

Abstract
Conditionally replicative adenoviruses are promising agents for oncolytic virotherapy. Various approaches have been attempted to retarget adenoviruses to tumor-specific antigens to circumvent deficiency of receptor for adenoviral binding and to provide an additional level of tumor specificity. Functional incorporation of highly specific targeting molecules into the viral capsid can potentially retarget adenoviral infection. However, conventional antibodies are not compatible with the cytoplasmic adenovirus capsid synthesis. The goal of this study was to evaluate the utility of single variable domains derived from heavy chain camelid antibodies for retargeting of adenovirus infection. We have combined transcriptional targeting using a tumor-specific promoter with transductional targeting through viral capsid incorporation of antihuman carcinoembryonic antigen single variable domains. Obtained data demonstrated that employment of a single variable domain genetically incorporated into an adenovirus fiber increased specificity of infection and efficacy of replication of single variable domain-targeted oncolytic adenovirus. The double targeting, both transcriptional through the C-X-C chemokine receptor type 4 promoter and transductional using the single variable domain, is a promising means to improve the therapeutic index for these advanced generation conditionally replicative adenoviruses. A successful strategy to transductional retargeting of oncolytic adenovirus infection has not been shown before and therefore we believe this is the first employment of transductional targeting using single variable domains derived from heavy chain camelid antibodies to enhance specificity of conditionally replicative adenoviruses.
Introduction
A growing body of evidence demonstrates the promise of gene therapy and oncolytic virotherapy in preclinical and clinical studies. Among the oncolytic viruses, adenoviruses were the first and most frequently used vectors in clinical trials.1 Recently, the first oncolytic adenovirus H101 has been approved for the treatment of head and neck carcinoma in combination with chemotherapy.2 In most trials with adenovirus vectors, only mild adverse events were seen and no dose-limiting toxicity was reported. However, the antitumor efficacy of these Ad vectors was only modest, therefore, improvement of the efficacy of the treatment is needed.3 Oncolytic conditionally replicative adenoviruses (CRAds) are novel therapeutic agents for targeted-therapy based on the cytolytic effect of replicating viruses which results in tumor cell death. Replicative specificity of the virus is the basis for the targeted action of CRAds and results in tumor-specific cytotoxicity. This tumor selectivity is established via the use of tumor specific promoters (TSPs) to achieve conditional replication. In this regard, TSPs restrict viral replication by replacing the native viral E1a promoter. The use of TSPs in CRAd designs has therefore represented the principle means to limit viral replication to tumor cells.
Despite controlled replication via the above mentioned TSP approach, off-target infection and replication may provide the basis for limiting toxicity.4 Thus, additional levels of control of virus infection and replication are therefore desirable. Another strategy to achieve CRAd specificity is controlling adenovirus infection at the level of target cell attachment. Transductional targeting is the strategy of altering viral binding to achieve target cell specific binding.5 Importantly, such an approach could potentially synergize with transcriptional targeting.
In this regard, adenoviral tropism is dictated by the interaction of the adenoviral fiber with the native receptor—coxsackievirus and adenovirus receptor (CAR).6 Various approaches have been attempted to retarget adenovirus serotype 5 (Ad5) towards cancer cells, including the use of adaptor molecules and genetic modifications of the adenovirus capsid.7,8 Although the adapter approach has proven the feasibility of retargeting adenovirus vectors, a “single-unit” molecule is preferred for the purpose of therapeutic use. Thus, genetic modification of the fiber potentially provides a way to redirect the adenovirus to an alternative cellular receptor. The goal of this study was to design, develop, and characterize a clinically relevant oncolytic virotherapy agent. A central facet of our approach is to confer the CRAd capacity to specifically infect tumor-associated targets in a CAR-independent manner.
The camelid family heavy-chain-only antibodies possess ideal characteristics for a CRAd retargeting strategy including cytosolic stability to allow functional incorporation into the CRAd capsid as well as compatibility with phage biopanning selection to allow tumor cell specificity.9 In addition, engineered single domain antibody (sdAb) proteins have demonstrated effective targeting in model systems.10,11 Based on these useful attributes, we have endeavored a proof-of-principle study to evaluate the utility of sdAb as a candidate antibody for oncolytic CRAd retargeting. In this study, we have explored the utility of capsid incorporated sdAb against the human carcinoembryonic antigen (hCEA) to achieve targeted CRAd-mediated oncolysis. In addition, in order to confer the CRAd replication specificity to cancer cells, we have employed the promoter element of the C-X-C chemokine receptor type 4 (CXCR4) gene. We evaluated CRAd vectors using a panel of human cancer cells. Obtained data demonstrated that employment of fiber displaying anti-hCEA sdAb increased CRAd infection in hCEA-overexpressed tumor cells. These findings highlight the improved antitumor activities that may accrue with advancements in the design of CRAd agents for oncolytic virotherapy and show the first successful employment of transductional retargeting using sdAb in a CRAd design.
Results
Expression of the hCEA-targeted fiber-fibritin-sdAb protein
For this study, we produced a panel of Ad5 based vectors expressing the E1a gene under transcriptional control of the CXCR4 promoter element, including Ad.CXCR4E1 with wild-type Ad5 fiber, Ad.CXCR4E1.B2 vector with a fiber-fibritin chimera expressing anti-hCEA sdAb (clone B2), as well as replication-deficient recombinant adenoviruses, Ad.CXCR4Luc and Ad.CMVLuc encoding the firefly luciferase (Luc) gene under control of the CXCR4 or human cytomegalovirus (CMV) promoter, respectively. Wild-type Ad5 was used as control virus. A simplified schematic diagram of recombinant adenovirus genomes used in this study are shown in Figure 1a. To confirm the incorporation of the chimeric fiber-fibritin-sdAb protein into Ad.CXCR4E1.B2, boiled and unboiled purified adenovirus vectors were analyzed by western blotting using an antifiber mAb. As expected, the chimeric fiber-fibritin-sdAb in Ad.CXCR4E1.B2 is slightly larger (with predicted molecular weight 67.7 kDa for fiber-fibritin-sdAb monomer) than the native Ad5 (molecular weight of wild-type Ad5 fiber protein is 61.6 kDa) and fiber displayed in Ad.CXCR4E1 and Ad5. Genetic incorporation of sdAbs produced a stable fusion with fiber-fibritin molecules that maintained the trimerization potential of chimeric fiber-fibritin-sdAb proteins under native conditions (Figure 1b).
