

Abstract
Oncolytic adenoviruses (Onc.Ads) produce significant antitumor effects but as single agents they rarely eliminate tumors. Investigators have therefore incorporated sequences into these vectors that encode immunomodulatory molecules to enhance antitumor immunity. Successful implementation of this strategy requires multiple tumor immune inhibitory mechanisms to be overcome, and insertion of the corresponding multiple functional genes reduces the titer and replication of Onc.Ads, compromising their direct ant-tumor effects. By contrast, helper-dependent (HD) Ads are devoid of viral coding sequences, allowing inclusion of multiple transgenes. HDAds, however, lack replicative capacity. Since HDAds encode the adenoviral packaging signal, we hypothesized that the coadministration of Onc.Ad with HDAd would allow to be amplified and packaged during replication of Onc.Ad in transduced cancer cells. This combination could provide immunostimulation without losing oncolytic activity. We now show that coinfection of Onc.Ad with HDAd subsequently replicates HDAd vector DNA in trans in human cancer cell lines in vitro and in vivo, amplifying the transgenes the HDAd encode. This combinatorial treatment significantly suppresses the tumor growth compared to treatment with a single agent in an immunocompetent mouse model. Hence, combinatorial treatment of Onc.Ad with HDAd should overcome the inherent limitations of each agent and provide a highly immunogenic oncolytic therapy.
Introduction
Oncolytic adenoviruses (Onc.Ads) can selectively replicate in cancer cells1,2, and have demonstrated safety as well as therapeutic responses in some patients with malignant diseases.3 However, oncolytic antitumor activity alone rarely eliminates patient tumors, and investigators have therefore incorporated sequences encoding “immunomodulatory molecules,” such as proinflammatory cytokines, into OncAds.2 This strategy has successfully enhanced antitumor immunity in host animals as well as in patients with malignancies.4,5 Importantly, local administration of an Onc.Ad expressing an immunomodulatory gene (“armed” Onc.Ad) can induce an adaptive immune response to cancer cells, that can eliminate uninfected tumor cells both locally and in distant metastases1, and several “armed” Onc.Ads have been developed for clinical use to augment the antitumor responses seen with unmodified Onc.Ad.2 Although recruitment of the host immune system can usefully complement the direct oncolytic activity of adenoviruses, multiple tumor immune inhibitory mechanisms must be overcome if antitumor immunity is to reach its full potential.6 Therefore, multiple immunomodulatory molecules will likely need to be encoded by a single Onc.Ad, a requirement that compromises the titer and oncolytic activity of Onc.Ad, because of the limited capacity (circa 2 kb) of Onc.Ad packaging.7
Helper-dependent adenoviral vectors (HDAds) are devoid of all viral-coding sequences, affording them a large transgene coding capacity (up to 32 kb) that is suited to insertion of multiple transgenes in a single vector. Although advances in HDAd production techniques have significantly simplified manufacture, and made this class of agents a realistic consideration for clinical development8,9, their lack of in vivo replicative capacity means that they are of limited value as single agents for the treatment of human tumors. Since HDAd vector DNA contains only the cis-acting Ad sequences necessary for vector DNA replication (inverted terminal repeats, ITRs) and packaging (Φ), a helper virus is required for their propagation.10 We hypothesized that the coadministration of HDAd with Onc.Ad expressing functional viral genes should allow HDAd to be amplified within cancer cells during replication of Onc.Ad, leading to multiple cycles of production and release of both the oncolysis and the HDAd-derived immunogenic components (Figure 1). In other words, the combination of Onc.Ad with HDAd could be complementary, and overcome the inherent limitations of each agent alone—the small coding capacity of Onc.Ad and the lack of a replicative/lytic effect of HDAd.
Figure 1.
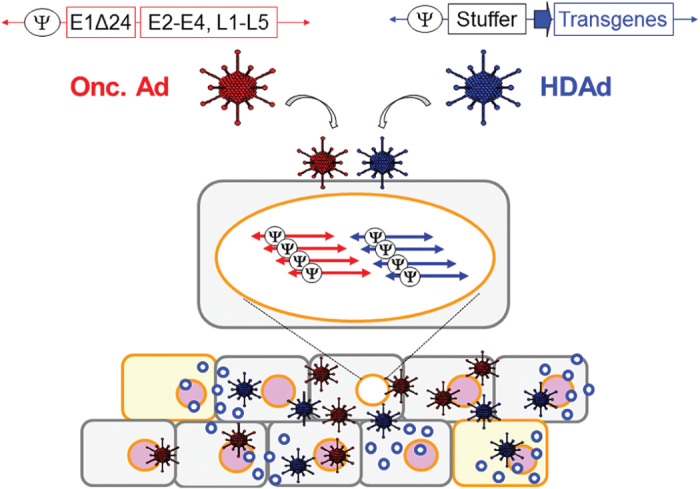
Concept of combinatorial treatment of Onc.Ad with HDAd. Replication machinery of Onc.Ad recognizes coinfected HDAd vector DNA in transduced cancer cells and replicates HDAd vector DNA in trans, leading to multiple cycles of production and release of both the oncolysis and the transgenes encoded in an HDAd. These vectors are packaged into newly synthesized Ad particles and distribute to surrounding cells including tumor stroma. Transgenes (blue) encoded in HDAd are expressed in transduced cells.
Here, we demonstrate that coinfection of Onc.Ad with HDAd can indeed amplify the transgene products (including reporter and cytokine genes) encoded in HDAd in human cancer cell lines in vitro and in vivo and that such combination therapy enhances the therapeutic effects compared to treatment with HDAd or Onc.Ad alone in an immunocompetent mouse model.
