

Abstract
Carbonic anhydrase IX (CAIX) is a tumor-associated antigen and marker of hypoxia that is overexpressed on > 90% of clear-cell type renal cell carcinoma (RCC) but not on neighboring normal kidney tissue. Here, we report on the construction of two chimeric antigen receptors (CARs) that utilize a carbonic anhydrase (CA) domain mapped, human single chain antibody (scFv G36) as a targeting moiety but differ in their capacity to provide costimulatory signaling for optimal T cell proliferation and tumor cell killing. The resulting anti-CAIX CARs were expressed on human primary T cells via lentivirus transduction. CAR-transduced T cells (CART cells) expressing second-generation G36-CD28-TCRζ exhibited more potent in vitro antitumor effects on CAIX+ RCC cells than first-generation G36-CD8-TCRζ including cytotoxicity, cytokine secretion, proliferation, and clonal expansion. Adoptive G36-CD28-TCRζ CART cell therapy combined with high-dose interleukin (IL)-2 injection also lead to superior regression of established RCC in nude mice with evidence of tumor cell apoptosis and tissue necrosis. These results suggest that the fully human G36-CD28-TCRζ CARs should provide substantial improvements over first-generation mouse anti-CAIX CARs in clinical use through reduced human anti-mouse antibody responses against the targeting scFv and administration of lower doses of T cells during CART cell therapy of CAIX+ RCC.
Introduction
Carbonic anhydrases (CA) are a family of zinc metalloenzymes, which catalyze reversible hydration of carbon dioxide in order to maintain pH balance in living organisms. CAIX is a transmembrane glycoprotein with molecular weight of 54/58kDa. Structurally, CAIX consists of four domains: an N-terminal proteoglycan-like domain (PG) (aa 53-111), a CA catalytic domain (CA) (aa 135 -391), a transmembrane helical segment (aa 415-434), and a short intracytoplasmic tail (aa 434-459). In hypoxic conditions, the CAIX gene is directly activated at the transcriptional level by hypoxia inducible transcription factor HIF-1α, leading to transport of protons to the extracellular medium and lowering of pH.1 Thus, CAIX expression can be regarded as a surrogate marker for hypoxia in various tumors.2 The resulting acidification of the tumor microenvironment by CA activity and the keratin sulfate unit in the O-linked glycan structure in the PG domain of CAIX are presumed to play an important role in the processes of cell adhesion and tumor progression.3
CAIX is considered a tumor-associated antigen and its overexpression is found among several solid tumor types, particularly in clear cell type renal cell carcinomas (RCC)4 as well as carcinomas of several histologic types including ovarian, breast, esophageal, bladder, colon, non-small cell lung, dysplasia of the cervix and others.5 CAIX expression has been suggested to serve as a marker for cancer diagnosis and early detection of carcinogenesis6; it is also a prognostic marker for favorable response in interleukin (IL)-2-treated patients of melanoma and kidney cancer, leading to high response rates and low toxicity.7 Immunostaining and western blot studies have shown that a high level of CAIX expression is restricted to the majority of primary RCC (clear cell type with granular or spindle cell, papillary type of chromophilic cell and collecting duct except for chromphobic cell), cystic RCCs, and metastatic RCCs but is not observed in normal kidney tissues, benign epithelial cystic lesions, or non-renal cell clear cell adenocarcinoma.2,6
RCC is one of two immunogenic tumor types, besides melanoma, that exhibits evidence of spontaneous regression of metastatic lesions after nephrectomy8 and of being responsive to immunomodulating therapies such as cancer vaccines and IL-2.9 Adoptive T cell therapy for metastatic melanoma and RCC patients using ex vivo expanded tumor-infiltrating lymphocytes has shown some success.10 Recently, T-cell receptor (TCR)-modified T cells (TCR-α and -β chains) were also used to provide an effective tumor targeting T-cell repertoire.11 However, post-targeting antitumor activity can be hampered by deficiencies that involve downregulation at all levels of the major histocompatibility complex class I-restricted antigen presentation machinery,12 induced anergy due to the loss of expression of costimulatory molecules on the tumor13 as well as shedding of molecules and secretion of cytokines with immunosuppressive activity by tumors.14,15
Chimeric antigen receptors (CARs) were designed to consist of a single-chain antibody (scFv) coupled to signaling modules of a TCR complex, such as the CD3ζ chain.16 Expression of CAR on T cells (CART cells) enables them to redirect T cells against preselected tumor antigens by an major histocompatibility complex-independent, antibody-type recognition with potent TCR cytotoxicity. In one example, murine mAbG250, which recognizes the PG domain on CAIX was used to construct a first-generation single-chain antibody chimeric receptor scFv(G250)-CD4-FcϵRIγ which was then retrovirus transduced and expressed on autologous T cells for adoptive immunotherapy in conjunction with low-dose IL-2 treatment in three metastatic RCC patients. However, two out of the three patients suffered from liver toxicity, necessitating lower CART dosing and pretreatment with CAIX monoclonal antibody G250 to prevent liver toxicities.17 Thus, improvement in both safety and efficacy is required for this cellular therapy against RCC to be moved into further clinical trials.
Recently, the focus of CAR designs has shifted to incorporation of endodomains from T cell costimulatory molecules, such as CD28, 4-1BB, and OX40, to overcome problems with inefficient effector function and anergic status of antitumor CART cells. Incorporation of CD28 costimulatory molecule into CARs generates CART cells with superior antitumor activities including tumor-induced proliferation, cytotoxicity, cytokine secretion, clonal expansion, and tumor regression in animal tumor models. Two encouraging preclinical findings have established the advantages of CART therapy over traditional adoptive T cell therapy. First, injection of CART cells required as little as 1 week for tumor regression/eradication in leukemia18 and metastistic cancer19 without IL-2, and for established tumors in mice with IL-2.20 Second, in vivo persistence of CART cells in hosts after treatment was strong and antitumor activity was maximized. CART cells incorporating CD137 costimulatory molecule could be detected for at least 6 months in NOD-SCID-β2−/− mouse bearing human pre-B acute lymphoblastic leukemia (ALL).18 In addition, in a recent clinical trial, complete remission and long-term survival of effector memory T cells was observed in two out of three patients with advanced chemotherapy-resistant chronic lymphocytic leukemia (CLL) who had received adoptive immunotherapy using second-generation CAR (4-1BB) targeting CD19, illustrating successful translation of such a therapeutic approach.21 Similarly, second-generation CD28-encoded CARs targeting CD19 have shown promising results in CLL, indolent B-cell lymphoma, and ALL.22
Here, we characterize the antibody-dependent cellular cytotoxicity (ADCC) of five CAIX-specific scFv antibodies isolated from a human phage display library. The lead scFv (G36) was chosen for incorporation into two CAR constructs: first-generation scFv-CD8-TCRζ (CD8z) and second-generation scFv-CD28-TCRζ (CD28z). We compared the in vitro antitumor responses of these two types of scFvG36-retargeted CART cells and show superiority of G36-CD28z CART cells by multiple parameters. In conjunction with high-dose IL-2 injection, G36-CD28z CART cells showed marked tumor regression in nude mice bearing subcutaneous established CAIX+ RCC. In addition, in situ distribution of G36-CD28z CART cells within tumors of these mice provide evidence of the enhanced antitumor activity.
Results
ADCC-mediated killing of anti-CAIX antibodies and choice of CAR-targeting moiety
We have previously reported on a panel of high-affinity human anti-CAIX antibodies that differed in their epitope mapping, expression levels, and ability to internalize CAIX.23 Our first aim was to investigate the antitumor activity of five of these anti-CAIX single-chain antibodies as candidates for CAR construction. To test for anti-CAIX mAb-mediated ADCC, the scFvs were converted to scFv-Fc (hIgG1) minibodies.23 We found that all scFv-Fcs exhibited antigen-specific tumor lysis. For tumor cell line sk-rc-09 with high CAIX+ expression, specific lysis ranged from 40 to 57% and for sk-rc-52 with moderate CAIX+ expression, specific lysis ranged from 46 to 60%, with background of lysis of < 5% for the CAIX− tumor cell line sk-rc-59. For negative control scFv-Fcs such as anti-CXCR4 48-Fc23 and anti-SARS 11A-Fc,24 only background levels of cell lysis were seen (Figure 1). Based on ADCC killing and other published analyses, scFvG36 was chosen for further evaluation as the CAR-targeting moiety.
Figure 1.