Figure 1.
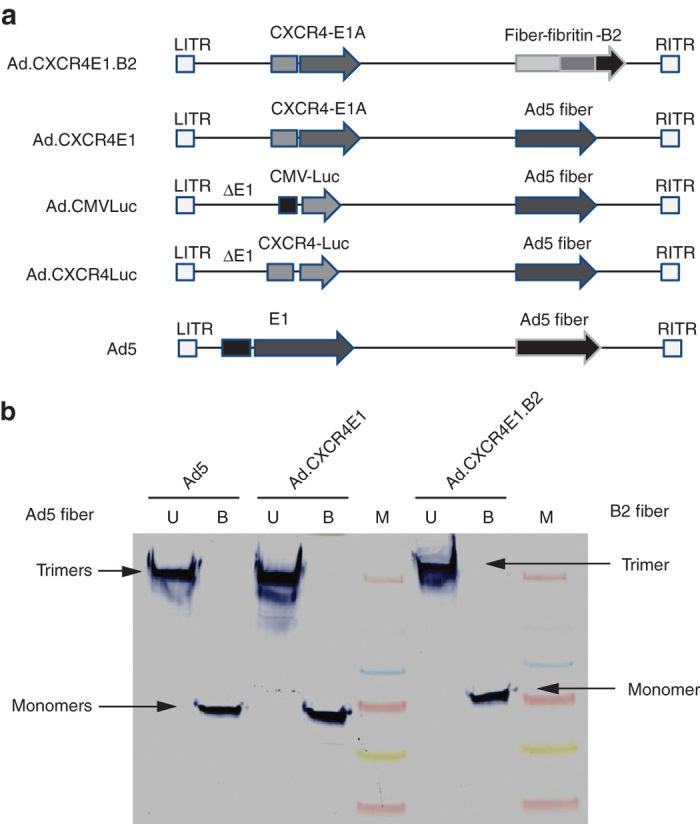
Validation of incorporation of sdAb-targeted chimeric fiber protein in CRAds. (a) Simplified schematic overview of adenovirus vectors used in this study. Only relevant genomic regions are shown. (b) Validation of incorporation of fiber-fibritin-sdAb into the viral capsid using western blot analysis. Equal amounts (5 × 109 vp) of purified viral particles from Ad5, Ad.CXCR4E1 and Ad.CXCR4E1.B2 were loaded in sample buffer in each lane without (lane 1, 3, and 6) or with boiling (lane 2, 4, and 7). Proteins were separated on a SDS-PAGE gel followed by western blot transfer to a PVDF membrane. Fiber protein expression was detected using antifiber mAb. Predicted molecular weight (MW) of wild-type Ad5 fiber monomers is 61.6 kDa and MW 67.7 kDa for fiber-fibritin-sdAb. One representative of three different experiments is shown. B, boiled; LITR, left inverted terminal repeat; M, marker; PVDF, polyvinylidene difluoride; RITR, right inverted terminal repeat; U, unboiled; ΔE1, E1 deleted.
Validation of vector thermostability
Next, we confirmed the structural integrity of the capsid-modified CRAd, as modifications in fiber proteins can affect viral capsid stability. Based on results of preliminary studies that show a temperature-dependent decrease of adenovirus-mediated gene transfer,12 the viruses were preincubated at 45 °C for different times before infection of hCEA(+) LS174T human colorectal adenocarcinoma cells. The relative cytotoxicity was obtained by comparing with the cytotoxicity of untreated CRAds. The oncolytic effect of Ad.CXCR4E1 and Ad.CXCR4E1.B2 was not significantly reduced by thermal treatment when tested up until 30 minutes incubation at 45 °C (data not shown).
Ad.CXCR4E1.B2 demonstrates hCEA-selective binding
To evaluate specificity of Ad.CXCR4E1.B2 transduction, MC38 and MC38CEA murine colon adenocarcinoma cells were used. As expected, FACS analysis showed no hCAR expression in both cell lines and no hCEA expression in the MC38 cells (Table 1 and Supplementary Figure S1a) in contrast to the high levels of hCEA expression in MC38CEA cells (Table 1 and Supplementary Figure S1b). Both cell lines were infected with Ad.CXCR4E1.B2 or Ad.CXCR4E1 for 1 hour, washed, total DNA was extracted and subjected to quantitative real-time PCR (qPCR) analysis. Cell binding by Ad.CXCR4E1.B2 was strongly enhanced in the hCEA expressing MC38CEA cells compared to the control vector, while both CRAds had limited binding to the hCEA(−)/hCAR(−) MC38 cell line. As shown in Figure 2, Ad.CXCR4E1.B2 binding to hCEA(+) MC38CEA cells was significantly higher (about 25-fold; P < 0.01) compared to binding to the hCEA(−) MC38 cells. In contrast, Ad.CXCR4E1 with wild-type Ad5 fiber demonstrated negligible change in binding to MC38CEA cells in comparison with MC38 cells. Also, MC38 cell binding by Ad.CXCR4E1.B2 was slightly higher (about twofold) compared to Ad.CXCR4E1, probably due to structural difference of wild-type Ad5 fiber and fiber-fibritin fusion proteins. Thus, Ad.CXCR4E1.B2 demonstrates hCEA-specific cell binding validating that specificity of the B2 sdAb is maintained in the CRAd context.
Table 1. Flow cytometry analysis of hCEA and hCAR surface expression.
| Cell line | % of positive cells (mean of fluorescence intensity)a |
|
|---|---|---|
| hCEA | hCAR | |
| MC38 | 1 + 3 (4 + 1) | 8 + 7 (2 + 2) |
| MC38CEA | 95 + 8 (48 + 19) | 4 + 3 (2 + 1) |
| LS174T | 67 + 14 (12 + 9) | 61 + 17 (28 + 16) |
| HS766T | 35 + 17 (9 + 8) | 2 + 1 (3 + 3) |
| U118MG | 2 + 1 (2 + 3) | 2 + 3 (5 + 7) |
| U118-hCAR | 1 + 1 (1 + 3) | 99 + 5 (581 + 76) |
| THLE-3 | 10 + 9 (4 + 3) | 55 + 11 (44 + 17) |
Human and mouse cells were stained with saturating amounts of antibody recognizing hCEA or hCAR and expression was evaluated by FACS analysis. Immunoglobulin G isotype primary antibodies were used as a negative control. Data are the means ± SD of three or four independent experiments.