Results
Cotransduction of Onc.Ad and HDAd amplifies transgene encoded in the HDAd in human prostate cancer cells
To follow the amplification of both HDAd and Onc.Ad in transduced human cancer cells, we constructed HDAd and Onc.Ad encoding EGFP and RFP respectively and confirmed the expression of each transgene in A549 cells (Figure 2a). To test whether coinfection of Onc.Ad with HDAd could sequentially amplify HDAd in transduced human cancer cells, we coinfected HDAdEGFP and Onc.AdRFP into human cancer cells (primary infection) and then harvested the cell lysate and added it to untreated cancer cells (secondary infection). This lysate was in turn added to a third set of cancer cells (tertiary infection) (Figure 2b). To determine whether the ratio of OncAd:HDAd affected the amplification of each component, we initially infected the human prostate cancer cell line DU-145 with 20 viral particles (Vp) per cell at ratios of Onc.Ad to HDAd of 0:10, 3:10, 7:10, 10:10, 10:7, 10:3, and 10:0 (Figure 2c). The cells were analyzed 48 hours after each round of infection. As anticipated, cells infected with Onc.Ad or HDAd alone (10:0 or 0:10) during the primary infection showed only RFP or EGFP signal respectively. Cells coinfected with the Onc.Ad/HDAd mix had a signal derived from both vectors and also had a higher proportion of (HDAd-derived) EGFP positive populations, including the EGFP and RFP double positive population, compared to cells initially infected with HDAd alone, suggesting that the Onc.Ad replication machinery amplifies the HDAd within transduced cancer cells. Moreover, since HDAd lacks replicative ability, there were no EGFP positive cells after secondary and tertiary rounds of infection in the group treated with lysate from cells infected with HDAd alone. By contrast, cells treated with lysate from cells infected with Onc.Ad/HDAd mixes showed both EGFP and RFP positive populations after secondary and tertiary rounds of infection, suggesting that the Onc.Ad replication machinery acts in trans to produce both Onc.Ad and freshly amplified HDAd within infected cancer cells, resulting in multiple cycles of production of both Ads.
Figure 2.

Coinfection of human prostate cancer cells with Onc.Ad and HDAd can continuously amplify the transgenes encoded in the HDAd. (a) Schematic structure of adenoviral vectors. Onc.Ad5Δ24RFP encodes a monomeric RFP transgene in the E3 region. HDAd5EGFP encodes an EGFP expression cassette. EGFP is driven by the CMV promoter. (b) Schematic flow of in vitro coinfection experiment. Onc.Ad is mixed with HDAd at multiple ratios to a total 20 Vp/cell and used to infect human cancer cell lines (first infection). Cells were harvested at 48 hours after infection and subjected to analysis (e.g., Flow cytometry). After the first infection, a portion of the cell lysates (1/10) were added to untreated cells after three freeze-thaw cycles (representing the second infection). Forty-eight hours after this second infection, the cells were harvested and subjected to analysis. After lysis by three freeze-thaw cycles, a portion of the lysate (1/10) was added to untreated cells (third infection). Forty-eight hours after the third round of infection, the target cells were analyzed. (c) Human prostate cancer cell line, DU-145 was infected with a total 20 Vp/cell of Onc.Ad/HDAd mix at different ratios of Onc.Ad to HDAd = 0:10, 3:10, 7:10, 10:10, 10:7, 10:3, and 10:0. Cells were harvested at 48 hours after infection, and analyzed by flow cytometry to determine the populations of RFP (Onc.Ad) and EGFP (HDAd) positive cells. The EGFP single, RFP single or EGFP-RFP double positive populations were plotted separately. The experiment was repeated triplicate with similar results.
Coinfection with Onc.Ad and HDAd continuously amplifies HDAd vector DNA in human cancer cell lines
We next verified the continuous amplification of the HDAd transgene by quantifying HDAd and Onc.Ad vector copies using primer sets for each backbone in DU-145 cells infected with each vector alone or with the vector combination at a ratio of Onc.Ad to HDAd of 3:10 (the optimum ratio for HD production from Figure 2c). Figure 3a shows that cells infected with the Onc.Ad/HDAd mix had a 1,000-fold higher level of HDAd vector copies compared to cells infected with HDAd alone during the primary infection, confirming that the Onc.Ad replication machinery recognizes coinfected HDAd vector DNA and replicates it in trans. On secondary and tertiary infection, HDAd vector copies declined to basal level in cells treated with lysate from the cells infected with HDAd alone, while fresh DU-145 cells treated with lysates from cells infected with the Onc.Ad/HDAd mix, had a sustained level of HDAd vector copies through all rounds of infection. Hence, the Onc.Ad replication machinery both replicates HDAd vector DNA and produces new Ads containing HDAd vector DNA. These results suggest that DNA binding proteins (e.g., protein VII) produced from coinfected Onc.Ad bind to both Onc.Ad DNA and HDAd vector DNA and protect HDAd vector DNA from activating the DNA damage response in transduced cells.11
Figure 3.

During Onc.Ad replication, there is amplification-in-trans of coinfected HDAd vector DNA in human cancer cell lines. The human prostate cancer cell line DU-145 was infected with a total 20 Vp/cell of HDAd alone, Onc.Ad alone or with an Onc.Ad/HDAd mix (Onc.Ad to HDAd; 3:10). DNA samples were extracted 48 hours after infection at each infection, and Onc.Ad and HDAd vector copy numbers were measured by quantitative PCR using primer sets for vector backbone (a). Data was normalized with human genomic GAPDH. Data are presented as means ± SD (n = 6). *P < 0.001, **P < 0.03. (b) Human small cell lung carcinoma cell line A549 was infected with a total of 20 Vp/cell of HDAd alone, Onc.Ad alone or with an Onc.Ad/HDAd mix (Onc.Ad to HDAd; 3:10). Cells were harvested 48 hours after infection at each infection, and populations of EGFP (HDAd) and RFP (Onc Ad) positive cells were analyzed via Flow cytometry. The EGFP single and EGFP-RFP double positive populations were plotted as EGFP positive population. The RFP single and EGFP-RFP double positive populations were plotted as RFP positive population. The experiment was repeated triplicate with similar results. Total DNA samples were extracted from A549 cells 48 hours after infection at each infection, and Onc.Ad and HDAd vector copy numbers were measured by quantitative PCR using primer sets for each vector backbone (c). Data was normalized with human genomic GAPDH. Data are presented as means ± SD (n = 6). *P < 0.03, **P < 0.05, ***P < 0.001.
