging hydrocupration step. Moreover, satisfactory protocols that utilize electrophilic aminating agents to transfer aromatic amines or ammonia (or its equivalents) have not yet been realized. Finally, while published methods often include requirements for long reaction times and the rigorous exclusion of air and moisture, solutions to these issues will be the subject of a soon to be submitted manuscript.
Scheme 1.

Two possible regiochemical outcomes with the hydroamination of olefins.
Acknowledgments
Research reported in this publication was supported by the National Institutes of Health under award no. GM58160. (M. T. P. and Y.-M. W. thank the National Institutes of Health for postdoctoral fellowships (GM113311 and GM112218, respectively). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We are grateful to Dr. Jeffrey Bandar for help with the preparation of this manuscript.
Biographies
Michael Pirnot was born in Allentown, PA and raised in Kutztown, PA. He received his BS degree in Chemistry from the University of Delaware in 2009 and his Ph. D. in 2014 at Princeton University studying photoredox organocatalysis under the tutelage of Professor David W. C. MacMillan. Michael is currently a NIH postdoctoral fellow at MIT in Professor Stephen L. Buchwald’s lab.
Yi-Ming Wang was born in Shanghai, China, and grew up in Boulder, Colorado. He graduated with an A.B./A.M. degree in chemistry/physics and mathematics from Harvard University in 2008 and obtained his Ph.D. under the guidance of Professor F. Dean Toste at the University of California, Berkeley in 2013. He is currently conducting research as an NIH postdoctoral fellow at MIT in Professor Stephen L. Buchwald’s lab.
Stephen L. Buchwald is the Camille Dreyfus Professor of Chemistry at MIT where he has been a faculty member since 1984. He has received numerous awards of which the Linus Pauling Award and the BBVA Frontiers of Knowledge Award in Basic Sciences are two of the most recent.
References
- 1.General references on amine synthesis: Li W, Zhang X, editors. Topics in Current Chemistry. Vol. 343. Springer; Berlin: 2014. Stereoselective Formation of Amines.Nugent TC, editor. Chiral Amine Synthesis. Wiley; Weinheim: 2010. Lawrence SA. Amines. Cambridge University Press; Cambridge: 2006.
- 2.Ricci A, editor. Modern Amination Methods. Wiley; Weinheim: 2008. [Google Scholar]
- 3.General reviews on metal-catalyzed alkene hydroamination: Huang L, Arndt M, Gooβen K, Heydt H, Gooβen LJ. Chem Rev. 2015;115:2596–2697. doi: 10.1021/cr300389u.Coman SM, Parvulescu VI. Org Process Res Dev. 2015 doi: 10.1021/acs.oprd.5b00010.Bernoud E, Lepori C, Mellah M, Schulz E, Hannedouche J. Catal Sci Technol. 2015;5:2017–2037.Patton DA, Cremeens ME. Rev J Chem. 2014;4:1–20.Schafer LL, Yim JC-H, Yonson N. In: Metal-Catalyzed Cross-Coupling Reactions and More. de Meijere A, Bräse S, Oestreich M, editors. Chapter 15. Wiley; Weinheim: 2014. pp. 1135–1258.Nishina N, Yamamoto Y. Top Organomet Chem. 2013;43:115–144.Julian LD. Top Heterocycl Chem. 2013;32:109–156.Reznichenko AL, Hultzsch KC. Top Organomet Chem. 2013;43:51–114.Yadav JS, Antony A, Rao TS, Reddy BVS. J Organomet Chem. 2011;696:16–36.Hesp KD, Stradiotto M. ChemCatChem. 2010;2:1192–1207.Taylor JG, Adrio LA, Hii KK. Dalton Trans. 2010;39:1171–1175. doi: 10.1039/b918970j.Dzhemilev UM, Tolstikov GA, Khusnutdinov RI. Rus J Org Chem. 2009;45:957–987.Müller TE, Hultzsch KC, Yus M, Foubelo F, Tada M. Chem Rev. 2008;109:3795–3892. doi: 10.1021/cr0306788.Lee AV, Schafer LL. Eur J Inorg Chem. 2007:2243–2255.Hultzsch KC. Adv Synth Catal. 2005;347:367–391.Hultzsch KC, Gribkov DV, Hampel F. J Organomet Chem. 2005;690:4441–4452.Hong S, Marks TJ. Acc Chem Res. 2004;37:673–686. doi: 10.1021/ar040051r.Seayad J, Tillack A, Hartung CG, Beller M. Adv Synth Catal. 2002;344:795–813.Reviews on enantioselective alkene hydroamination: Reznichenko AL, Nawara-Hultzsch AJ, Hultzsch KC. Top Curr Chem. 2014;343:191–260. doi: 10.1007/128_2013_500.Hannedouche J, Schulz E. Chem Eur J. 2013;19:4972–4985. doi: 10.1002/chem.201203956.Zi G. J Organomet Chem. 2011;696:68–75.Zi G. Dalton Trans. 2009:9101–9109. doi: 10.1039/b906588a.Chemler SR. Org Biomol Chem. 2009;7:3009–3019. doi: 10.1039/B907743J.Aillaud I, Collin J, Hannedouche J, Schulz E. Dalton Trans. 2007:5105–5118. doi: 10.1039/b711126f.Hii Pure KK. Appl Chem. 2006;78:341–349.