Figure 2.
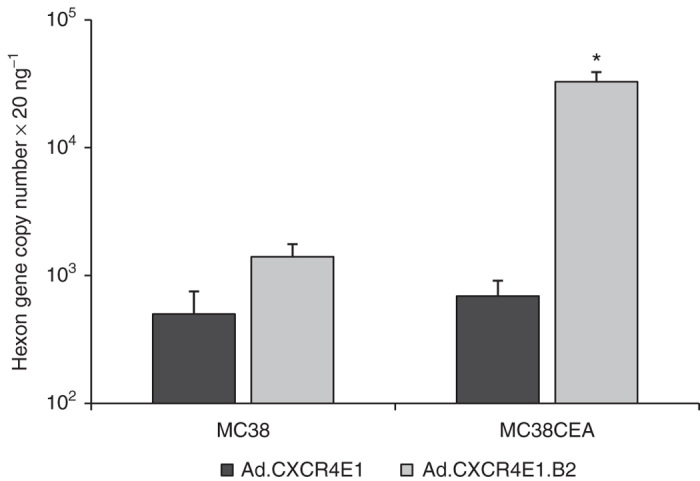
Evaluation of binding specificity of sdAb-targeted CRAds. MC38 and MC38CEA murine colon adenocarcinoma cells were incubated with 1 × 103 vp per cell of the indicated vector for 1 hour. Total DNA was isolated and hexon gene copy number was obtained using quantitative PCR. Data are presented as mean ± SD (*P < 0.01 versus MC38 cells).
Evidence of CRAd replication in a human colorectal adenocarcinoma cell line
To evaluate whether sdAb-targeted CRAds are able to replicate after infection of hCEA(+) cells, a replication assay was performed. CXCR4 promoter activity was evaluated for different cell lines by infection with Ad.CMVLuc and Ad.CXCR4Luc, encoding the Luc gene under control of the CMV or CXCR4 promoter, respectively (Figure 3a). Levels of Luc expression varied in different cell lines in proportion to viral doses of infection (results not shown). Infection with Ad.CXCR4Luc yielded lower Luc expression in comparison with Ad.CMVLuc. Additionally, we calculated ratios of Luc expression in cancer cells following Ad.CMVLuc and Ad.CXCR4Luc infection. Average ratios for all the individual sets of numbers for different cancer cells were compared. As shown in Figure 3b, HS766T cells demonstrated high CXCR4-to-CMV ratio of Luc expression in comparison with LS174T and THLE-3 cells, whereas U118MG and U118-hCAR cells showed the lowest CXCR4-to-CMV ratios. Thus, all tested cells demonstrated levels of CXCR4 activity suitable to facilitate replication of CXCR4-driven CRAds.
Figure 3.

In vitro characterization of CRAd replication. (a) Relative Luc expression following infection of human cancer cells with either Ad.CMVLuc or Ad.CXCR4Luc. Luciferase activity was measured in cell lysates at 48 hours after infection. Data are presented as mean ± SD. RLUs, relative light units. (b) The CXCR4-to-CMV ratios of Luc expression in human cells following infection with Ad.CMVLuc or Ad.CXCR4Luc. Data points represent the mean ± SD of a representative experiment. (c) Evaluation of CRAd replication. Human colorectal adenocarcinoma LS174T cells were infected with 1 × 103 vp per cell and harvested on indicated time points. Total DNA was isolated and hexon gene copy number was obtained using quantitative PCR. Data are presented as mean ± SD.
FACS analysis of human colorectal adenocarcinoma LS174T cells revealed relatively high levels of hCAR and hCEA expression (Table 1 and Supplementary Figure S1c). Taking into consideration the results of previous experiments, LS174T cells were selected for subsequent evaluation of Ad.CXCR4E1.B2 and Ad.CXCR4E1 replication. For this study, LS174T cells were infected with either Ad.CXCR4E1.B2 or Ad.CXCR4E1, then cells and media were collected at 1, 24, 48, 76, and 120 hours after infection. Replication was measured by evaluating the presence of the adenoviral hexon gene with qPCR. Both vectors show efficient replication, with the hexon gene copy number increasing ~1,000-fold in the first 24 hours after infection (Figure 3c). Thus, these data demonstrate that retargeting through incorporation of sdAb allows Ad.CXCR4E1.B2 to replicate in tumor cells. Of note, the level of replication achieved compared to Ad.CXCR4E1 with wild-type fiber.
Ad.CXCR4E1.B2 selectively induces tumor cell lysis
To evaluate whether specific replication in hCEA positive, CXCR4 positive tumor cells resulted in subsequent cytolysis by Ad.CXCR4E1.B2, a cytotoxicity assay was performed. Different cancer cell lines were evaluated for hCAR and hCEA surface expression using FACS analysis (Table 1 and Supplementary Figure S1c–f). Based on these findings, colorectal adenocarcinoma LS174T, pancreatic carcinoma HS766T, glioma U118MG and U118-hCAR cells were infected with Ad.CXCR4E1.B2, Ad.CXCR4E1 or wild-type Ad5. Five days after infection viable cells were evaluated using a crystal violet staining assay. As shown in Figure 4, infection with Ad.CXCR4E1.B2 resulted in increased cytotoxicity in hCEA(+) LS174T and HS766T cells in comparison with hCEA(−) U118MG and U118-hCAR cells, while the control Ad.CXCR4E1 and wild-type Ad5 viruses were able to produce cell killing in hCAR(+) LS174T and U118-hCAR cells. In contrast, no cytolysis for either of the vectors was observed in human glioma U118MG cells deficient for hCEA and hCAR expression (Figure 4). Of interest, Ad.CXCR4E1 infection resulted in a modest increase of HS766T cell killing in comparison with U118MG cells (both cell lines demonstrate a low levels of hCAR expression), probably due to different levels of CXCR4 promoter activity in these cells, CXCR4-to-CMV ratio of Luc expression in HS766T and U118MG cells was 0.14 ± 0.009 and 0.02 ± 0.011, respectively (Figure 3b). Taken together, these findings indicate that infection with Ad.CXCR4E1.B2 induces efficient cytolysis uniquely in hCEA expressing tumor cells.