To test whether the Onc.Ad replication machinery can replicate and package HDAds in other human cancer cell lines, the small cell lung carcinoma cell line A549 was infected with HDAd, Onc.Ad or an Onc.Ad/HDAd mix (ratio of Onc.Ad to HDAd = 3:10), and transgene expression at each infection was evaluated (Figure 3b). Cells infected with an Onc.Ad/HDAd mix showed the same continuous EGFP expression (HDAd transgene) throughout the sequential infection that we observed in DU-145. We also quantified the vector copies of each Ad in these A549 cells, measuring both vector copies after infection with HDAd, Onc.Ad, or an Onc.Ad/HDAd mix. Figure 3c shows the Onc.Ad/HDAd combination has similar effects as in DU-145, and these results could also be replicated with a human prostate cancer cell line LNCaP and a human hepatocellular carcinoma cell line HepG2 (Supplementary Figure S1). Hence, Onc.Ad dependent HDAd amplification appears to be a generalizable property for cancer cell lines.
Coinfection of Onc.Ad with HDAd can amplify HDAd in vivo
We next looked for evidence of Onc.Ad mediated amplification of HDAd in vivo. Nude mice were subcutaneously transplanted with A549 cells, and the tumor was injected with Onc.Ad and HDAd encoding RFP and luciferase respectively (Onc.AdRFP and HDAdLuc) (Supplementary Figure S2). Preliminary experiments showed that the optimum ratio of OncAd:HDAd for maximum replication of HDAd was lower in vivo than in vitro, so all experiments with xenograft models were subsequently conducted at a ratio of 1:20 OncAd to HDAd. To analyze the effects of the combination therapy on viral replication, we injected the A549 tumors when they reached approximately 100 mm3. Each tumor received a total of 1 × 108 Vp of Onc.AdRFP, HDAdLuc, or an Onc.AdRFP/HDAdLuc mix (ratio of Onc.Ad to HDAd = 1:20), and we monitored expression of each reporter gene (Figure 4a). Mice injected with an Onc.Ad/HDAd mix initially had significantly higher Luc activity in tumors than mice injected with HDAdLuc alone, suggesting that Onc.Ad replication machinery can amplify coinjected HDAd in vivo as it does in vitro. From day 12 onwards, however, the Luc activity of tumors injected with an Onc.AdRFP/HDAdLuc mix decreased to the same level as tumors injected with HDAdLuc alone. This reduction in Luc signal was not caused by the progressive loss of HDAdLuc sequences in tumors, as even on day 15, mice injected with the Onc.Ad/HDAd mix had a >1,000-fold higher level of HDAdLuc vector copies than mice treated with HDAdLuc alone (Figure 4b). Moreover, the HDAdLuc remained infectious. When A549 cells were treated in vitro with the tumor lysates from each treatment group, they showed a high level of both RFP (Onc.Ad derived) and Luc (HDAd derived) positivities by 24 hours after exposure. (Figure 4c). Hence, the loss of luciferase expression from Onc.Ad/HDAd injected tumors in vivo likely indicates inherent restrictions that prevent the continuous broadening of the biodistribution of Ads within tumor masses that is attributable to extensive necrosis and disruption of tumor vasculature by the oncolytic process.12,13
Figure 4.

Coinfection of Onc.Ad amplified HDAd vector DNA and transgene product following intratumoral injection. (a) A549 cells were transplanted subcutaneously into the right flanks of nude mice, and a total of 1 × 108 Vp of Onc.Ad alone, HDAd alone or an Onc.Ad/HDAd mix (Onc.Ad to HDAd; 1:20) was injected intratumorally. Luciferase (HDAd) and RFP (Onc Ad) activity in tumors were measured at 0, 2, 4, 6, 8, 11, and 14 days after injection. Data are presented as means ± SD (n = 6). The experiment was repeated twice with similar results. *P < 0.001, **P < 0.004, ***P < 0.01, ****P < 0.002, *****P < 0.03. (b) Tumors were collected and harvested at 15 days after injection. DNA was extracted from each tumor sample, and the copy number of each vector determined by quantitative PCR using primer sets for each vector backbone. Data was normalized with human genomic GAPDH. Data are presented as means ± SD (n = 6). *P < 0.001, **P < 0.03, ***P < 0.01. (c) Homogenized tumor samples were lysed via 3 freeze-thaw cycles, and a portion of the tumor lysate was added to untreated A549 cells in vitro. RFP fluorescence (Onc Ad) was captured at 24 hours after treatment, and the population of RFP positive cells were analyzed by Flow cytometry. The luciferase activity (HDAd) of cells treated with tumor lysates was also measured at 24 hours after treatment. Data are presented as means ± SD (n = 6). *P < 0.003, **P < 0.001.
We repeated this in vivo tumor injection study, substituting DU-154 tumors, and obtained similar results (Supplementary Figure S3), so that the Onc.Ad replication machinery can also amplify the HDAd vector DNA and transgene product in a second in vivo tumor model.
Coinfection of Onc.Ad with HDAd increases GM-CSF and IL-12p70 expression from a single HDAd vector in vitro and in vivo
To discover whether combinatorial treatment of an Onc.Ad with an HDAd can amplify multiple transgenes encoded in a single HDAd, we constructed an HDAd encoding human GM-CSF and human IL-12p70 in a single vector (HDAdcyt) (Figure 5a). IL-12 and GM-CSF activate different, but complementary, arms of the immune response14, and preclinical studies have shown that an Ad expressing both GM-CSF and IL-12p70 produced synergistic antitumor effects compared to Ad encoding a single cytokine alone.14,15
Figure 5.