- 4.Leading references on Brønsted acid catalyzed hydroamination: Schulummer B, Hartwig JF. Org Lett. 2002;4:1471–1474. doi: 10.1021/ol025659j.Anderson LL, Arnold J, Bergman RG. J Am Chem Soc. 2005;127:14542–14543. doi: 10.1021/ja053700i.Shapiro ND, Rauniyar V, Hamilton GL, Wu J, Toste FD. Nature. 2011;470:245–249. doi: 10.1038/nature09723.Lin JS, Yu P, Huang L, Zhang P, Tan B, Liu XY. Angew Chem Int Ed. 2015;54:7847–7851. doi: 10.1002/anie.201501762.
- 5.Leading references on radical-based hydroamination processes: Kemper J, Studer A. Angew Chem Int Ed. 2005;44:4914–4917. doi: 10.1002/anie.200463032.Guin J, Mück-Lichtenfield C, Grimme S, Studer A. J Am Chem Soc. 2007;129:4498–4503. doi: 10.1021/ja0692581.Nguyen TM, Nicewicz DA. J Am Chem Soc. 2013;135:9588–9591. doi: 10.1021/ja4031616.Musacchio AJ, Nguyen LQ, Beard GH, Knowles RR. J Am Chem Soc. 2014;136:12217–12220. doi: 10.1021/ja5056774.
- 6.Leading references on hydroamination processes based on pericyclic reactions: Beauchemin AM, Moran J, Lebrun ME, Séguin C, Dimitrijevic E, Zhang L, Gorelsky SI. Angew Chem Int Ed. 2008;47:1410–1413. doi: 10.1002/anie.200703495.Brown AR, Uyeda C, Brotherton CA, Jacobsen EA. J Am Chem Soc. 2013;135:6747–6749. doi: 10.1021/ja402893z.
- 7.Miki Y, Hirano K, Satoh T, Miura M. Angew Chem Int Ed. 2013;52:10830–10834. doi: 10.1002/anie.201304365.Zhu S, Niljianskul N, Buchwald SL. J Am Chem Soc. 2013;135:15746–15749. doi: 10.1021/ja4092819.For a highlight, see: Hesp KD. Angew Chem Int Ed. 2014;53:2034–2036. doi: 10.1002/anie.201309262.
- 8.Chen J-X, Daeuble JF, Brestensky DM, Stryker JM. Tetrahedron. 2000;56:2153–2166. [Google Scholar]
- 9.Wurtz A. Ann Chim Phys. 1844;11:250–251. [Google Scholar]
- 10.Whitesides GM, Filipo JS, Stredronsky ER, Casey CP. J Am Chem Soc. 1969;91:6542–6544.Solid state structure of [(Ph3P)CuH]6: Churchill MR, Bezman SA, Osborn JA, Wormald J. J Am Chem Soc. 1971;93:2063–2065.