Figure 4.
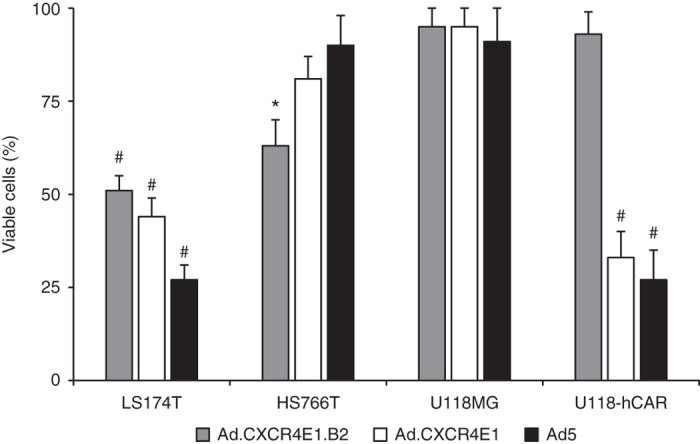
hCEA-targeted CRAd specifically kills hCEA positive tumor cells and mitigates off-target cytotoxicity. Human colorectal adenocarcinoma LS174T, pancreatic carcinoma HS766T, glioma U118MG and U118-hCAR cancer cells were infected with 1 × 103 vp per cell of Ad.CXCR4E1.B2, control vector Ad.CXCR4E1 or wild-type Ad5. Cytotoxicity was determined at 120 hours after infection using a crystal violet staining assay. Number of viable cells is given as percentage of the cell number of uninfected control. The hCEA and hCAR expression status of the cell lines is as follows: LS174T: hCEA(+)/hCAR(+); HS766T: hCEA(+)/hCAR(−); U118MG: hCEA(−)/hCAR(−); U118-hCAR: hCEA(−)/hCAR(+). Data are presented as mean ± SD (*P < 0.05 versus U118MG cells; #P < 0.01 versus U118MG cells).
Ad.CXCR4E1.B2 adds an additional level of specificity to limit off-target cytotoxicity in normal cells in vitro
For this analysis, we evaluated the hCAR and hCEA surface expression of normal immortalized liver THLE-3 cells using FACS. As shown in Table 1, THLE-3 cells resembled a “normal cell phenotype”: hCAR positive and hCEA negative. To demonstrate the additional level of specificity of sdAb-targeted CRAds compared to wild-type fiber containing CRAds, THLE-3 cells were infected with increasing concentrations of either Ad.CXCR4E1.B2 or Ad.CXCR4E1. Cytotoxicity was determined five days after infection, using a crystal violet staining. As shown in Figure 5, in contrast to the CAR-dependent Ad.CXCR4E1 vector, Ad.CXCR4E1.B2 demonstrated low levels of cytotoxicity at all indicated concentrations. These data indicate that the sdAb-mediated transductional retargeting adds an additional level of specificity to CRAds, thereby limiting off-target cytotoxicity.
Figure 5.
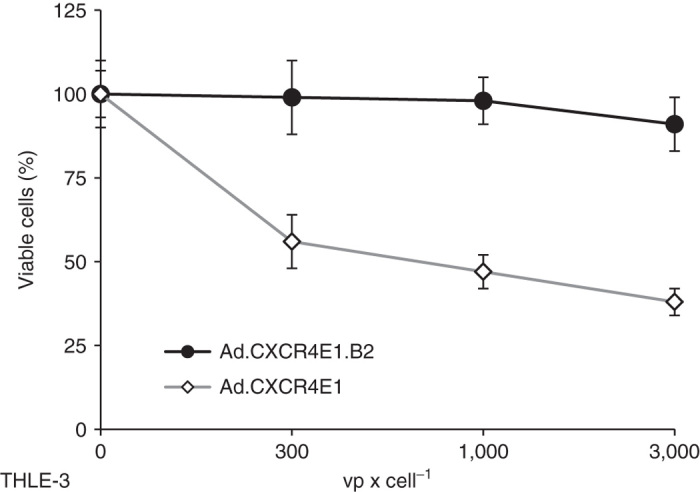
hCEA-targeted CRAd does not kill immortalized normal liver cells. Human immortalized normal liver THLE-3 cells were infected with increasing concentrations of either Ad.CXCR4E1.B2 or control vector Ad.CXCR4E1. Cytotoxicity was determined at 120 hours after infection using a crystal violet staining assay. Number of viable cells is given as percentage of the cell number of uninfected samples.
Cytotoxicity by Ad.CXCR4E1.B2 is hCAR-independent and inhibited by soluble hCEA
To demonstrate that Ad.CXCR4E1.B2 infection is hCAR-independent and hCEA-dependent, competition experiments were performed. Preincubation of tumor cells with soluble Ad5 knob protein was not able to block tumor cell cytolysis in Ad.CXCR4E1.B2 infected cells. However, cytotoxicity of control Ad.CXCR4E1 vector was efficiently blocked by incubation with the Ad5 knob protein (Figure 6a). Preincubation of the vectors with hCEA protein was able to efficiently block tumor cell death for Ad.CXCR4E1.B2 but not for control Ad.CXCR4E1 vector (Figure 6b). Collectively, these data show that Ad.CXCR4E1.B2 has enhanced tumor specificity for hCEA positive tumor cell lines compared to endogenous targeted CRAds and is able to cause subsequent oncolysis.
Figure 6.