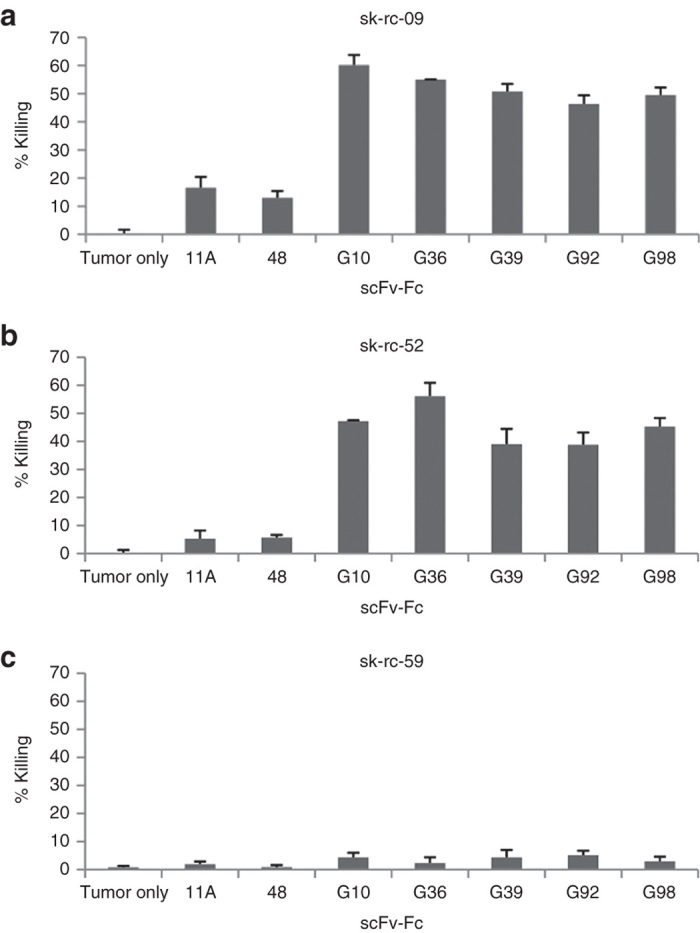
Antibody-dependent cellular cytotoxicity of carbonic anhydrase IX (CAIX)-specific Antibodies. 1 µg/ml CAIX-specific scFv-Fc minibodies were added to the target tumor cells in the presence of human peripheral blood mononucleated cell (E:T 25:1). Similar results were obtained in two experiments. Irrelevant anti-SARS scFv-Fc (11A) and anti-CCR4 scFv-Fc (48) minibodies were used as negative controls. (a) CAIX+ sk-rc-09 cells; (b) CAIX+ sk-rc-52 cells; (c) CAIX- sk-rc-59 cells.
Construction and expression of CAIX-specific chimeric receptors
Two generations of anti-CAIX CARs were constructed: first-generation G36 CD8 CAR, with scFvG36 linked to CD8, truncated extracellular, hinge, and transmembrane domains plus signaling domain of TCRζ (G36-CD8z). To deliver costimulatory signals, second-generation CD28 CAR was generated, consisting of scFvG36 fused to truncated extracellular, transmembrane and intracellular domains of CD28 plus signaling domain of TCRζ (G36-CD28z) (Figure 2a). Irrelevant second-generation CD28 CAR was made by using anti-HIV CCR5 (clone A8) scFv.25 In order to detect the expression of these constructs, human rhodopsin C9 tag were inserted between the scFv and CD8 or CD28 domains, respectively and ZsGreen was expressed after the IRES sequence. High concentrations of viral stocks were obtained at comparable levels among the different constructs that were tested by cotransfection of vector plasmids into 293T cells (data not shown).
Figure 2.

Construction and expression of carbonic anhydrase IX (CAIX)-specific CARs. (a) Construction: the first-generation CAR, scFv-CD8-TCRζ (CD8 CAR), is composed of a specific anti-CAIX scFv that is coupled to truncated human CD8α extracellular and hinge domains, transmembrane (TM), and intracellular (IC) regions, then to the signaling domain of human TCRζ. The second-generation CAR, scFv-CD28-TCRζ (CD28 CAR), contains anti-CAIX scFv fused with human CD28 extracellular, TM, and intracellular signaling domain to TCRζ. Both anti-CAIX CARs were cloned into a bicistronic self-inactivating (SIN) lentiviral vector with expression driven by an internal eF1α promoter. The CAR control construct contains an irrelevant anti-HIV CCR5 specific A8 scFv substitution. (b) FACS analysis: Reporter gene ZsGreen was used to quantitate primary T cell transduction efficiency by the lentiviral CAR constructs. In addition, anti-CAIX scFv CARs were stained with CAIX-Fc fusion protein and C9-tag (TETSQVAPA) was stained with 1D4 antibody. Untransduced activated T-cells, LAK only were served as unstained cell control (i) or stained with second antibody ((ii) PE-anti-human IgG and (iii) APC-anti-mouse IgG) were used as staining controls. (c) Western blot: molecular sizes of monomer/dimer structures of anti-CAIX (clone G36) CD28 and anti-CCR5 (clone A8) CD28 CARs, as well as endogenous TCRζ chain of untransduced T cells were indicated.
For transduction, PHA mitogen was used to stimulate peripheral blood lymphocytes for 3 days. Concentrated lentivirus supernatants were used to infect human primary T cells in the presence of cationic reagent diethylaminoethyl-dextran (DEAE-dextran) as it increased the transduction rate of 1.5–2×-fold as compared with polybrene (data not shown). The transduction rate of primary T cells ranged from 17 to 45% by ZsGreen expression in fluorescence-activated cell sorting (FACS) analysis. A representative experiment showing ZsGreen expression in circa 25% by primary CART cells following lentivirus transduction is shown in Figure 2b, left column. CAIX-Fc fusion protein can bind to the G36-CD8z and -CD28z CART cells but not to control A8-CD28z CART cells (Figure 2b, middle column). C9-tag expression was only detected at circa one-third the level of the CAIX-Fc protein (Figure 2b, right column), which is likely related to the finding that mAb 1D4 preferentially recognizes the rhodopsin nonapeptide C9 when presented as a carboxy-terminal versus internal polypeptide sequence (data not shown). Transduced cells that were cultured in vitro for 6 weeks maintained their expression of ZsGreen.
On western blot under reducing conditions, G36 and A8 CD28z CARs migrated with a molecular weight of circa 53kD whereas endogenous TCRζ was 16 kDa. G36-CD8z CAR migrated with a molecular weight of circa 48 kD. Under nonreducing conditions, these two CD28z CARs formed homodimers (Figure 2c, data of CD8z CAR not shown).
Enhanced cytokine secretion by transduced T cells on contact with CAIX+ tumor
A study was performed to compare the reported superior effects of using second-generation G36-CD28z CART cells that incorporate signaling components of the costimulatory molecule CD28 to bypass major histocompatibility complex presentation and enhance T cell effector functions versus first-generation G36-CD8z CART cells. As seen in Figure 3a, after incubation with CAIX+ sk-rc-52 cells overnight, only low levels of type I cytokines IL-2, interferon (IFN)-γ, and IL-17 secretion were seen with control A8 CD28z CART cells or LAK cells alone. In contrast, both first- and second-generation G36 expressing CART cells showed elevated levels of cytokine secretion with second-generation G36-CD28z CART cells secreting higher amounts of type I cytokines, which reflects their higher activation status compared to first-generation G36-CD8z CART cells. Specifically, G36-CD28z CART cells secreted 6.5×, 2.3×, and 4× more IL-2, IFN-γ, and IL-17, respectively than G36-CD8z CART cells. Specificity of cytokine secretion induction by the two G36 CART cells is seen by their minimal stimulation with CAIX− sk-rc-59 cells.
Figure 3.

Effector functions of carbonic anhydrase IX (CAIX)-specific CARTs. (a) Cytokine secretion. Anti-CAIX CART, irrelevant CART, or activated control T cells (LAK) were cocultivated overnight with kidney cancer cell lines sk-rc-52 (CAIX+) and sk-rc-59 (CAIX−) for cytokine production. One representative out of two to three results is shown. (b) ELISPOT. G36 CART or control A8 CART cells were added to tumor cells overnight. IFN-γ or granzyme B secreting T cells detected by ELISPOT. Similar results were obtained in two to three experiments. (c) Specific antitumor cytotoxicity of CAIX-specific CART cells, control A8 CART cells, or LAK cells were incubated in a 4-hour cytotoxicity assay at different amounts of target tumor cell at the ratios as indicated. One out of two experiments is shown. Clone 4-1 is a in vivo passaged subclone of sk-rc-52.