- 11.Mahoney WS, Brestensky DM, Stryker JM. J Am Chem Soc. 1988;110:291–293. [Google Scholar]
- 12.Brunner H, Miehling W. J Organomet Chem. 1984;275:C17–C21. [Google Scholar]
- 13.Mahoney WS, Stryker JM. J Am Chem Soc. 1989;111:8818–8823. [Google Scholar]
- 14.Lipshutz BH, Keith J, Papa P, Vivian R. Tetrahedron Lett. 1998;39:4627–4630. [Google Scholar]
- 15.a) Appella DH, Moritani Y, Shintani R, Ferreira EM, Buchwald SL. J Am Chem Soc. 1999;121:9473–9474. [Google Scholar]; b) Moritani Y, Appella DH, Jurkauskas V, Buchwald SL. J Am Chem Soc. 2000;122:6797–6798. [Google Scholar]; c) Yun J, Buchwald SL. Org Lett. 2001;3:1129–1131. doi: 10.1021/ol015577f. [DOI] [PubMed] [Google Scholar]; d) Jurkauskas V, Buchwald SL. J Am Chem Soc. 2002;124:2892–2893. doi: 10.1021/ja025603k. [DOI] [PubMed] [Google Scholar]; e) Rainka MP, Aye Y, Buchwald SL. Proc Natl Acad Sci USA. 2004;101:5821–5823. doi: 10.1073/pnas.0307764101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a) Lipshutz BH, Noson K, Chrisman W. J Am Chem Soc. 2001;123:12917–12918. doi: 10.1021/ja011529e. [DOI] [PubMed] [Google Scholar]; b) Lipshutz BH, Noson K, Chrisman W, Lower A. J Am Chem Soc. 2003;125:8779–8789. doi: 10.1021/ja021391f. [DOI] [PubMed] [Google Scholar]; c) Lipshutz BH, Shimizu H. Angew Chem Int Ed. 2004;43:2228–2230. doi: 10.1002/anie.200353294. [DOI] [PubMed] [Google Scholar]
- 17.Noh D, Chea H, Ju J, Yun J. Angew Chem Int Ed. 2009;48:6062–6064. doi: 10.1002/anie.200902015.. For the use of vinylazaarenes in a CuH-catalyzed process, see: Rupnicki L, Saxena A, Lam HW. J Am Chem Soc. 2009;131:10386–10387. doi: 10.1021/ja904365h.
- 18.Boche G, Mayer N, Bernheim M, Wagner K. Angew Chem Int Ed Engl. 1978;17:687–688. [Google Scholar]
- 19.Erdik E, Ay M. Synth React Inorg Met-Org Chem. 1989;19:663–668. [Google Scholar]
- 20.Berman AM, Johnson JS. J Am Chem Soc. 2004;126:5680–5681. doi: 10.1021/ja049474e. [DOI] [PubMed] [Google Scholar]
- 21.Campbell MJ, Johnson JS. Org Lett. 2007;9:1521–1524. doi: 10.1021/ol0702829. [DOI] [PubMed] [Google Scholar]
- 22.a) Matsuda N, Hirano K, Satoh T, Miura M. Angew Chem Int Ed. 2012;51:3642–3645. doi: 10.1002/anie.201108773. [DOI] [PubMed] [Google Scholar]; b) Rucker RP, Whittaker AM, Dang H, Lalic G. Angew Chem Int Ed. 2012;51:3953–3956. doi: 10.1002/anie.201200480. [DOI] [PubMed] [Google Scholar]; c) Matusda N, Hirano K, Satoh T, Miura M. Angew Chem Int Ed. 2012;51:11827–11831. doi: 10.1002/anie.201206755. [DOI] [PubMed] [Google Scholar]
- 23.Rucker RP, Whittaker AM, Dang H, Lalic G. J Am Chem Soc. 2012;134:6571–6574. doi: 10.1021/ja3023829. [DOI] [PubMed] [Google Scholar]
- 24.Matsuda N, Hirano K, Satoh T, Miura M. J Am Chem Soc. 2013;135:4934–4937. doi: 10.1021/ja4007645. [DOI] [PubMed] [Google Scholar]
- 25.a) Sakae R, Matsuda N, Hirano K, Satoh T, Miura M. Org Lett. 2014;16:1228–1231. doi: 10.1021/ol5001507. [DOI] [PubMed] [Google Scholar]; b) Sakae R, Hirano K, Satoh T, Miura M. Angew Chem Int Ed. 2015;54:613–617. doi: 10.1002/anie.201409104. [DOI] [PubMed] [Google Scholar]
- 26.Sakae R, Hirano K, Miura M. J Am Chem Soc. 2015;137:6460–6463. doi: 10.1021/jacs.5b02775. [DOI] [PubMed] [Google Scholar]
- 27.For a discussion of the factors dictating the regioselectivity of the related borocupration process, see: Dang L, Zhao H, Marder TB. Organometallics. 2007;26:2824–2832.