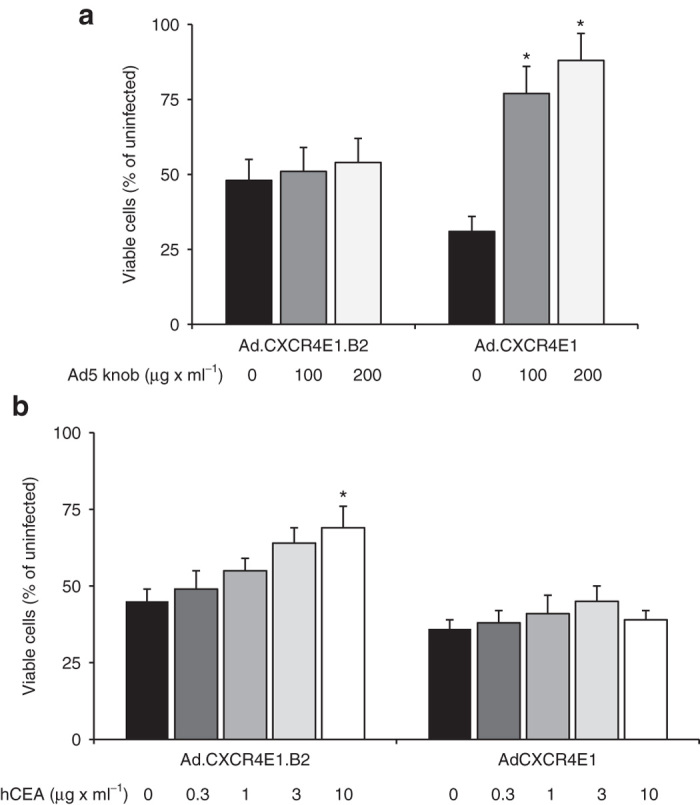
Ad.CXCR4E1.B2 induces hCEA-dependent and hCAR-independent oncolysis. (a) Human colorectal adenocarcinoma LS174T cells were preincubated with soluble Ad5 knob protein at indicated concentrations and infected with 2 × 103 vp per cell of Ad.CXCR4E1.B2 or Ad.CXCR4E1. Cytotoxicity was determined at 120 hours after infection using a crystal violet staining assay. Number of viable cells is given as percentage of the cell number of uninfected samples. Data are presented as mean ± SD (*P < 0.01 versus no treatment). (b) Inhibition of Ad.CXCR4E1.B2 mediated cytotoxicity. Ad.CXCR4E1.B2 and Ad.CXCR4E1 were incubated with hCEA at indicated concentrations. LS174T cells were infected with 2 × 103 vp per cell of Ad.CXCR4E1.B2 or Ad.CXCR4E1. Cytotoxicity was determined at 120 hours after infection using a crystal violet staining assay. Number of viable cells is given as percentage of the cell number of uninfected samples. Data are presented as mean ± SD (*P < 0.05 versus uninfected).
Discussion
A very promising targeted oncolytic therapy for the treatment of neoplastic diseases is the utilization of CRAd agents. CRAds can only replicate in tumor cells; however, their target cell binding still depends on the native tropism of the virus. In this early proof-of-concept study, we evaluated the utility of employing novel anti-hCEA camelid single domain antibodies for transductional targeting of oncolytic CRAd agents. To develop a hCEA-targeted recombinant CRAd vector, an anti-hCEA sdAb was genetically incorporated into a deknobbed Ad5 fiber-fibritin protein. Additionally, the CXCR4 promoter was employed for conditional replication of the CRAd. We found that the fiber-fibritin-sdAb chimera used in this study did not disrupt the trimerization capability of the adenovirus fiber and the hCEA antigen recognition. We demonstrated the ability of an anti-hCEA sdAb fused to a fiber-fibritin chimera to provide specific targeting and efficient oncolysis of hCEA-expressing tumor cells.
Over the last decades, substantial progress has been made in approaches to improve CRAd specificity. These efforts for development of oncolytic virotherapy agents can be illustrated by the transcriptional and transductional approaches used for targeting of CRAd vectors. CRAds were first based on deletion of mutation of early E1 genes, which allowed replication in specific tumors only.13–15 The first improvements embodied the use of tumor specific promoters (TSPs).16,17 The most important characteristics of these promoters are their “tumor on” and “liver off” expression profile.18 Although several of these promoters have been identified and used in a CRAd setting, the provided level of tumor cell specificity may not be sufficient. To this end, mRNA translation has been exploited as an additional strategy to control CRAd replication. The use of long, highly structured untranslated regions (UTR) in the adenoviral genome allows viral translation only in tumor cells with upregulated translation factors.19 Other research groups have recently exploited the use of miRNA to limit off-target adenoviral replication.20,21 However, even though these approaches have been able to partially restrict viral tropism, off-target toxicity remains an important issue to be solved in the field. A successful strategy to transductional retarget CRAds has not been shown before and therefore we believe this is the first employment of transductional targeting to enhance CRAd specificity.
The means to transductional retarget the CRAd has been feasibilized by two approaches recently developed. The first embodies a fiber modification which maintains trimerization of the fiber and allows incorporation of large and complex targeting moieties.22 However, conventional antibodies are shown to be incompatible with capsid formation and therefore unsuitable as targeting moieties.23 In this regard, we employed a distinct antibody species derived from the camelid family which consist of a heavy chain only. The single variable domain of these antibodies, also called single domain antibody (sdAb), is highly stable in the cytosolic environment. On this basis, we produced a replication-deficient adenoviral vector retargeted by an anti-hCEA-sdAb and showed hCEA-specific and hCAR-independent infection.11 This breakthrough preceded our current study and allowed us to test this system in a CRAd context.
The expression profile of hCEA which is up-regulated in a wide range of malignant tumors, including colon, gastric, lung, breast, and pancreatic cancer24–27 makes this protein an attractive tumor-cell specific target for oncolytic virotherapy. To initially validate the compatibility of this transductional retargeting in CRAds with a transcriptional targeting approach, we derived an infectivity-enhanced CRAd with replicative specificity achieved by replacement of the adenoviral E1a promoter with the CXCR4 promoter element. It was shown that CXCR4, a cell surface chemokine receptor, is implicated in the growth, invasion, angiogenesis, and metastasis (reviewed in refs. 28,29). In this study, we show that genetic incorporation of a hCEA-specific sdAb in the viral capsid of CRAds provides an additional level of specificity for tumor cell targeting. Our study confirmed that transductional targeting resulted in hCEA-dependent binding. Next, we compared replication with a control CRAd displaying the native Ad5 fiber in the permissive hCEA(+) and hCAR(+) human colorectal adenocarcinoma LS174T cell line. Ad.CXCR4E1.B2 and control vector Ad.CXCR4E1 displayed a similar replication pattern, showing that genetic incorporation of the sdAb in the CRAd viral capsid did not impede replication. To confirm whether Ad.CXCR4E1.B2 replication led to tumor cell death, cytotoxicity assays were performed. We characterized hCAR and hCEA expression in different tumor cell lines and tested cytotoxicity in hCAR(+), hCEA(+), as well as a double positive and double negative cells. Obtained data demonstrate that the hCEA-targeted Ad.CXCR4E1.B2 is able to produce cytotoxicity in hCEA(+) cancer cells, irrespective of the hCAR-status of the cells, in contrast to the wild-type Ad5 and Ad.CXCR4E1 which induced cell killing of hCAR(+) cells. Also, the results of the in vitro inhibition assay confirmed hCEA-dependent and hCAR-independent Ad.CXCR4E1.B2-mediated cytolysis.