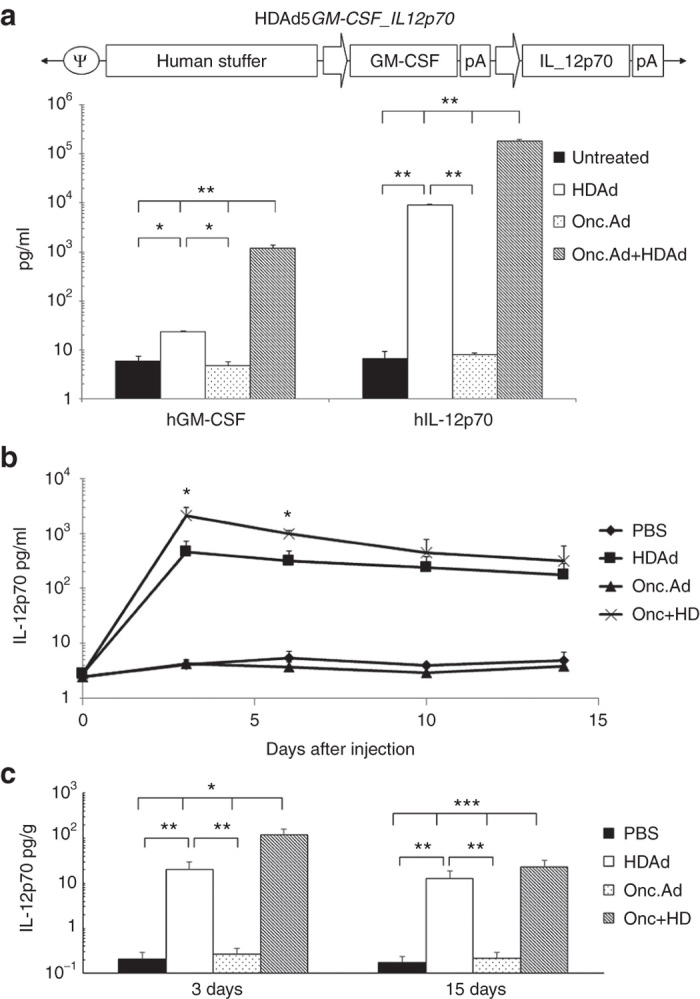
Cytokines encoded in HDAd were amplified by coinfection of Onc.Ad in A549 cells and in A549 tumors. (a) HDAd encoding human GM-CSF and human IL-12p70 expression cassettes were constructed. GM-CSF is driven by hamster GRP78 ubiquitous promoter, and IL-12p70 is driven by human GRP94 ubiquitous promoter (Invivogen, San Diego, CA). A549 cells were infected with a total of 20 Vp/cell of HDAd alone, Onc.Ad alone or with an Onc.Ad/HDAd mix (3:10). Medium was collected 48 hours after infection, and human GM-CSF and IL-12p70 levels were measured. Data are presented as means ± SD (n = 4). *P < 0.01, **P < 0.001. The experiment was repeated twice with similar results. (b) A549 cells were transplanted subcutaneously in the right flank of nude mice, and a total of 1 × 108 Vp of Onc.Ad alone, HDAd alone or an Onc.Ad/HDAd mix (1:20) was injected intratumorally. Blood samples were collected at 0, 3, 7, 10, and 14 days after injection, and human IL-12p70 in serum samples was measured. Data is presented as means ± SD (n = 5). *P < 0.001. (c) Tumors were collected at 3 and 15 days after injection, and human IL-12p70 level in each tumor lysate were measured. Human IL-12p70 level in each tumor lysate was calculated per gram (g) of total protein in lysate and plotted. Data are presented as means ± SD (n = 5). *P < 0.01, **P < 0.009, ***P < 0.03.
A549 cells were infected with 20 Vp/cell of Onc.Ad, or HDAdcyt, or with an Onc.Ad/HDAdcyt mix (ratio of Onc.Ad to HDAd = 3:10); the levels of each cytokine in the medium were measured 48 hours after infection (Figure 5a). Cells infected with an Onc.Ad/HDAdcyt mix had 100-fold higher expression of each cytokine in media compared to cells infected with HDAdcyt alone, showing that that Onc.Ad dependent HDAd amplification is not impaired by the transgene sequence or an increased number of expression cassettes in the HDAdcyt vector DNA. Similar results were obtained in DU-145 cells (data not shown). To detect HDAdcyt amplification in vivo, 1 × 108 Vp of Onc.Ad, HDAdcyt or an Onc.Ad/ HDAdcyt mix (Onc.Ad:HDAd of 1:20) were injected intratumorally into nude mice bearing an A549 tumor. Blood samples were collected at multiple times after injection, and serum IL-12p70 levels were measured (Figure 5b).
Mice receiving the Onc.Ad/HDAdcyt mix initially showed significantly higher IL-12p70 levels in serum compared to mice injected with HDAdcyt alone. By 10 days after injection, IL12 levels were essentially identical in the Onc.Ad/HDAdcyt mix and HDAd alone groups, indicating the same termination of Ad spread as observed with the marker studies (Figure 4). We also harvested these tumors on days 3 and 15 after injection from a subset of the mice and measured the IL-12p70 levels in tumors (Figure 5c). At 3 and 15 days after injection, mice treated with an Onc.Ad/ HDAdcyt mix respectively had ten- and twofold higher IL-12p70 levels in tumors compared to those injected with HDAd alone. Similar results were obtained when we measured human GM-CSF levels in the tumors (Supplementary Figure S4).
Onc.Ad replication machinery can amplify HDAd in the mouse breast cancer cell line 4T1 and enhanced antitumor activity in a syngeneic mouse model
Because the nude mouse model cannot be used to evaluate the immunostimulatory effects of the secreted cytokines, we used an immunocompetent mouse bearing the murine breast cancer cell line 4T1 which has been reported to replicate human Onc.Ad.16,17 We first tested whether Onc.Ad can replicate HDAd in murine 4T1 by coinfecting the line with HDAdEGFP and Onc.Ad. EGFP positivity was observed at 72 hours after infection, and cells coinfected with the Onc.Ad/HDAd mix had stronger EGFP signal compared to cells infected with HDAd alone (Figure 6a). We confirmed that the stronger signal was due to increased HDAd replication by quantifying vector copy numbers; cells infected with the Onc.Ad/HDAd mix had a tenfold higher HDAd vector copies than cells infected with HDAd alone (Figure 6a). Although Onc.Ad replication machinery can amplify HDAd vector DNA in 4T1 cells, the replication efficiency of HDAd in 4T1 cells was lower than that in human cancer cells. This could be dependent on species-specific Ad replication machinery18, so that mouse cells may lack (or poorly express) the functional proteins required for human Ad replication.19
Figure 6.