In an Elispot study, after interaction with CAIX+ sk-rc-52 tumors, G36-CD28z CART cells became high-capacity IFN-γ producing cells (Figure 3b). G36-CD28z CART cells produced 6 times more spots than seen for G36-CD8z CART cells upon interaction with CAIX+ sk-rc 52 tumor cells and 12 times more spots than seen after interaction with CAIX− sk-rc-59 tumor cells. Similarly, G36-CD28z CART cells had a higher amount of granzyme B-secreting spots after contact with CAIX+ tumors as compared with G36-CD8z CART cells and control T cells. PMA and ionomycin stimulated T cells yielded the highest amount of IFN-γ and granzyme B secreting T cells. These studies demonstrate both specificity and high capacity of G36-CD28z CART cells to be activated by contact with CAIX+ tumor cells.
Specific cytotoxicity via CAR signaling in transduced T cells
An in vitro cytotoxicity assay was established to further evaluate the killing activity of the different G36 CART cells. Using different ratios of effector-to-target, G36-CD28z CART cells and its twice in vivo passaged subclone 4-1 exhibited the highest amount of cytolysis of CAIX+ tumor sk-rc-52 (Figure 3c). With high ratio of more than 25:1, G36-CD28z CART cells showed two- to threefold higher cytotoxicity than G36-CD8z CART cells and with low ratio of 5:1, G36-CD28z CART cells showed eight- to ninefold higher lysis than G36-CD8z CART cells. However, G36-CD8z CART cells still exhibited good cytotoxicity with up to more than 60% tumor lysis using 100:1 of E:T ratio. Irrelevant A8-CD28z CART cells and control T cell LAK showed the background nonspecific tumor lysis with around 20% lysis when using the highest 100:1 of E:T ratio. In all cases of using CAIX− tumor sk-rc-59, transduced and untransduced T cells showed background lysis.
Improved in vitro proliferation in CART cells with prolonged CAIX+ tumor
Besides enhanced cytokine secretion and cytotoxicity on short-term CAIX+ tumor cell contact, incorporation of the CD28 costimulatory molecule into the CAR construct demonstrated improved proliferation upon prolonged contact with antigen-specific tumor cells. Untransduced and transduced (around 20%) T cells were mixed with freshly irradiated tumor cells weekly in the presence of 100 units/ml human IL-2. To test the different levels of antigen stimulation to a fixed amount of T cells, we used tumor cell to T cell ratios of 1:8, 1:4, and 1:2. T cell numbers were counted by trypan exclusion and CART cell fractions were examined by flow cytometry. Under culture with CAIX− sk-rc-59 tumor cells, the number of transduced and untransduced T cells was maintained (Figure 4a, right panel). The lack of basal level of proliferation of control T cells might be due to the high amount of suppressive cytokines secreted by the tumor cell line. In contrast, after 2 weeks of culture with CAIX+ sk-rc-52 tumor cells, at ratio 1:8, the population of G36-CD28z CART cells increased to 30-fold and G36-CD8z CART cells proliferated up to 17-fold whereas at a ratio of 1:4, the number of G36-CD28z CART cells increased 19-fold and G36-CD8z CART cells proliferated 4-fold. With higher amounts of tumor cells, neither G36-CD28z or G36-CD8z CART cells could proliferate. Irrelevant A8-CD28z CART cells and control T cell LAK showed no proliferation with tumor cells (Figure 4a, left panel).
Figure 4.

Clonal expansion of chimeric antigen receptor-transduced T cells (CART) cells after tumor contact. (a) Proliferation. CAR-transduced T cells or untransduced T cells (LAK) were plated with irradiated tumor cells (CAIX+ sk-rc-52 and CAIX− sk-rc-59) weekly at three different ratios of tumor to T cells as indicated. Number of T cells was counted every 3–4 days in triplicate from two separate wells. Similar results were obtained in two experiments. (b) Clonal enrichment. In tumor stimulation experiments, cultures from CART− and LAK cells were assayed on 1 week and 2 weeks by flow cytometry for expression of CART and (c) T-cell subsets. One representative of two results is shown.
Proliferating T cells were also harvested to examine their enrichment on CAIX+ tumor cell contact. On CAIX− tumor contact, there was no change in the percentage of any CART cells within the population. However, on contact with CAIX+ sk-rc-52 tumor cells, there was enrichment in both populations of G36 CART cells. For G36-CD28z CART cells, the positive population was enriched from 18% on day 0 to 52% on day 8 to 88% on day 16. Expression of G36-CD8z CART cells was enriched from 19% on day 0 (same levels at T cells only) to 32% on day 8, and to 72% on day 16. No expansion of A8-CD28z CART cells was seen over the 2-week study (Figure 4b). The percentage of CD8 cells remained constant throughout the 16-day study under all conditions (Figure 4c).
Persistent effector function of CART cells after recontact with tumor
Transduced T cells that were in contact with irradiated tumor cells for 1 or 2 weeks were also tested for cytokine secretion after 24 hours of contact with fresh nonirradiated tumor cells. Upon contact with CAIX+ tumor (sk-rc-52) for 1 or 2 weeks, G36-CD28z and G36-CD8z CART cells showed similar IFN-γ secretion levels although costimulatory signaling through G36-CD28z CAR yielding 2× to 2.5× more IFN-γ secretion than seen for G36-CD8z CAR (Table 1). For IL-2 secretion, 2 weeks of tumor contact for G36-CD28z and G36-CD8z CART cells exhibited more IL-2 secretion than 1 week of contact. G36-CD28z CART cells yielding 5× more IL-2 than G36-CD8z CART cell on 1 week of contact and 2.5× more on contact for 2 weeks. In addition, G36-CD28z CART cells in contact with tumor cells for 2 weeks secreted 3.3× more IL-2 than one time tumor contact whereas G36-CD8z CART gave 6.8× more IL-2 secretion after 2 weeks compared to after 1 week of tumor contact. These results indicate that the transduced CART cells did not become exhausted and maintain functional activity after a second tumor stimulation. Only background levels of INF-γ and IL-2 secretion were seen with A8-CD28z, LAK, and G36 CART cell treatments on contact with CAIX− sk-rc59 cells.
Table 1. Cytokine secretion after 1 or 2 weeks of contact with tumor cellsa.
| CART cells |
IFN-γ (pg/ml) |
IL-2 (pg/ml) |
||
|---|---|---|---|---|
| One week | Two weeks | One week | Two weeks | |
| RC-SK-52 (CAIX+) cells | ||||
| G36-CD28z | 25,788 | 28,192 | 7,524 | 24,937 |
| G36-CD8z | 13,096 | 10,961 | 1,470 | 10,029 |
| A8-CD28z | 55 | 55 | 9 | 13 |
| LAK | 68 | 58 | 9 | 13 |
| RC-SK-59 (CAIX−) cells | ||||
| G36-CD28z | 31 | 29 | 5 | 4 |
| G36-CD8z | 27 | 38 | 8 | 10 |
| A8-CD28z | 56 | 55 | 7 | 8 |
| LAK | 49 | 56 | 10 | 8 |
Transduced T cells were incubated with irradiated tumor cells for 1 or 2 weeks then harvested, washed, and incubated with fresh nonirradiated tumor cells overnight and supernatants collected after 24 hours for cytokine analysis. For T cell cultures that did not interact with tumor cells, only background level of cytokines were detected at levels < 50 pg/ml interferon-γ and <10 pg/ml interleukin-2.
Suppression of established tumor by CART cells
We next tested CART cells for their ability to inhibit established tumor cell growth in nude mice that were inoculated with sk-rc-52 tumor cells on left flank and sk-rc-59 tumor cells on right flank that had been established to yield similar tumor curves. On day 7 after tumor implantation, with typical tumor size of ~6 × 6 mm, 50 million G36-CD28z CART cells, A8-CD28z CART cell, or untransduced T cells (LAK) were injected intravenously. Adoptive T-cell therapy was performed in two separate experiments with group sizes of n = 7 in the first trial and n = 8 in the second trial, in the presence of high-dose IL-2 (2 × 105 IU) via intraperitoneal injection. No T-cell treatment was included in order to compare the growth of tumor and the effect of cell-therapy.
In trial one, treated and untreated CAIX− sk-rc-59 tumors had average size of 6.09 ± 0.02 mm on day 4 and 9.29 ± 0.12 mm on day 25 (within four tested groups). They exhibited the same tumor growth rate in control groups and T-cell-treated groups. Untreated CAIX+ tumors that received no T cells showed similar tumor size as CAIX− tumors, with an average size of 6.09 ± 0.13 mm on day 4 and 9.15 ± 0.11 mm on day 25. However, the tumor size of G36-CD28z CART cell-treated mice showed statistically significant reduction in size compared to no T-cell-treated mice at every time point that was examined over the 25-day study (Figure 5). G36-CD28z CART treatment also led to a greater reduction in tumor size than seen with A8-CD28z CART cell and LAK-treated mice on day 7 (P < 0.05) and on day 25 (P < 0.001), as calculated by two-tailed t-test. In trial two, tumor size of G36-CD28z CART cell-treated mice was significant smaller than that of no T-cell-treated mice through the 29-day experiment. G36-CD28z CART cell-treated mice also had smaller tumors than were seen with A8 CD28z CART cell- and LAK-treated mice on day 8 to day 26 with P < 0.01 and on day 29 with P < 0.001 (Figure 5).