We hypothesize that the addition of sdAb-based transductional targeting will improve the tumor cell selectivity and efficacy of the transcriptional restricted CRAd agent thereby allowing its progress into development for oncolytic virotherapy. Thus, a major goal of this study was to construct an sdAb targeted CRAd agent, based upon the Ad.CXCR4E1 viral backbone, and to validate its targeting specificity for CEA(+) cancer cells in vitro. Our experiments sought to test specific replication and subsequent cytotoxicity of our sdAb-targeted CRAd in a permissive in vitro setting. However, additional in vivo experiments that are beyond the scope of this study are required to investigate the biodistribution of sdAb-targeted CRAds and optimize tumor-specific targeting of oncolytic virotherapy. The ultimate read out would be to test the off-target effect of our system in a replication permissive in vivo model for CRAds. Syrian golden hamsters are known to be a permissive model to study CRAd targeting.30 However, antibodies for this species are not available yet. Therefore, our approach cannot be tested in an adequate in vivo model system at this stage, but this would be of great interest as soon as the required methods are available.
Although Ad.CXCR4E1.B2 is promising as an agent to achieve selective targeting and replication in specific target cells, further optimization of our sdAb-targeted CRAd may be required to obtain a suitable viral vector for therapeutic use in humans. Foremost, systemic delivery is desired in the clinical setting, as patients may have tumors that are inaccessible or are already metastatic at the time of detection. The major problem of systemic adenoviral delivery is sequestration of the virus by the liver.31 Several studies have shown that this off-target effect is not abolished by hCAR detargeting and interactions of the hexon capsid protein with blood factors seem to be involved.32,33 It was shown that substitution of the Ad5 hypervariable region 7 (HVR7) with HVR7 from adenovirus serotype 3 (Ad3) decreased liver tropism following systemic administration.34 Also, shielding of the viral capsid using monovalent (e.g., PEG) or multivalent (e.g., HPMA) polymers has been proposed to circumvent liver sequestration.35 The hexon interaction with blood factors is not the only way of adenoviral lever sequestration. The RGD motif present in the penton base is thought to facilitate binding to liver sinusoidal endothelial cells (LSECs).36 Therefore, the next step in this retargeted virus design would include both hexon and penton base modifications against liver sequestration.
In conclusion, we successfully developed a transductional retargeted Ad.CXCR4E1.B2 vector, providing an additional level of specificity over early generation CRAd vectors. Ad.CXCR4E1.B2 is able to efficiently kill tumor cells overexpressing hCEA. To our knowledge, this is the first time that infection and replication of an sdAb-targeted CRAd is shown. The application of this additional level of transductional control allows specific targeting of tumor cells and limits off-target toxicity. The double targeting employing transcriptional and transductional approaches is a promising means to improve the therapeutic index for these advanced generation CRAds. This study, therefore, highlights an important improvement in CRAd design for future applications in cancer therapy.
Materials and Methods
Adenoviral vectors
The fiber-fibritin-hCEA protein was created as described before.11 Shortly, alpacas were immunized with soluble human CEA (ProNique Scientific, Castle Rock, CO) and sdAbs against hCEA were acquired by phage biopanning. Of all screened sdAb clones B2 was the most efficient in binding hCEA. B2 was fused in single open reading frame with a chimeric fiber-fibritin protein which contained the N-terminal Ad5 fiber tail region fused to the trimerizing domain of the fibritin protein of bacteriophage T4 followed by a peptide linker (G-G-G-S) connected to the B2 sdAb as described previously.22 The fiber-fibritin-B2 (FFB2) protein was retrieved from pKan566FFB2 using EcoR I and Sal I restriction sites. Recombinant adenovirus genomes were generated by homologous DNA recombination in E. coli BJ5183 between the restricted FFB2 and Ad5 fiber gene deleted pVK500C.CXCR4E1, resulting in pVK500C.CXCR4E1.B2. Insertion of the fiber gene was confirmed by PCR and partial sequence analysis. The plasmid was linearized using Pac I restriction and transfected into 293F28 cells using SuperFect Transfection Reagent (Qiagen, Chatsworth, CA). 293F28 cells stably express the native Ad5 fiber; therefore, a mixture of fibers was present on the viruses rescued at this point. After an additional round of amplification in 293F28 cells, viruses were amplified in Ad5-fiber negative HEK293 cells to obtain viral particles containing only the B2-fiber. Viruses were propagated in HEK293 cells and purified twice by CsCl gradient centrifugation. Viral particles were dialyzed against 10% glycerol in phosphate-buffered saline (PBS). Viral particles (vp) were quantified by measuring absorbance of the dissociated virus at A260 nm using a conversion factor of 1.1 × 1012 vp per absorbance unit.
The Ad.CXCR4E1 conditionally replicative vector and replication deficient Ad.CMVLuc and Ad.CXCR4Luc vectors were created as described before.11 Wild-type Ad5 was kindly provided by Dr H Ugai (Washington University in St Louis, St Louis, MO). A schematic overview of the vectors used in this study is presented in Figure 1a.