Human Onc.Ad can amplify coinfected HDAds in mouse breast cancer cell line 4T1 and enhance antitumor activity in a syngeneic mouse model. (a) 4T1 cells were infected with a total of 100 Vp/cell of HDAdEGFP alone, Onc.Ad alone or an Onc.Ad/HDAd mix (Onc.Ad to HDAd; 5:10). EGFP fluorescent images were captured at 72 hours after infection, and total DNA was extracted. Onc.Ad and HDAd vector copy numbers were quantified by PCR using primer sets for each vector backbone. Data was normalized with mouse genomic GAPDH. Data are presented as means ± SD (n = 4). *P < 0.03, **P < 0.05, ***P < 0.001. (b) HDAd encoding mouse GM-CSF and mouse IL-12p70 expression cassettes were constructed. Mouse GM-CSF is driven by hamster GRP78 ubiquitous promoter, and mouse IL-12p70 is driven by human GRP94 ubiquitous promoter (Invivogen, San Diego, CA). 4T1 cells were infected with a total of 100 Vp/cell of HDAd alone, Onc.Ad alone or an Onc.Ad/HDAd mix (5:10). Medium was collected at 72 hours after infection, and mouse GM-CSF and IL-12p70 levels were measured. Data are presented as means ± SD (n = 4). *P < 0.009, **P < 0.001. The experiment was repeated with similar results. (c) Mouse breast cancer cells 4T1 were transplanted into the right flank with 1 × 106 cells to a WT Balb/c mouse. After the tumors reached about 100 mm3, 3 × 108 Vp/tumor of Onc.Ad alone, HDAd alone or an Onc.Ad/HDAd mix (3:10) were intratumorally injected into the right flank tumors on days 0, 2, and 4 (total dose; 1 × 109 Vp). Tumor volumes were measured at different time points. Data are presented as means ± SD (n = 10). *P < 0.001. The experiment was repeated twice with similar results. (d) Kaplan–Meier survival curve after administration. The end point was established at tumor volume of >1,500 mm3. Data are presented as means ± SD (n = 10). *P < 0.001.
We next constructed an HDAd vector encoding the murine cytokines GM-CSF and IL-12p70 expression cassettes (HDAdmcyt) (Figure 6b). We coinfected this HDAdmcyt with Onc.Ad into 4T1 cells and measured the cytokine levels in the media at 72 hours after infection (Figure 6b). Cells coinfected with an Onc.Ad/HDAdmcyt mix had >50-fold higher levels of each cytokine compared to cells infected with HDAdmcyt alone, corresponding to the results obtained from human cancer cells infected with OncAd/HDAdcyt encoding human cytokines (Figure 5).
We generated a syngeneic mouse model bearing 4T1 tumors. After the tumor reached about 100 mm3, we injected it with a total of 1 × 109 Vp of Onc.Ad, or HDAdmcyt or with an Onc.Ad/HDAdmcyt mix (ratio of Onc.Ad to HDAd = 3:10 (optimal ratio for maximum replication of HDAd in 4T1 tumor)). We measured the change in size of the tumors following injection (Figure 6c), and observed that only mice receiving the Onc.Ad/HDAd mix had a significant reduction in tumor size. Of note, injection of Onc.Ad or HDAd alone had minimal effects on the growth of the injected tumor (Figure 6c). Treatment of Onc.Ad/HDAd mix also prolonged animal survival compared to a single agent (HDAd alone, Onc.Ad alone) (Figure 6d). These results suggest that a combination of oncolysis and immunostimulatory cytokine secretion is necessary for optimum tumor growth suppression and prolongation of survival. We also examined whether Onc.Ad/HDAd mix (combination of oncolysis with cytokines) alters the systemic toxicity of Ad (Supplementary Figure S5). We compared markers of hepatotoxicity (AST, ALT) in wild-type Balb/c mice at different time points. Although mice treated with HDAd alone or Onc.Ad/HDAd mix had slightly higher AST and ALT than control mice at 24 hours after infection, these enzymes declined to basal levels by 72 hours after infection. In contrast, mice treated with Onc.Ad alone had significantly higher ALT and AST compared to other groups at 72 hours after infection, as previously reported.20 Hence, combinatorial treatment (combination of oncolysis with cytokines) is potentially a less toxic and more effective means of delivering Ad-based cancer gene therapy compared to single agent treatments (HDAd alone or Onc.Ad alone).
Discussion
In this study, we have demonstrated that coinfection of Onc.Ad with HDAd will amplify the transgene products (including reporter and cytokine genes) encoded in the HDAd by enabling replication of the HDAd vector DNA in human cancer cell lines. This effect can augment the antitumor effect of each agent alone and thereby reduce the tumor growth in a syngeneic mouse model.
Cotransduction of Onc.Ad and HDAd into human cancer cells produces a 1,000-fold increase in HDAd vector copies compared to HDAd alone (Figure 3). Optimal output of HDAd following coinfection with an Onc.Ad requires a ratio of HDAd:Onc.Ad between 3:1 and 20:1, indicating that Onc.Ad preferentially amplifies the Onc.Ad vector DNA over the HDAd vector DNA in vitro and in vivo. Since the HDAd and Onc.Ad used in this study share the same viral capsid (serotype 5), both Ad should have the same transduction efficiency into the cell and translocation efficiency into the nucleus. It has been suggested that efficient synthesis and packaging of functional Ad viral vectors may require Ad derived sequences to be present in vector DNA, although the precise molecular mechanism has not yet been elucidated.11,21 Certainly, the preferential replication of Ad itself over HDAd was also observed during large-scale production of HDAd, where optimal HDAd production also required a ratio of HDAd:Helper virus of 3:1.8 Irrespective of the mechanism favoring selective replication of Onc.Ad, adjustment of the OncAd:HDAd ratio allows high level HDAd replication and translation of encoded genes, so that Onc.Ad induced amplification of HDAd allows the combination of Onc.Ad and HDAd to have higher activity than HDAd alone, which may obviate the need to administer extremely high doses of HDAd as a single agent. However, Onc.Ad dependent amplification (overexpression) of secreted immunostimulatory molecules (e.g., IL-12p70) encoded in HDAd vector DNA may induce toxicity as shown in clinical trials of recombinant IL-12.22 This issue could be addressed by optimizing the ratio of Onc.Ad to HDAd to reduce the circulating immunostimulatory molecules in the blood of patients.