Figure 5.

Regression of established human renal cell carcinoma (RCC) xenografts by chimeric antigen receptor-transduced T cells (CART) cells. Athymic null mice were inoculated subcutaneously with 7.5 × 106 sk-rc-52 and 5 × 106 sk-rc-59 RCC tumor cells at left and right flank respectively. After 6 days of tumor implantation, mice were injected intravenously with 50 × 106 G36 CD28 CART cells, A8 CD28 CART cells (≥ 20% CAR+), LAK, or phosphate-buffered saline alone. High dose of IL-2 (1 × 105 U/ml) was injected every 2–3 days. Tumor size was measured by caliper every 2–3 days. Experiment 1, n = 7 and experiment 2, n = 8. Tumor size of these two experiments was shown separately. +P < 0.05; *P < 0.01; **P < 0.001 in groups of G36 Tandem-treated mice versus control no T-cell-treated mice in these two trials. Other statistic calculations are reported in the text.
Partial regression of the CAIX+ tumor was considered when the tumor size was smaller than 30% volume of control CAIX− tumor in a same mouse receiving the same T-cell treatment. Partial tumor regression was observed in a high percentage of cases using G36-CD28z CART cells (10 out of 15 (67%)), but only infrequently in irrelevant target A8-CD28z CART cells (1 out of 15 (7%)) and in activated T cell LAKs (2 out of 15 (13%)) (Table 2). Frequency of partial regression response was found to be statistically significant for mice treated with G36-CD28z CART cells versus control A8-CD28z CART cells and LAKs at P < 0.001 and P < 0.005, respectively by Fisher test.
Table 2. Frequency of partial regression of CAIX+ tumors by G36-CD28z CART cells.
| Gene construct | LAK | A8-CD28z | G36-CD28z | Statistics |
|---|---|---|---|---|
| Target antigen | None | Irrelevant | Specific | — |
| Costimulatory | None | Two signals | Two signals | — |
| Partial response | 2 | 1 | 10 | P < 0.005*; p < 0.001** |
| Nonpartial response | 13 | 14 | 5 | N.S. |
Mice from experiments reported in Figure 5 (experiment 1, n = 7 and experiment 2, n = 8) were scored for response at day 10. Partial response is defined as the regression of tumor to smaller than 30% volume of control tumor (same T-cell treatment in the same mouse bearing left flank of sk-rc-52 and right flank of control tumor sk-rc-59).
N.S., no statistically significant relationship between number of tumors and partial response between T cells transduced with LAK and with A8-CD28z.
Fisher test results—*G36-CD28z versus LAK; **G36-CD28z versus A8-CD28z.
In situ cytotoxicity by CART cells
A sample of the whole population of transduced T cells used for the in vivo study were prestained with Far red dye and the CART cells expressing ZsGreen protein within the population were analyzed by confocal microscopy. These results demonstrated circa 30% transduction efficiency, which is in agreement with our FACS analysis (Figure 6a).
Figure 6.

In vivo antitumor activity of chimeric antigen receptor (CAR+) T-cells. (a) Expression of ZsGreen by CART cells is shown in the upper panel. CART cells were prestained with Far Red dye, cytospun and examined by fluorescent microscopy (lower panel). (b) In situ staining of G36 CD28 CART cells in regressing tumor. CART-cells were intravenously injected into renal cell carcinoma established mice and tumor tissue was collected on day 1–3. Confocal microscopy was used to measure apoptosis of tumor cells by TUNNEL assay with PE-Cy5 dye (shown as red). Transduced T cells were shown by ZsGreen. Nuclei were counterstained with DAPI. Two representative slides were shown to indicate the apoptosis of tumor cells at the edge of tumor (upper panel) and inside the tumor bed (middle panel), respectively. The magnified image (lower panel) demonstrates CART cells interacted with multiple tumors while a few surrounding tumor cells were dying (left column, merged staining). (c) Granzyme B+ T cells and tumor necrosis. After the treatment with CART cells, the regressing CAIX+ sk-rc-52 tumors were stained by granzyme B antibody (brown) and H&E. The higher magnification view (middle and lower panels of sections a and b in upper panel) shows the locations of granzyme B+ T cells (shown by arrows) and the corresponding H&E slide shows the tumor necrosis (shown by n). Granzyme B+ T cells are distributed at the edge of tumor (middle panel) and inside the tumor (lower panel).
To provide evidence that G36-CD28z CART cell treatment of CAIX+ sk-rc-52 tumor cells in vivo resulted in killing by apoptosis, tumor sections were stained by Tunnel assay. On day 3 after adoptive T cell treatment, Tunnel staining identified apoptotic tumor cells (red) at the edge of tumor (Figure 6b, upper row) and inside the tumor bed (Figure 6b, middle row). The apoptoic tumor cells lost the DAPI nuclear staining. Shown in the enlarged graph (Figure 6b, bottom row) is a ZsGreen expressing CART cell interacting with two tumor cells that were going apoptosis.
Due to the limitation of fluorescent signal, ZsGreen expressing CART cells could not be observed from the whole tissue section. Therefore, on day 3 after G36-CD28z CART cell or LAK treatment, the tumors were harvested and sections were also stained with granzyme B antibody to locate the activated T cells. In Figure 6c, the dark brown areas of staining show granzyme B+ T cells that are seen infiltrating into the CAIX+ sk-rc-52 tumor sections (Figure 6c, upper left). These granzyme B+ T cells were seen surrounding the tumor (Figure 6c, upper left (a) and middle) and inside the tumor (Figure 6c, upper left (b) and lower). Tumors with necrotic areas were shown in H&E stained slides (labeled as n inside Figure 6c, right middle and lower) and lie at locations near to the granzyme B+ T cells. In contrast, the CAIX+ sk-rc-52 tumors treated with control activated T cells (LAK) (Supplementary Figure S1) did not show any granzyme B+ T cells. Similarly, CAIX− sk-rc-59 treated with G36-CD28z CART cells (Supplementary Figure S2) or treated with LAK (Supplementary Figure S3) showed a low-background staining while tumor was proliferating. For positive control of granzyme B staining, CART cells was locally injected into the established sk-rc-52 tumor in mice. After 1 day, the mice was sacrificed and tumor tissue was sectioned for this staining (Supplementary Figure S4).
Discussion
In this study, we characterized a panel of five human scFv-Fcs against the catalytic domain of carbonic anhydrase CAIX for ADCC activity. All anti-CAIX mAbs showed potent ADCC activity suggesting that the CAIX epitope(s) are properly oriented with respect to the plasma membrane for FcγR-mediated killing to occur. Antibody scFvG36 was chosen for targeting the anti-CAIX CARs based on its killing activity, good expression levels, and ability to mediate CAIX internalization.23 We constructed self-inactivating lentiviral vectors expressing first- and second-generation fully human G36 CARs and performed extensive in vitro comparisons of the two. We are also the first to report on the in vivo antitumor activity of G36 CART cells against RCC.
Consistent with previous reports with CARs containing costimulatory signaling motifs, second-generation G36-CD28z CART cells possessed superior effector functions compared with first-generation G36-CD8z CART cells in all in vitro assays including IFN-γ, IL-2, and IL-17 cytokine secretion (Figure 3a), cytolytic activity (Figure 3b,c), proliferation (Figure 4a), and clonal expansion (Figure 4b). IL-2 and IFN-γ have been shown to promote antitumor activity of CD8+ T cells and are likely involved in the tumorocidal activity that was observed in vivo. However, these same cytokines have also been implicated in the recruitment of NK cells to tumors.26,27 The role of IL-17 in antitumor immunity is controversial28 and this cytokine may have different functions depending on the tumor type and stage.19,29 IL-17 secreting CD8+ T cells have been shown to have enhanced antitumor immunity in vivo30 and a mechanistic link between IL-17 and NK activity has been recently demonstrated.29
Second-generation G36-CD28z CART cells also showed superior activity in vivo in inhibiting CAIX+ tumor cell growth compared to irrelevant CART or LAK cells (Figure 5). In situ studies provided evidence of tumor cell apoptosis (Figure 6b) as well as the presence of granzyme B+ T cells and tumor necrosis (Figure 6c). The greater in vivo effector functions mediated by G36-CD28z CART cells may be due to several mechanisms. First, G36-CD28z CART cells showed a greater capacity to proliferate on repeated contact with CAIX+ tumor cells than did G36-CD8z CART cells (Figure 4b). They showed this same augmented capacity to secrete IFN-γ and IL-2 on recontact with CAIX+ tumor cells (Table 1). Second, in addition to enhanced proinflammatory cytokine secretion, the G36-CD28z CART cells may be more resistant to activation-induced cell death resulting from tumor antigen contact than with G36-CD8z CART that receive T-cell receptor (signal 1)-only stimulation.31 This activation-induced cell death resistance is due to upregulation of expression of antiapoptotic proteins such as cFLIP, bcl-xL, and bcl-2.32 The CD28 cytoplasmic domain can also serve to enhance cellular activation through protein kinase C-θ-mediated signaling.33 Third, G36-CD28z CART cells may be better protected from suppression by regulatory T cells (Tregs) and their inhibitory cytokine, transforming growth factor-β, due to the enhanced NF-κB activation through the phosphoinositide 3-kinase-Akt pathway.34 Indeed, expansion of Tregs was found to be significantly higher in DC-vaccination and cytokine therapy nonresponders than in responding patients.35 These resistances provide extra benefits for G36-CD28z CART immunotherapy.