- 28.Miki Y, Hirano K, Satoh T, Miura M. Org Lett. 2014;16:1498–1501. doi: 10.1021/ol5003219. [DOI] [PubMed] [Google Scholar]
- 29.Niljianskul N, Zhu S, Buchwald SL. Angew Chem Int Ed. 2015;54:1638–1641. doi: 10.1002/anie.201410326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.For an overview of organosilicon compounds in medicinal chemistry, see: Franz AK, Wilson SO. J Med Chem. 2013;56:388–405. doi: 10.1021/jm3010114.Catalytic, enantioselective approaches to α-aminosilanes: Hensel A, Nagura K, Delvos LB, Oesterich M. Angew Chem Int Ed. 2014;126:5064–5067.Mita T, Sugawara M, Saito K, Sato Y. Org Lett. 2014;16:3028–3031. doi: 10.1021/ol501143c.
- 31.For a discussion of this electronic effect, see: Brook MA. Silicon in Organic, Organometallic, and Polymer Chemistry. John Wiley; New York: 2000.
- 32.Thomas SP, Aggarwal VK. Angew Chem Int Ed. 2009;48:1896–1898. doi: 10.1002/anie.200805604. [DOI] [PubMed] [Google Scholar]
- 33.Zhu S, Buchwald SL. J Am Chem Soc. 2014;136:15913–15916. doi: 10.1021/ja509786v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi S-L, Buchwald SL. Nat Chem. 2015;7:38–44. doi: 10.1038/nchem.2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.a) Semba K, Fujihara T, Xu T, Terao J, Tsuji Y. Adv Synth Catal. 2012;354:1542–1550. [Google Scholar]; b) Whittaker AM, Lalic G. Org Lett. 2013;15:1112–1115. doi: 10.1021/ol4001679. [DOI] [PubMed] [Google Scholar]
- 36.Niu D, Buchwald SL. J Am Chem Soc. 2015 doi: 10.1021/jacs.5b05446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hydroformylation of internal alkenes: Sakai N, Nozaki K, Takaya H. J Chem Soc – Chem Commun. 1994:295–296.Gadzikwa T, Bellini R, Dekker HL, Reek JN. J Am Chem Soc. 2012;134:2860–2863. doi: 10.1021/ja211455j.
- 38.Chain walking isomerization in reported processes: Pereira S, Srebnik M. J Am Chem Soc. 1996;118:909–910.van der Veen LA, Kamer PCJ, wan Leeuwen PWNM. Angew Chem Int Ed. 1999;38:336–338. doi: 10.1002/(SICI)1521-3773(19990201)38:3<336::AID-ANIE336>3.0.CO;2-P.Obligacion JV, Chirik PJ. J Am Chem Soc. 2013;135:19107–19110. doi: 10.1021/ja4108148.
- 39.Nugent TC, El-Shazly M. Adv Synth Catal. 2010;352:753–819. [Google Scholar]
- 40.Yang Y, Shi SL, Niu D, Liu P, Buchwald SL. Science. 2015;349:62–66. doi: 10.1126/science.aab3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.a) Whitesell JK. Chem Rev. 1989;89:1581–1590. [Google Scholar]; b) Walsh P, Kowzlowski M. Fundamentals of Asymmetric Catalysis. University Science Books; Sausalito: 2009. [Google Scholar]
- 42.a) Abel-Magid AF, Mehrman SJ. Org Process Res Dev. 2006;10:971–1031. [Google Scholar]; b) Baxter EW, Reitz AB. Organic Reactions. Vol. 59. Wiley; Weinheim: 2004. Reductive Aminations of Carbonyl Compounds with Borohydride and Borane Reducing Agents. [Google Scholar]