Several clinical trials with Onc.Ads have shown the benefits of combining replicative oncolysis with GM-CSF (CG0070, CGTG-102) mediated stimulation of granulocyte and monocyte induced inflammation and adaptive immunity against tumor antigens.23 Importantly, local administration of such an Onc.Ad can induce an adaptive immune response to cancer cells, thereby eliminating uninfected tumor cells both locally and in distant metastases in animal models and in patients with malignant disease.4 Although recruitment of the host immune system can usefully complement the direct oncolytic activity of Onc.Ads, the range and complexity of tumor immune escape mechanisms6 means that multiple immunomodulatory molecules will likely be required to optimally enhance the development of antitumor immunity following administration of Onc.Ad. IL-12 and GM-CSF activate different, but complementary, arms of the immune response14, and preclinical studies have shown that immunocompetent mice injected with an Ad expressing both GM-CSF and IL-12p70 effectively induced the infiltration of immune cells, leading to a more potent adaptive antitumor immune response than Ad encoding a single cytokine.14,15 We found that both GM-CSF and IL-12 were highly expressed in the tumors of mice receiving the combination of HDAdcyt and Onc.Ad combinatorial treatment (Figure 5, Supplementary Figure S4), and that there was also a rise in serum IL12 levels. Production of these cytokines from the injected HDAd significantly suppressed the long-term tumor growth and prolonged animal survival in a syngeneic mouse model (Figure 6c,d). The development of this antitumor immunity may also be promoted by Onc.Ad dependent oncolysis, since this will expose tumor-associated antigens to cross presentation to the GM-CSF and IL-12 activated host immune system.6,14 Certainly the combination of first-generation Ad (FGAd) expressing thymidine kinase (TK) driven by a tumor specific promoter with FGAd expressing fms-like tyrosine kinase-3 ligand (Flt3L: a potent dendritic cell growth factor) established immunological memory to cancer cells after administration of ganciclovir more effectively than treatment with a FGAd encoding a single transgene (TK or Flt3L) alone and led to long term survival of immunocompetent tumor bearing mice and rats.24
In conclusion, combinatorial treatment of Onc.Ad with HDAd led to multiple cycles of production and release of both the oncolysis and the immunogenic components encoded in an HDAd, resulting in augmentation of the antitumor effect of each agent alone in a syngeneic mouse model. Our data suggests that combinatorial treatment of Onc.Ad with HDAd can potentiate to provide a highly replicative, highly immunogenic oncolytic adenoviral therapy for cancer.
Materials and Methods
Viral vectors (HDAds and Onc.Ads)
HDAd HDΔ28E4Luc, and HDAd HDΔ28E4EGFP constructs containing the Firefly luciferase or EGFP transgene driven by the CMV promoter (HDAdLuc, HDAdEGFP) were produced as described elsewhere.25 HDAd-hGM-CSF_hIL-12p70 and HDAd-mGM-CSF_mIL-12p70 were generated and amplified using standard HDAd preparation techniques.8 The human/murine GM-CSF complementary DNA and human/murine IL-12p70 complementary DNA (Invivogen, San Diego, CA) were inserted into pVIVO1 containing 2 different mammalian ubiquitous promoters with polyA signal sequences (Invivogen). After confirmation of sequence, these expression cassettes were inserted into pHDΔ25E4 vector described elsewhere.26 HDΔ25E4hGM-CSF_hIL-12p70 and HDΔ25E4mGM-CSF_mIL-12p70 virus genome were transfected to 116 cells for amplification and rescuing.8
Onc.Ad5Δ24RFP was generated and amplified using standard adenovirus preparation techniques.4,27 In order to insert RFP gene into E3 region, the monomeric RFP complementary DNA was inserted into pTHSN.4,28 pAd5Δ24RFP was generated by homologous recombination in E. coli between linearized pTHSNRFP and linearized pAd5Δ24. Ad5Δ24RFP virus genome was transfected to A549 cells for amplification and rescue.4
Cell lines and adenovirus infection
Human prostate cancer cell lines DU-145, LNCaP, human squamous carcinoma cell line A549, human hepatocellular carcinoma cell line HepG2, and mouse breast cancer cell line 4T1 were obtained from ATCC (Manassas, VA). Cells were cultured under recommended conditions. Human cancer cells were infected with 20 viral particles (Vp)/cell of Ads described in Results section and harvested at 48 hours after infection. Mouse cancer cells were infected with 100 Vp/cell of Ads described in Results section and harvested at 72 hours after infection. A portion of cells and/or media was submitted for analysis as described below. Remaining cell lysates were subjected to Freeze-Thaw cycles 3 times, and 1/10th volume of cell lysates were submitted for a second infection after centrifugation at 1,500 rpm for 5 minutes (Figure 1b).
Animals and adenovirus injection
NU/J mice and Balb/c mice were purchased from Jackson laboratories (Bar Harbor, ME). All of the mice were housed under pathogen-free conditions; food and water were provided ad libitum. All experimental procedures were conducted in accordance with institutional guidelines for animal care and use.
After counting the harvested cancer cells, one million human cancer cells (DU-145, A549) were resuspended in a volume of 100 µl of PBS and subcutaneously injected at 5–6 weeks of age into NU/J male mice. After the tumor size reached 100 mm3, a total of 1 × 108 Vp of Ads described in Results section were intratumorally injected in a volume of 20 μl. One million (right flank) 4T1 cells were subcutaneously injected at 5–6 weeks of age into wild-type Balb/c female mice (Figure 6a). After the tumor size reached approximately 100 mm3, a total of 1 × 109 Vp of Ads described in Results section were intratumorally injected in a volume of 20 μl. Tumor size was followed and volumes were calculated using the formula: width2 × length × 0.5.29
Flow cytometry
Cells were infected with Ads in Results section and harvested at 48 hours after infection (Figure 1b). Fluorescent gene expression (EGFP and RFP) in Ad infected cancer cells was analyzed using a Gallios flow cytometer with Kaluza software (BD, Franklin Lakes, NJ) according to manufacturer’s instructions.