To avoid the development of unwanted immunogenic response following the introduction of CAR-specific antibodies during adoptive transfer using chimeric TCR receptors, the use of a humanized or a human scFv had been suggested.36 It has been shown that the scFv component of the CAR derived from murine monoclonal antibody MOv18 resulted in detectable levels of human anti-mouse antibodies (HAMA).37 HAMA might inhibit redirected T cell function by blocking the interaction between CAR and target antigen.38 The immunogenicity of CARs can restrict the duration of treatment and thus limit the overall antitumor effect. Indeed, when murine anti-CAIX mAb G250 was used as a first-generation CAR in a RCC clinical trial both anti-idiotypic antibodies and anti-CAR cell-mediated immunity to the scFv component developed that neutralized and resulted in CART cell clearance.36 In addition, so-called “on-target” liver toxicity cells was observed that was attributed to CAIX expression on bile duct epithelial cells.17 Indeed, the expression of CAIX on normal tissues by immunohistochemical staining has been documented using murine anti-CAIX mAbs M75 and G250 that are directed to the N-terminal PG domain. This staining is largely restricted to the apical surface of cells of the stomach and bile duct mucosa39,40 and small intestine.4,39,41 Thus, this autoreactivity could be either induced directly by the anti-CAIX CART cells recognizing normal tissues expressing the target antigen or indirectly by antibodies or cell-mediated immunity directed to the anti-CAIX CART cells that have localized to this tissue. In contrast, mAb G36 recognizes the centrally located catalytic domain and does not recognize CAIX by immunohistochemical staining (data not shown), and therefore, it remains to be determined if similar crossreactivity and toxicity would occur in vivo with G36-CD28z CART cells. However, because of the superior effective antitumor functions of these second-generation CART cells and the fully human format of scFv, this G36-CD28z CAR may be a preferred agent for use in future clinical trials, which we propose should require a lower dose of infused CART cells and have reduced immunogenicity due to absence of HAMA and cell-mediated immunity (CMI) responses to the scFv and CART cells.
As in the cases of other cell surface-expressed tumor-associated antigens, CAIX is shed from the tumor cell surface to the blood stream and urine.42 It is not known whether the circulating CAIX will compete with cell surface CAIX on tumor cells for binding to the CART cells. Also another potential complication is that soluble serum CAIX will cause widespread triggering of infused genetically redirected T cells which in turn might lead to massive cytokine release including IFN-γ. Fortunately, several studies have documented that the soluble serum antigen did not inhibit CART cell responses as indicated by the results of cytokine release and cytotoxicity analysis.43
In summary, we demonstrate that second-generation G36-CD28z CART cells possess superior antitumor responses as evidenced by the combined effect of stronger cytotoxic potency, increased cytokine secretion, enhanced proliferation and clonal expansion in vitro, and improved suppression of tumors in vivo with IL-2 provision. Higher response rate and low toxicity were also demonstrated in patients treated with first-generation anti-CAIX CART cells that were pretreated with chimeric CAIX Abs, a strategy that could be similarly developed here with fully human anti-CAIX mAbs.17 In addition, as other costimulatory molecules including 4-1BB and OX40 continue to be assessed in CAR constructs in order to maximize effective activity of modified T cells18,19,31,44–47 opportunities may arise to reduce the number of cells required for efficacious adoptive T-cell therapy. Importantly, the human G36-CD28z CART cells should not elicit HAMA and CMI responses during treatment cycles. We envision that these CART cells used either alone or together with other immunotherapic48,49 agents including those that are focused on costimulatory blockade such as CTLA4 and PD1/PDL1 axis may provide a next generation treatment for RCC.
Materials and Methods
Cells, culture media, and reagents
Human CAIX+ RCC cell lines sk-rc-52, sk-rc-09, and CAIX− sk-rc-59 were obtained from Dr Gerd Ritter, Memorial Sloan-Kettering Cancer Center, New York. They were cultured at 37 °C with 5% CO2 in R-10 complete medium containing RPMI 1640 medium (Life Technologies, Grand Island, NY) supplemented with 10% FCS, 2 mmol/l L-glutamine, 100 U/ml penicillin, and 100 µg/ml streptomycin (Sigma, St Louis, MO). Primary human T cells were maintained in R-10 with 10% human serum and 100 IU/ml recombinant human IL-2 (Chiron, Emeryville, CA). Human embryonic kidney cell line 293T (ATCC, Manassas, VA) and mouse fibroblast NIH3T3 cells (ATCC) were grown in D-10 complete medium (Life Technologies) containing Dulbecco's modified Eagle's medium medium with 10% FCS, 100 U/ml penicillin, and 100 µg/ml streptomycin (Sigma). Leukopacks obtained from the blood bank of the Children’s Hospital Boston were collected from healthy volunteers with written informed consent.
scFv isolation and conversion of scFv to scFv-Fc
CAIX-specific scFv antibodies were isolated from a nonimmune human scFv phage library as previously reported and submitted to GenBank with accession numbers of GQ903548-GQ903561 (ref. 23). scFv-coding DNA fragments from the pFarber phagemid were digested with SfiI/NotI sites and subcloned into the mammalian expression vector pcDNA3.1-F105L-hinge-stuffer, which has a human IgG1 F105 leader sequence and the human IgG1 hinge-CH2-CH3 Fc portion to express scFv-Fc antibodies. Plasmids of scFv-Fc were transiently transfected into 293T cells by lipofectamine 2000 (Invitrogen, Carlsbad, CA), and expressed antibodies were purified using Sepharose protein A beads (Amersham Bioscience, GE Healthcare, Piscataway, NJ). Specific binding to CAIX was tested by staining with phage scFv antibodies or scFv converted into scFv-Fc format antibodies by incubation with CAIX-expressing 293T and sk-rc-52 cell lines, and with CAIX negative 293T and sk-rc-59 cell lines. In these experiments, irrelevant anti-HIV CCR5 antibody (clone A8)25 or anti-SARS antibody (11A)24 and fluorescently conjugated secondary antibodies alone were used as negative controls.
Construction of scFv-CD8-TCRζ and scFv-CD28-TCRζ constructs
Pz1, scFv-CD8-TCRζ, and P28z, scFv-CD28-TCRζ, DNA constructs in phagemid vector pSL1180 were obtained from Dr Michel Sadelain, Memorial Sloan-Kettering Cancer Center, New York. In Pz1, the scFv and TCRζ intracellular domain are appended to N- and C-terminus of human CD8α chain, respectively. Similarly, in P28z, the scFv and TCRζ sequences are appended to the N- and C-terminus of human CD28, respectively. The amino acid sequence of human CD8α is 71 residues in length, consisting of 47 (aa 137–183), 23 (aa 184–206), and 2 (aa 207–208) residues of the CD8α extracellular and hinge, transmembrane, and cytoplasmic domains, respectively. The CD28 sequence in P28z is 107 residues in length, consisting of 40 (aa 114–153), 23 (aa 154–176), and 44 (aa 177–220) residues of the CD28 extracellular, transmembrane, and cytoplasmic domains respectively. The human CD3ζ intracellular domain common to both CARs consists of 112 amino acids (aa 52–163).