Cell lines
Human colorectal adenocarcinoma LS174T and human glioma U118MG cells were purchased from ATCC (Manassas, VA). Human pancreatic carcinoma HS766T cells were kindly provided by Dr PG Oliver (University of Alabama at Birmingham, Birmingham, AL). Human glioma U118-hCAR cells expressing hCAR were kindly provided by Dr JT Douglas (University of Alabama at Birmingham). For propagation of our vector we used HEK293 cells, purchased from Microbix Biosystems (Mississauga, Ontario, Canada), and 293F28 cells expressing wild-type Ad5 fiber protein, which have been described previously.37 All abovementioned cell lines were cultured in DMEM/F12 (Mediatech, Nerndon, VA) medium supplemented with 10% FBS, 100 IU/ml penicillin and 100 µg/ml streptomycin.
The murine colon adenocarcinoma MC38 and MC38CEA cells have been described previously.38 MC38CEA cells expressing hCEA were generated by retroviral transduction with hCEA cDNA. MC38 and MC38CEA cells were provided by Dr HR Herschman (University of California, Los Angeles, CA) and cultured in DMEM medium supplemented with 10% FBS, 0.1 mmol/l NaPyruvate, 100 IU/ml penicillin, and 100 µg/ml streptomycin.
Immortalized primary human liver THLE-3 cells were purchased from ATCC and cultured in accordance with vendor instructions.
FACS analysis
To determine the levels of hCEA surface expression, approximately 1 × 106 cells were collected, washed with PBS, and stained with anti-hCEA rabbit IgG (Millipore, Billerica, MA) and antirabbit FITC-labeled goat IgG (Millipore) for 1 hour at 4 °C. Levels of hCAR surface expression were measured with anti-hCAR mAb (RcmB), kindly provided by Dr J Douglas (University of Alabama at Birmingham) and antimouse FITC-labeled goat IgG (Molecular Probes, Eugene, OR). Mouse IgG1 negative control (Millipore) and rabbit IgG isotype control (Thermo Scientific, Rockford, IL) were used as isotype controls. After washing in PBS for three times, cells were resuspended in FACS buffer. Approximately 1 × 104 cells were illuminated at 488 nm, detecting fluorescence in the FITC (525/20 nm) channel.
Western blotting assay
Fiber-fibritin-B2 expression was evaluated by western blot. Samples containing 5 × 109 viral particles were preincubated in Laemmli sample buffer for 10 minutes at 99 °C or 25 °C for seminative conditions. Proteins were separated using a 4–20% gradient polyacrylamide Precise Protein gel (Thermo Scientific, Wilmington, DE). The proteins were blotted onto polyvinylidene difluoride (PVDF) membranes and developed with the Sigma FAST 3,3′-diaminobenzidine system (Sigma-Aldrich, St Louis, MO) according to the manufacturer’s protocol. Anti-Ad5 fiber mAb (4D2, Thermo Scientific) and goat-anti-mouse Ig-HRP (DakoCytomation Denmark A/S, Glostrup, Denmark) were used for Ad5 fiber protein detection.
Luciferase assay
Cells were seeded at 5 × 104 cells per well in a 24-well plate and grown overnight. The next day, medium was removed and cells were infected with 1 × 103 vp per cell of Ad.CMVLuc or Ad.CXCR4Luc in triplicate. After 1 hour, cell culture medium was removed and fresh medium was added. After 48 hours, medium was removed, and cells were lysed. The Luciferase Assay System (Promega, Madison, WI) and Femtomaster FB12 luminometer (Zylux, Oak Ridge, TN) were used for Luc activity evaluation. Luc activity was normalized by the protein concentration of the cell lysate using DC Protein Assay (Bio-Rad, Hercules, CA). Data expressed as relative light units (RLU) per 1 × 104 cells.
Crystal violet cell viability assay
For the measurement of cell viability, cell culture medium was removed after a given time point and surviving cells were fixed and stained with 1% crystal violet (Sigma-Aldrich, St Louis, MO) in 70% ethanol for at least 3 hours at 25 °C. The plates were extensively washed in tap-water, air-dried and optical density was measured at 595 nm using an EL 800 Universal Microplate Reader (BIO-TEK Instruments, Winooski, VT). The percentage viable cells was calculated for infected cells relative to uninfected cells.
Thermostability assay
Cells were seeded 2 × 104 cells per well in a 96-well plate and grown overnight. Cells were infected with 2 × 103 vp per cell of Ad.CXCR4E1.B2 or control vector Ad.CXCR4E1 which were incubated at 45 °C for 0, 10, 20, 30, or 40 minutes beforehand. After 72 hours, cell culture medium was removed and surviving cells were fixed and stained with 1% crystal violet and the percentage viable cells was calculated for infected cells relative to uninfected cells as described above.
In vitro binding assay
Cells were seeded 3 × 105 cells per well in a six-well tissue culture plate and grown overnight. The next day medium was removed and cells were infected with 1 × 103 vp per cell of Ad.CXCR4E1 or Ad.CXCR4E1.B2. After incubation at 37 °C for 1 hour, cells were washed in PBS and collected by trypsinization. The DNA was isolated from the cells using a QIAamp DNA mini Kit (Qiagen, Chatsworth, CA).
Ad5 hexon expression was measured using quantitative real-time PCR. Serial tenfold dilutions (from 1 × 109 to 10 viral particles per reaction) of viral control DNA were included to establish a standard curve. The following primers were used for Ad5 hexon gene detection: Ad5Hexon-fwd: 5′-TAC GCA CGA CGT GAC CAC A-3′, Ad5Hexon-rev: 5′-ATC CTC ACG GTC CAC AGG G-3′ and the following TaqMan probe was used: Ad5Hexon-probe: 5′-6FAM-ACC GGT CCC AGC GTT TCA CGC-BHQ1-3′. Mouse β-actin gene expression was used to normalize the samples. The following mouse β-actin probes were used: mβ-actin-fwd: 5′-AGC TGG AGG ACT TCC GAG ACT-3′, mβ-actin-rev: 5′-TGG CAC TTC TCC TGC ACC TT-3′, and mβ-actin-probe: 5′-HEX-TAG ACG CCT GCA CAA GCC GCC-BHQ1-3′.
In each reaction, 20 ng of total DNA was added to a total of 10 µl of reaction mixture containing 2× Fast Start TaqMan Probe Master Mix (Roche Applied Science, Indianapolis, IN), 333 nmol/l of each primer and fluorogenic probe. Reactions were carried out in triplicates in a 96-well reaction plate (PE Applied Biosystems, Grand Island, NY) in a spectrofluorimetric thermal cycler (LightCycler 480 Real-Time PCR system, Roche Applied Science). The following program was used: denaturation (2 minutes at 95 °C) and amplification with 45 cycles (15 seconds at 90 °C and 60 seconds at 60 °C). The level of binding to MC38 and MC38CEA cells was determined as the Ad hexon gene copy number per 20 ng total DNA.