Cytokine analysis
Human cancer cells were infected with 20 Vp/cell of Ads described in Results section, and media were collected at 48 hours after infection. Media samples were frozen and stored at −80 °C until analysis. After intratumoral injection of a total of 1 × 108 Vp of Ads described in Results section, blood was collected retro-orbitally at different time points, and serum samples were frozen and stored at −80 °C until analysis. Tumors were harvested at 3 and 15 days after injection and washed with PBS. Tumors were homogenized with micropestles (VWR, Radnor, PA) at 4 °C, and supernatants of tumor lysates were isolated via centrifugation at 2,000 rpm for 5 minutes. Human GM-CSF and IL-12p70 in media, sera, and tumor lysates were assayed by using the BD cytokine multiplex bead array system (BD Biosciences, San Jose, CA), and analyzed using a BD Facs Array instrument (BD Biosciences) according to manufacturer’s instructions.30,31 Total protein concentration of each tumor lysate was measured by using Micro BCA protein assay kit (Thermo Scientific, Rockford, IL), and human GM-CSF and human IL-12p70 levels in tumor lysates were calculated per gram of total protein concentration. Murine GM-CSF and IL-12p70 in media of 4T1 cell cultures were measured by a similar manner.
Luciferase analysis in vitro
A549 cells were treated with lysates from tumors injected with Ads described in Results section. Cells were washed with PBS and harvested with reporter lysis buffer (Promega, Madison, WI) at 24 hours after treatment. Luciferase activity of each sample was measured with Luciferase assay system (Promega).
In vivo imaging (Luciferase and RFP)
Tumors were injected with a total of 1 × 108 Vp of Ads described in Results section, and expression of reporter genes (RFP and Luciferase) was captured at time points indicated in Figures 23456. RFP activity in tumors of mice was captured using the IVIS imaging system series 100, using 535 nm excitation with a DsRed emission filter (Xenogen, Alameda, CA). For measuring luciferase activity in tumors, 1.5 mg of D-Luciferin (GoldBio Technology, St Louis, MO) was intraperitoneally injected to each group of mice. After 10 minutes, images were captured with the IVIS imaging system. Photon emission values of RFP and Luciferase in tumors were calculated using Living Image v4.3 software (Xenogen).
Quantification of vector genome DNA in Ad infected cells and in Ad injected tumors
Cells were infected with 20 Vp/cell of Ads and harvested at 48 hours after infection. Tumors were injected with a total of 1 × 108 Vp of Ads and harvested at time points described in Results section. Total DNA was extracted from infected cells or tumors using a DNeasy Blood and Tissue Kit (QIAGEN, Valencia, CA).32 DNA samples were analyzed by quantitative real-time PCR (10 minutes at 95 °C and then 45 cycles of 10 seconds at 95 °C, 15 seconds at 55 °C, and 30 seconds at 72 °C) using a Bio-Rad iQ5 Real-time PCR detection system (Bio-Rad, Hercules, CA), and Applied Biosystems SYBR green PCR master mix (Life technologies, Grand Island, NY) with primer sets shown in Supplementary Table S1.
Statistical analysis
Data was analyzed by t-test or one-way ANOVA analysis of variance followed by Shapiro-Wilk’s protected least significant difference test (SigmaPlot).
Acknowledgments
We thank MK Brenner, in the Center for Cell and Gene Therapy at Baylor College of Medicine for his valuable critiques of this manuscript. This work was support by the National Institutes of Health (R00HL098692) to M.S., and Cancer Prevention and Research Institute of Texas (RP101017) to B.L.
Footnotes
H.A. is shareholder in Oncos Therapeutics Ltd, and employee and shareholder in TILT Biotherapeutics Ltd. The other authors declared no conflict of interest.
References
- Cerullo, V, Koski, A, Vähä-Koskela, M and Hemminki, A (2012). Chapter eight–Oncolytic adenoviruses for cancer immunotherapy: data from mice, hamsters, and humans. Adv Cancer Res 115: 265–318. [DOI] [PubMed] [Google Scholar]
- Choi, JW, Lee, JS, Kim, SW and Yun, CO (2012). Evolution of oncolytic adenovirus for cancer treatment. Adv Drug Deliv Rev 64: 720–729. [DOI] [PubMed] [Google Scholar]
- Nokisalmi, P, Pesonen, S, Escutenaire, S, Särkioja, M, Raki, M, Cerullo, V et al. (2010). Oncolytic adenovirus ICOVIR-7 in patients with advanced and refractory solid tumors. Clin Cancer Res 16: 3035–3043. [DOI] [PubMed] [Google Scholar]
- Koski, A, Kangasniemi, L, Escutenaire, S, Pesonen, S, Cerullo, V, Diaconu, I et al. (2010). Treatment of cancer patients with a serotype 5/3 chimeric oncolytic adenovirus expressing GMCSF. Mol Ther 18: 1874–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesonen, S, Diaconu, I, Kangasniemi, L, Ranki, T, Kanerva, A, Pesonen, SK et al. (2012). Oncolytic immunotherapy of advanced solid tumors with a CD40L-expressing replicating adenovirus: assessment of safety and immunologic responses in patients. Cancer Res 72: 1621–1631. [DOI] [PubMed] [Google Scholar]
- Mellman, I, Coukos, G and Dranoff, G (2011). Cancer immunotherapy comes of age. Nature 480: 480–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bett, AJ, Prevec, L and Graham, FL (1993). Packaging capacity and stability of human adenovirus type 5 vectors. J Virol 67: 5911–5921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, M, Cela, R, Clarke, C, Bertin, TK, Mouriño, S and Lee, B (2010). Large-scale production of high-quality helper-dependent adenoviral vectors using adherent cells in cell factories. Hum Gene Ther 21: 120–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer, D and Ng, P (2003). Improved system for helper-dependent adenoviral vector production. Mol Ther 8: 846–852. [DOI] [PubMed] [Google Scholar]