The nucleic acid sequence encoding an internal C9-tag (a nine-amino acid peptide of human rhodopsin, TETSQVAPA) with a GGGGS linker was amplified by PCR and was fused upstream with CD8-TCRζ and CD28-TCRζ sequences with 5′ NotI site and 3′ PacI sites. The primers used for cloning chimeric TCRζ constructs are 5′ TAG GGC GCG GCC GCa acc gag acc agc cag gtg gcg ccc gcc GGG GGA GGA GGC AGC CCC ACC ACG ACG CCA GCG CCG CGA 3′ (forward primer for CD8 construct where italic is the NotI site, upper case is the C9 tag sequence, and underlining indicates the GGGGS linker), 5′ TAG GGC GCG GCC GCa acc gag acc agc cag gtg gcg ccc gcc GGC GGA GGA GGC AGC ATT GAA GTT ATG TAT CCT CCT CCT 3′ (forward primer for CD28 construct) and reverse primer for both constructs CTA GCC TT AAT TAA,TTA GCG AGG AGG GGG CAG GGC CTG CAT, italic is Pac I site. These DNA fragments encoded functional features which are arranged in accordance with the following sequence: NotI – C9tag (TETSQVQPQ) – GGGGS – CD8 or CD28 – TCRζ – PacI. The chimeric TCR constructs tagged with internal C9 peptide were cloned into the pcDNA3.1-F105L-hinge stuffer vector containing anti-CXCR4 scFv-Fc, clone 48, using NotI and PacI restriction sites. This design allowed us to insert chimeric TCR receptor constructs to replace Fc portion fragment. Later, anti-CAIX scFv (clone G36) and anti-CCR5 scFv (clone A8, as irrelevant scFv control) antibody fragments were cloned to replace anti-CXCR4 scFv at SfiI/NotI sites to create CAIX-specific chimeric TCR constructs.
The lentivirus vector pHAGE-CMV-DsRed-IRES-ZsGreen, and four HIV helper plasmids pHDM-Hgpm2 (HIV gag-pol), pMD-tat, pRC/CMV-rev, and an Env VSV-G pseudotype were obtained from Dr Richard Mulligan, of the Virus Production Core at The Harvard Gene Therapy Initiative in Boston. The CMV promoter in pHAGE-CMV-IRES-ZsGreen was replaced by an eF1α promoter derived from the pSIN lentivirus vector at SpeI/NotI sites. One of the five scFv-Fc antibodies, G36, which possess high affinity to CAIX+ cells and high ADCC only against CAIX+ tumor cells, was cloned into pHAGE-eF1α lentivirus vector at AscI/BamHI to replace the first cassette of the DsRed protein.
Lentivirus production and transduction of human primary T cells
Lentivirus was produced by five plasmid transient transfection into 293T cells using lipofectamine 2000 as per the manufacturer’s instructions (Invitrogen). Cells were prepared for 80% confluence in 15 cm Petri dishes (Nalge Nunc, Rochester, NY) and transfected with 30 µg of total plasmid DNA. The ratio of vector plasmids (pHDM-Hgpm2 (HIV gag-pol): pMD-tat: pRC/CMV-rev: Env VSV-G pseudotype) was 20:1:1:1:2. After changing to D-10 medium, virus supernatant was harvested on day 3, filtrated through a 0.45-µm filter, and concentrated by ultracentrifugation (Beckman Coulter, Fullerton, CA) for 90 minutes at 16,500 rpm (48,960 × g, Beckman SW28 rotor) and 4 °C. The virus pellets were resuspended in R-10 medium and kept frozen at −80 °C.
Human peripheral blood mononucleated cells were isolated by ficoll density gradient separation and were activated with 2 µg/ml phytohemagglutinin (PHA) (Sigma) plus 100 IU/ml human IL-2 for 4 days. The cells were infected with two or three rounds of lentivirus transduction at multiplicity of infection of 10–20 in the presence of 10 µg/ml DEAE-dextran. Three days after transduction, transduced T cells were collected for phenotypic and functional analyses in vitro, or were expanded for in vivo experiments.
Flow cytometric analysis
Transduction efficiency of human primary T cells was assessed by expression of a reporter gene (ZsGreen, Clontech, Mountain View, CA). The CAIX-Fc protein was expressed from a pcDNA3.1 plasmid that encoded amino acids 38–397 of CAIX followed by human IgG1 hinge, CH2, and CH3 domains; the CAIX signal peptide (aa 1-37) was replaced with Ig leader sequence. Expression of scFv(G250) on transduced T cells was tested by staining the cells with 1 µg CAIX-Fc protein, and then APC-conjugated mouse anti-human IgG antibody (Jackson ImmunoResearch, West Grove, PA). Additionally, expression of the internal rhodopsin nonapeptide (TETSQVAPA) C9 tag of the scFv domain of TCR constructs on transduced T cells was detected by staining with 5 µg mouse 1D4 antibody followed by APC-conjugated goat anti-mouse IgG antibody (Jackson ImmunoResearch). For analysis, the subsets of human cells in culture during clonal expansion experiment were stained with fluorescence conjugated mouse anti-human antibodies (Invitrogen) against CD3 (clone S4.1), CD4 (clone S3.5), or CD8 (clone 3B5). In all cell staining, five hundred thousand cells were stained with antibodies at recommended concentration according to company’s instruction. The matched isotype control antibodies for each sample were used and the cells were analyzed using a FACSCalibur cytometer (Becton-Dickinson, Franklin Lakes, NJ).
ADCC and cytotoxicity assay of lentivirus transduced T cells
Cytotoxicity assays were performed using the DELFIA EuTDA Cytotoxicity kit (Perkin Elmer, Boston, MA) in accordance with the manufacturer’s instructions. Briefly, target tumor cells were labeled with a fluorescent ligand (BATDA) for 30 minutes at 37 °C and 1 × 104 labeled cells were loaded per well in 96-well U-bottom plate. For ADCC assay, a panel of anti-CAIX scFv-Fc antibodies or irrevelant scFv-Fc antibody at a concentration of 1 or 5 µg/ml was added separately. The assay was set up with ratios of effector cells (human peripheral blood mononucleated cell) to target cells (E:T) at 50:1, 25:1, and 12.5:1. For the T cell cytoxicity assay, different ratios of effector cells (nontransduced or transduced T cells) to target cells (E:T) were prepared (100:1, 50:1, and 25:1). The culture was incubated for 4 hours in humidified 5% CO2 at 37 °C. After the plate was spun for 5 minutes at 500× g, 20 µl of supernatant was transferred to a flat-bottom plate. Two hundred microliters of Europium solution was added and the fluorescence released from the cells was read by fluorometer (Victor, PerkinElmer). The control for spontaneous release was prepared by culturing the labeling cells only and the control for maximum release was made by adding lysis buffer (kit provided) to the labeling cells.
Enzyme-linked immunosorbent assay, ELISPOT assays, and western blot
For cytokine secretion, RCC cell lines sk-rc-52 (CAIX+) or sk-rc-59 (CAIX−) were seeded overnight at 1 × 106 per well in a 24-well plate, followed by 1 × 106 untransduced or transduced T cells. Before coculture with tumor cells, T cells were washed with phosphate-buffered saline twice to remove human IL-2. After overnight incubation, the supernatant was harvested and analyzed for IL-2 and IFN-γ by enzyme-linked immunosorbent assay (eBioscience, San Diego, CA). In detecting T cells for the IFN-γ ELISPOT assay (eBioscience), a membrane was developed using AEC substrate solution and the number of spots was counted by ELISPOT plate reader (C.T.L. Cellular Technology, Shaker Heights, OH).
For western blot, preparation of untransduced and transduced T cells was described.50 One million cells were prepared in nonreducing and reducing buffer (0.1 mol/l dithiothreitol) and run on a 10–20% polyacrylamide gradient gel (Invitrogen). Proteins were transferred to polyvinylidence fluoride transfer membrane (NEN Life Science Products, Boston, MA) at 100 V, 4 °C overnight. The membrane was incubated with 1:2,000 primary antibody, anti-human ζ-chain monoclonal antibody 8D3 (BD Pharmingen, San Diego, CA) and then with 1:3,000 secondary antibody horseradish peroxidase (Caltag, Buckingham, UK). Immunodetection was performed using the ECL Plus Western blotting detection system (GE Healthcare, Piscataway, NJ) and X-ray film exposure.
Proliferation, clonal expansion, and cytokine secretion after tumor cell contact
Tumor cells were irradiated (3,000 rads) and seeded at 2.5 × 105 per well. T cells were added at 1 × 106 in culture medium containing R-10 plus 100 IU/ml human IL-2 for a week culture. T cells were split to maintain suitable density and restimulated with tumor cells weekly. The number of T cells was counted every 3 or 4 days for 2 weeks. The percentage expression of ZsGreen by transduced T cells and T cell subsets were determined weekly by FACS. For cytokine secretion studies after tumor cell contact, T cells that were in contact with irradiated tumor cells for 1 or 2 weeks were washed, incubated with fresh tumor cells overnight and culture supernatants were collected after 24 hours for analysis.