- Palmer, DJ and Ng, P (2005). Helper-dependent adenoviral vectors for gene therapy. Hum Gene Ther 16: 1–16. [DOI] [PubMed] [Google Scholar]
- Giberson, AN, Davidson, AR and Parks, RJ (2012). Chromatin structure of adenovirus DNA throughout infection. Nucleic Acids Res 40: 2369–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gau, CL, Rosenblatt, RA, Cerullo, V, Lay, FD, Dow, AC, Livesay, J et al. (2009). Short-term correction of arginase deficiency in a neonatal murine model with a helper-dependent adenoviral vector. Mol Ther 17: 1155–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerullo, V, McCormack, W, Seiler, M, Mane, V, Cela, R, Clarke, C et al. (2007). Antigen-specific tolerance of human alpha1-antitrypsin induced by helper-dependent adenovirus. Hum Gene Ther 18: 1215–1224. [DOI] [PubMed] [Google Scholar]
- Choi, KJ, Zhang, SN, Choi, IK, Kim, JS and Yun, CO (2012). Strengthening of antitumor immune memory and prevention of thymic atrophy mediated by adenovirus expressing IL-12 and GM-CSF. Gene Ther 19: 711–723. [DOI] [PubMed] [Google Scholar]
- Zhang, SN, Choi, IK, Huang, JH, Yoo, JY, Choi, KJ and Yun, CO (2011). Optimizing DC vaccination by combination with oncolytic adenovirus coexpressing IL-12 and GM-CSF. Mol Ther 19: 1558–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z, Hu, Z, Gupta, J, Krimmel, JD, Gerseny, HM, Berg, AF et al. (2012). Intravenous administration of adenoviruses targeting transforming growth factor beta signaling inhibits established bone metastases in 4T1 mouse mammary tumor model in an immunocompetent syngeneic host. Cancer Gene Ther 19: 630–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, W, Zhu, H, Zhang, L, Davis, J, Teraishi, F, Roth, JA et al. (2006). Combination effect of oncolytic adenovirotherapy and TRAIL gene therapy in syngeneic murine breast cancer models. Cancer Gene Ther 13: 82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jogler, C, Hoffmann, D, Theegarten, D, Grunwald, T, Uberla, K and Wildner, O (2006). Replication properties of human adenovirus in vivo and in cultures of primary cells from different animal species. J Virol 80: 3549–3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young, AM, Archibald, KM, Tookman, LA, Pool, A, Dudek, K, Jones, C et al. (2012). Failure of translation of human adenovirus mRNA in murine cancer cells can be partially overcome by L4-100K expression in vitro and in vivo. Mol Ther 20: 1676–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas, JJ, Guedan, S, Searle, PF, Martinez-Quintanilla, J, Gil-Hoyos, R, Alcayaga-Miranda, F et al. (2010). Minimal RB-responsive E1A promoter modification to attain potency, selectivity, and transgene-arming capacity in oncolytic adenoviruses. Mol Ther 18: 1960–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauschhuber, C, Noske, N and Ehrhardt, A (2012). New insights into stability of recombinant adenovirus vector genomes in mammalian cells. Eur J Cell Biol 91: 2–9. [DOI] [PubMed] [Google Scholar]
- Del Vecchio, M, Bajetta, E, Canova, S, Lotze, MT, Wesa, A, Parmiani, G et al. (2007). Interleukin-12: biological properties and clinical application. Clin Cancer Res 13: 4677–4685. [DOI] [PubMed] [Google Scholar]
- Miest, TS and Cattaneo, R (2014). New viruses for cancer therapy: meeting clinical needs. Nat Rev Microbiol 12: 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assi, H, Candolfi, M, Baker, G, Mineharu, Y, Lowenstein, PR and Castro, MG (2012). Gene therapy for brain tumors: basic developments and clinical implementation. Neurosci Lett 527: 71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guse, K, Suzuki, M, Sule, G, Bertin, TK, Tyynismaa, H, Ahola-Erkkilä, S et al. (2012). Capsid-modified adenoviral vectors for improved muscle-directed gene therapy. Hum Gene Ther 23: 1065–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerullo, V, Seiler, MP, Mane, V, Cela, R, Clarke, C, Kaufman, RJ et al. (2007). Correction of murine hemophilia A and immunological differences of factor VIII variants delivered by helper-dependent adenoviral vectors. Mol Ther 15: 2080–2087. [DOI] [PubMed] [Google Scholar]
- Cerullo, V, Diaconu, I, Romano, V, Hirvinen, M, Ugolini, M, Escutenaire, S et al. (2012). An oncolytic adenovirus enhanced for toll-like receptor 9 stimulation increases antitumor immune responses and tumor clearance. Mol Ther 20: 2076–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaconu, I, Cerullo, V, Hirvinen, ML, Escutenaire, S, Ugolini, M, Pesonen, SK et al. (2012). Immune response is an important aspect of the antitumor effect produced by a CD40L-encoding oncolytic adenovirus. Cancer Res 72: 2327–2338. [DOI] [PubMed] [Google Scholar]
- Guse, K, Diaconu, I, Rajecki, M, Sloniecka, M, Hakkarainen, T, Ristimäki, A et al. (2009). Ad5/3-9HIF-Delta24-VEGFR-1-Ig, an infectivity enhanced, dual-targeted and antiangiogenic oncolytic adenovirus for kidney cancer treatment. Gene Ther 16: 1009–1020. [DOI] [PubMed] [Google Scholar]
- Suzuki, M, Cerullo, V, Bertin, TK, Cela, R, Clarke, C, Guenther, M et al. (2010). MyD88-dependent silencing of transgene expression during the innate and adaptive immune response to helper-dependent adenovirus. Hum Gene Ther 21: 325–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, M, Cela, R, Bertin, TK, Sule, G, Cerullo, V, Rodgers, JR et al. (2011). NOD2 signaling contributes to the innate immune response against helper-dependent adenovirus vectors independently of MyD88 in vivo. Hum Gene Ther 22: 1071–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, M, Bertin, TK, Rogers, GL, Cela, RG, Zolotukhin, I, Palmer, DJ et al. (2013). Differential type I interferon-dependent transgene silencing of helper-dependent adenoviral vs. adeno-associated viral vectors in vivo. Mol Ther 21: 796–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.