Tumor establishment and T cell therapy
Due to immune-rejection of sk-rc-52 in 6–8-week-old female BALB/c nude mice and to accelerate in vivo growth properties, five million cells were subcutaneously inoculated into the mice, harvested, and expanded in vitro. The cell line was then passaged two more times in nude mice and the passaged cells were expanded for further experiments (subclone 4-1). For the therapeutic experiments, 5 million sk-rc-59 and 7.5 million passaged sk-rc-52 cells were subcutaneously inoculated on opposing flanks into nude mice to yield comparable tumor growth rates. After 7 days, tumors grew to the size of ~6 mm, and 50 million nontransduced or transduced T cells were injected intravenously. The mice were also treated with 20,000 IU human IL-2 by peritoneal injection every 2 days. Tumor size was measured by caliper in two dimensions and the mean of two tumor diameter was reported here. Animal experiments were performed in accordance with the guidelines of the Dana Farber Cancer Institute Animal Care Committee. Mice were sacrificed when tumors reached 15-mm diameter or 2,000 mm3 and tumors were harvested.
Immunohistochemistry and immunofluorescence staining
For in vitro examination of transduced T cells, the cultured T cells were washed twice using phosphate-buffered saline and resupended in 2µmol/l Far Red DDAO-SE CellTrace dye (Molecular Probes, Life Technologies) in phosphate-buffered saline for 15 minutes at 37 °C. Then, the cells were washed with culture medium twice and cytospun on the glass slide. Far red prestained CART cells with ZsGreen coexpression were visualized using confocal microscopy (Zeiss, Jena, Germany) at the Optical Imaging Core facility, Harvard NeuroDiscovery Center.
To examine the killing effect of transduced T cells in tumor bed in situ, tumors were prepared for frozen sections for ApopTag Peroxidase In Situ Apoptosis Detection kit (EMD Millipore, Billerica, MA). Cryosections were incubated with TdT enzyme (Millipore) for 1 hour. Rabbit anti-DIG (Dako, Carpinteria, CA) was added and incubated for 30 minutes and then Cy3-conjugated anti-rabbit antibody (Invitrogen) was added and incubated for 30 minutes. Sections were mounted with 4′,6-diamidino-2-phenylindole (DAPI) antifade mounting medium and fluorescent images were examined using confocal microscopy.
Xenograft tumors and mouse spleens were harvested, fixed in 10% formalin/phosphate-buffered saline solution, and submitted to the Harvard Medical School, Rodent Histopathology Core Facility. Paraffin-embedded sections were dewaxed with xylene and rehydrated through graded alcohols before staining. Immunohistochemistry staining was performed by incubating with anti-human granzyme B antibody (Dako, clone GrB-7 (1:200)) as a primary antibody for 1 hour followed by secondary anti-rabbit antibody (Pierce, ThermoScientific, Rockford, IL) or anti-mouse antibody (Dako) for 30 minutes. Sections were developed using DAB substrate and counterstained with hematoxylin.
Statistical analyses
Statistical significance was determined using the two-tailed Student’s t-test.
Acknowledgments
We thank Zeng Qing for technical assistance, XiaoYin Xu for assisting with the animal studies, Aimee Tallarico for assisting with DNA analysis of vectors used in this paper and for help with the final draft of the manuscript. We also thank De-Kuan Chang for immunohistochemical analysis of the tissue samples and Quan Karen Zhu for her overall support in the project. We are also grateful to David Cook for reviewing the manuscript. We thank Gerd Ritter, Department of Human Cancer Immunology, Ludwig Institute for Cancer Research for providing the RCC cell lines (sk-rc-52, sk-rc-44 and sk-rc-59), Richard Mulligan, Harvard Gene Therapy Initiative, Harvard Medical School for providing the pHAGE lentiviral vector, and Andrew Kung, Department of Pediatric Oncology, Dana-Farber Cancer Institute, for offering the retroviral supernatant expressing firefly luciferase. We thank Aimee St. Clair Tallarico for her assistance in graphic design. A.S.-Y.L. and W.A.M. designed research and wrote the paper; C.X. provided anti-CAIX scFvs; A.M. provided CAIX cDNA. This work was supported by NIH grant R21DK072282 to W.A.M.
The authors declare no conflict of interest.
References
- Ivanov, S, Liao, SY, Ivanova, A, Danilkovitch-Miagkova, A, Tarasova, N, Weirich, G et al. (2001). Expression of hypoxia-inducible cell-surface transmembrane carbonic anhydrases in human cancer. Am J Pathol 158: 905–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loncaster, JA, Harris, AL, Davidson, SE, Logue, JP, Hunter, RD, Wycoff, CC et al. (2001). Carbonic anhydrase (CA IX) expression, a potential new intrinsic marker of hypoxia: correlations with tumor oxygen measurements and prognosis in locally advanced carcinoma of the cervix. Cancer Res 61: 6394–6399. [PubMed] [Google Scholar]
- Hilvo, M, Baranauskiene, L, Salzano, AM, Scaloni, A, Matulis, D, Innocenti, A et al. (2008). Biochemical characterization of CA IX, one of the most active carbonic anhydrase isozymes. J Biol Chem 283: 27799–27809. [DOI] [PubMed] [Google Scholar]
- Oosterwijk, E, Ruiter, DJ, Hoedemaeker, PJ, Pauwels, EK, Jonas, U, Zwartendijk, J et al. (1986). Monoclonal antibody G 250 recognizes a determinant present in renal-cell carcinoma and absent from normal kidney. Int J Cancer 38: 489–494. [DOI] [PubMed] [Google Scholar]
- Liao, SY, Brewer, C, Závada, J, Pastorek, J, Pastorekova, S, Manetta, A et al. (1994). Identification of the MN antigen as a diagnostic biomarker of cervical intraepithelial squamous and glandular neoplasia and cervical carcinomas. Am J Pathol 145: 598–609. [PMC free article] [PubMed] [Google Scholar]
- Liao, SY, Aurelio, ON, Jan, K, Zavada, J and Stanbridge, EJ (1997). Identification of the MN/CA9 protein as a reliable diagnostic biomarker of clear cell carcinoma of the kidney. Cancer Res 57: 2827–2831. [PubMed] [Google Scholar]
- Atkins, M, Regan, M, McDermott, D, Mier, J, Stanbridge, E, Youmans, A et al. (2005). Carbonic anhydrase IX expression predicts outcome of interleukin 2 therapy for renal cancer. Clin Cancer Res 11: 3714–3721. [DOI] [PubMed] [Google Scholar]
- Lokich, J (1997). Spontaneous regression of metastatic renal cancer. Case report and literature review. Am J Clin Oncol 20: 416–418. [DOI] [PubMed] [Google Scholar]
- Chang, AE, Li, Q, Jiang, G, Sayre, DM, Braun, TM and Redman, BG (2003). Phase II trial of autologous tumor vaccination, anti-CD3-activated vaccine-primed lymphocytes, and interleukin-2 in stage IV renal cell cancer. J Clin Oncol 21: 884–890. [DOI] [PubMed] [Google Scholar]
- Dudley, ME, Wunderlich, JR, Robbins, PF, Yang, JC, Hwu, P, Schwartzentruber, DJ et al. (2002). Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 298: 850–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaft, N, Willemsen, RA, de Vries, J, Lankiewicz, B, Essers, BW, Gratama, JW et al. (2003). Peptide fine specificity of anti-glycoprotein 100 CTL is preserved following transfer of engineered TCR alpha beta genes into primary human T lymphocytes. J Immunol 170: 2186–2194. [DOI] [PubMed] [Google Scholar]
- Bubeník, J (2004). MHC class I down-regulation: tumour escape from immune surveillance? (review). Int J Oncol 25: 487–491. [PubMed] [Google Scholar]
- Gajewski, TF, Meng, Y, Blank, C, Brown, I, Kacha, A, Kline, J et al. (2006). Immune resistance orchestrated by the tumor microenvironment. Immunol Rev 213: 131–145. [DOI] [PubMed] [Google Scholar]
- Frigola, X, Inman, BA, Lohse, CM, Krco, CJ, Cheville, JC, Thompson, RH et al. (2011). Identification of a soluble form of B7-H1 that retains immunosuppressive activity and is associated with aggressive renal cell carcinoma. Clin Cancer Res 17: 1915–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grépin, R, Guyot, M, Giuliano, S, Boncompagni, M, Ambrosetti, D, Chamorey, E et al. (2014). The CXCL7/CXCR1/2 axis is a key driver in the growth of clear cell renal cell carcinoma. Cancer Res 74: 873–883. [DOI] [PubMed] [Google Scholar]
- Sadelain, M, Brentjens, R and Rivière, I (2009). The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol 21: 215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamers, CH, Sleijfer, S, van Steenbergen, S, van Elzakker, P, van Krimpen, B, Groot, C et al. (2013). Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther 21: 904–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milone, MC, Fish, JD, Carpenito, C, Carroll, RG, Binder, GK, Teachey, D et al. (2009). Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther 17: 1453–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkie, S, Picco, G, Foster, J, Davies, DM, Julien, S, Cooper, L et al. (2008). Retargeting of human T cells to tumor-associated MUC1: the evolution of a chimeric antigen receptor. J Immunol 180: 4901–4909. [DOI] [PubMed] [Google Scholar]
- Lo, AS, Ma, Q, Liu, DL and Junghans, RP (2010). Anti-GD3 chimeric sFv-CD28/T-cell receptor zeta designer T cells for treatment of metastatic melanoma and other neuroectodermal tumors. Clin Cancer Res 16: 2769–2780. [DOI] [PubMed] [Google Scholar]
- Kalos, M, Levine, BL, Porter, DL, Katz, S, Grupp, SA, Bagg, A et al (2011). T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Translat Med 3: 95ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegram, HJ, Park, JH and Brentjens, RJ (2014). CD28z CARs and armored CARs. Cancer J 20: 127–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, C, Lo, A, Yammanuru, A, Tallarico, AS, Brady, K, Murakami, A et al. (2010). Unique biological properties of catalytic domain directed human anti-CAIX antibodies discovered through phage-display technology. PLoS ONE 5: e9625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui, J, Aird, DR, Tamin, A, Murakami, A, Yan, M, Yammanuru, A et al. (2008). Broadening of neutralization activity to directly block a dominant antibody-driven SARS-coronavirus evolution pathway. PLoS Pathog 4: e1000197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzabekov, T, Kontos, H, Farzan, M, Marasco, W and Sodroski, J (2000). Paramagnetic proteoliposomes containing a pure, native, and oriented seven-transmembrane segment protein, CCR5. Nat Biotechnol 18: 649–654. [DOI] [PubMed] [Google Scholar]
- Wald, O, Weiss, ID, Wald, H, Shoham, H, Bar-Shavit, Y, Beider, K et al. (2006). IFN-gamma acts on T cells to induce NK cell mobilization and accumulation in target organs. J Immunol 176: 4716–4729. [DOI] [PubMed] [Google Scholar]
- Zeytin, H, Reali, E, Zaharoff, DA, Rogers, CJ, Schlom, J and Greiner, JW (2008). Targeted delivery of murine IFN-gamma using a recombinant fowlpox virus: NK cell recruitment to regional lymph nodes and priming of tumor-specific host immunity. J Interferon Cytokine Res 28: 73–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murugaiyan, G and Saha, B (2009). Protumor vs antitumor functions of IL-17. J Immunol 183: 4169–4175. [DOI] [PubMed] [Google Scholar]
- Bär, E, Whitney, PG, Moor, K, Reis e Sousa, C and LeibundGut-Landmann, S (2014). IL-17 regulates systemic fungal immunity by controlling the functional competence of NK cells. Immunity 40: 117–127. [DOI] [PubMed] [Google Scholar]
- Hinrichs, CS, Kaiser, A, Paulos, CM, Cassard, L, Sanchez-Perez, L, Heemskerk, B et al. (2009). Type 17 CD8+ T cells display enhanced antitumor immunity. Blood 114: 596–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hombach, AA, Rappl, G and Abken, H (2013). Arming cytokine-induced killer cells with chimeric antigen receptors: CD28 outperforms combined CD28-OX40 “super-stimulation”. Mol Ther 21: 2268–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mor, F and Cohen, IR (1996). IL-2 rescues antigen-specific T cells from radiation or dexamethasone-induced apoptosis. Correlation with induction of Bcl-2. J Immunol 156: 515–522. [PubMed] [Google Scholar]
- Isakov, N and Altman, A (2012). PKC-theta-mediated signal delivery from the TCR/CD28 surface receptors. Front Immunol 3: 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loskog, A, Giandomenico, V, Rossig, C, Pule, M, Dotti, G and Brenner, MK (2006). Addition of the CD28 signaling domain to chimeric T-cell receptors enhances chimeric T-cell resistance to T regulatory cells. Leukemia 20: 1819–1828. [DOI] [PubMed] [Google Scholar]
- Schwarzer, A, Wolf, B, Fisher, JL, Schwaab, T, Olek, S, Baron, U et al. (2012). Regulatory T-cells and associated pathways in metastatic renal cell carcinoma (mRCC) patients undergoing DC-vaccination and cytokine-therapy. PLoS ONE 7: e46600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamers, CH, Willemsen, R, van Elzakker, P, van Steenbergen-Langeveld, S, Broertjes, M, Oosterwijk-Wakka, J et al. (2011). Immune responses to transgene and retroviral vector in patients treated with ex vivo-engineered T cells. Blood 117: 72–82. [DOI] [PubMed] [Google Scholar]
- Miotti, S, Negri, DR, Valota, O, Calabrese, M, Bolhuis, RL, Gratama, JW et al. (1999). Level of anti-mouse-antibody response induced by bi-specific monoclonal antibody OC/TR in ovarian-carcinoma patients is associated with longer survival. Int J Cancer 84: 62–68. [DOI] [PubMed] [Google Scholar]
- Kershaw, MH, Westwood, JA, Parker, LL, Wang, G, Eshhar, Z, Mavroukakis, SA et al. (2006). A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res 12(20 Pt 1): 6106–6115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastorekova, S (2004). Carbonic anhydrase IX (CA IX) as a potential target for cancer therapy. Cancer Ther 2: 245–262. [Google Scholar]
- Pastoreková, S, Parkkila, S, Parkkila, AK, Opavský, R, Zelník, V, Saarnio, J et al. (1997). Carbonic anhydrase IX, MN/CA IX: analysis of stomach complementary DNA sequence and expression in human and rat alimentary tracts. Gastroenterology 112: 398–408. [DOI] [PubMed] [Google Scholar]
- Saarnio, J, Parkkila, S, Parkkila, AK, Waheed, A, Casey, MC, Zhou, XY et al. (1998). Immunohistochemistry of carbonic anhydrase isozyme IX (MN/CA IX) in human gut reveals polarized expression in the epithelial cells with the highest proliferative capacity. J Histochem Cytochem 46: 497–504. [DOI] [PubMed] [Google Scholar]
- Závada, J, Závadová, Z, Zat’ovicová, M, Hyrsl, L and Kawaciuk, I (2003). Soluble form of carbonic anhydrase IX (CA IX) in the serum and urine of renal carcinoma patients. Br J Cancer 89: 1067–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hombach, A, Koch, D, Sircar, R, Heuser, C, Diehl, V, Kruis, W et al. (1999). A chimeric receptor that selectively targets membrane-bound carcinoembryonic antigen (mCEA) in the presence of soluble CEA. Gene Ther 6: 300–304. [DOI] [PubMed] [Google Scholar]
- Carpenito, C, Milone, MC, Hassan, R, Simonet, JC, Lakhal, M, Suhoski, MM et al. (2009). Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci USA 106: 3360–3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill, S, Tasian, SK, Ruella, M, Shestova, O, Li, Y, Porter, DL et al (2014). Efficacy against human acute myeloid leukemia and myeloablation of normal hematopoiesis in a mouse model using chimeric antigen receptor-modified T cells. Blood 123: 2343–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hombach, AA, Heiders, J, Foppe, M, Chmielewski, M and Abken, H (2012). OX40 costimulation by a chimeric antigen receptor abrogates CD28 and IL-2 induced IL-10 secretion by redirected CD4(+) T cells. Oncoimmunology 1: 458–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, DG, Ye, Q, Carpenito, C, Poussin, M, Wang, LP, Ji, C et al. (2011). In vivo persistence, tumor localization, and antitumor activity of CAR-engineered T cells is enhanced by costimulatory signaling through CD137 (4-1BB). Cancer Res 71: 4617–4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedke, J and Stenzl, A (2013). Immunotherapeutic strategies for the treatment of renal cell carcinoma: where are we now? Expert Rev Anticancer Ther 13: 1399–1408. [DOI] [PubMed] [Google Scholar]
- Bailey, A and McDermott, DF (2013). Immune checkpoint inhibitors as novel targets for renal cell carcinoma therapeutics. Cancer J 19: 348–352. [DOI] [PubMed] [Google Scholar]
- Maher, J, Brentjens, RJ, Gunset, G, Rivière, I and Sadelain, M (2002). Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat Biotechnol 20: 70–75. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.