Quantitative virus replication
Cells were seeded at 3 × 105 cells per well in six-well tissue culture plates and grown overnight. The next day medium was removed and cells were infected with 1 × 103 vp per cell of Ad.CXCR4E1 or Ad.CXCR4E1.B2. After incubation at 37 °C for 1 hour, the medium was replaced. Cells were harvested 1, 24, 48, 72, and 120 hours after infection, subjected to three freeze-thaw cycles and centrifuged at 5,000 RPM for 5 minutes. DNA from infected cells was isolated using QIAamp DNA Mini Kit (Qiagen, Chatsworth, CA). qPCR was performed as described above. Human β-actin gene expression was used to normalize the samples. The following human β-actin primers and probes were used: β-actin-fwd: 5′GAG GCA TCC TCA CCC TGA AG-3′, β-actin rev: 5′TCC ATC TCG CAG TTG GT-3′, and β-actin probe: 5′-HEX-CCC CATCGA GCA CGG CAT CG-BHQ1-3′.
In vitro cytotoxicity assay
To measure cytotoxicity of the sdAb-retargeted CRAd, cells were seeded into 96-well tissue culture plates at 5 × 103 cells per well, incubated for 24 hours and infected with CRAd vectors at 1 × 103 vp per cell. After 120 hours, cell culture medium was removed and surviving cells were fixed and stained with 1% crystal violet and measured as described above.
Competitive inhibition assay
To block hCAR specific transduction, cells were seeded 1 × 105 cells per well in a 24-well tissue culture plate and incubated after one day with 100 or 200 μg/ml of soluble Ad5 knob protein for 1 hour at 4 °C before infection with Ad.CXCR4E1.B2 or Ad.CXCR4E1 at 2 × 103 vp per cell. After 120 hours, the cells were stained with crystal violet as described above. To block hCEA specific transduction, cells were seeded 1 × 105 cells per well in a 24-well tissue culture plate. Both Ad.CXCR4E1.B2 and Ad.CXCR4E1 were incubated with 0.3, 1, 3, or 10 μg/ml of recombinant hCEA protein (ab742, Abcam, Cambridge, MA) for 30 minutes at room temperature. Afterwards cells were infected with the virus-hCEA mixture at 2 × 103 vp per cell. After 120 hours, the cells were stained with crystal violet as described above.
Statistical analysis
In experiments where triplicates were run to reduce measurement error, the statistical analyses were conducted on the mean triplicate value. All error terms are expressed as the standard deviation of the mean. Significance levels for comparison of differences between groups in the in vitro experiments were analyzed by the Student’s t-test. The differences were considered significant when P value was <0.05. All reported P values are two-sided.
Acknowledgments
This work was supported in part by the Stichting Jo Kolk Studiefonds and the Dutch Cancer Society (EAE) and the National Institutes of Health under award numbers 5P50 CA101955 and 5R01 CA154697 (DTC).
The authors declare no conflict of interest.
References
- Pesonen, S, Kangasniemi, L and Hemminki, A (2011). Oncolytic adenoviruses for the treatment of human cancer: focus on translational and clinical data. Mol Pharm 8: 12–28. [DOI] [PubMed] [Google Scholar]
- Garber, K (2006). China approves world’s first oncolytic virus therapy for cancer treatment. J Natl Cancer Inst 98: 298–300. [DOI] [PubMed] [Google Scholar]
- Yamamoto, M and Curiel, DT (2010). Current issues and future directions of oncolytic adenoviruses. Mol Ther 18: 243–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oosterhoff, D and van Beusechem, VW (2004). Conditionally replicating adenoviruses as anticancer agents and ways to improve their efficacy. J Exp Ther Oncol 4: 37–57. [PubMed] [Google Scholar]
- Beatty, MS and Curiel, DT (2012). Chapter two–Adenovirus strategies for tissue-specific targeting. Adv Cancer Res 115: 39–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergelson, JM, Cunningham, JA, Droguett, G, Kurt-Jones, EA, Krithivas, A, Hong, JS et al. (1997). Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science 275: 1320–1323. [DOI] [PubMed] [Google Scholar]
- Noureddini, SC and Curiel, DT (2005). Genetic targeting strategies for adenovirus. Mol Pharm 2: 341–347. [DOI] [PubMed] [Google Scholar]
- Glasgow, JN, Everts, M and Curiel, DT (2006). Transductional targeting of adenovirus vectors for gene therapy. Cancer Gene Ther 13: 830–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revets, H, De Baetselier, P and Muyldermans, S (2005). Nanobodies as novel agents for cancer therapy. Expert Opin Biol Ther 5: 111–124. [DOI] [PubMed] [Google Scholar]
- Cortez-Retamozo, V, Backmann, N, Senter, PD, Wernery, U, De Baetselier, P, Muyldermans, S et al. (2004). Efficient cancer therapy with a nanobody-based conjugate. Cancer Res 64: 2853–2857. [DOI] [PubMed] [Google Scholar]
- Kaliberov, SA, Kaliberova, LN, Buggio, M, Tremblay, JM, Shoemaker, CB and Curiel, DT (2014). Adenoviral targeting using genetically incorporated camelid single variable domains. Lab Invest 94: 893–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaliberov, SA, Kaliberova, LN, Buchsbaum, DJ and Curiel, DT (2014). Experimental virotherapy of chemoresistant pancreatic carcinoma using infectivity-enhanced fiber-mosaic oncolytic adenovirus. Cancer Gene Ther 21: 264–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff, JR, Kirn, DH, Williams, A, Heise, C, Horn, S, Muna, M et al. (1996). An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science 274: 373–376. [DOI] [PubMed] [Google Scholar]
- Heise, C, Hermiston, T, Johnson, L, Brooks, G, Sampson-Johannes, A, Williams, A et al. (2000). An adenovirus E1A mutant that demonstrates potent and selective systemic anti-tumoral efficacy. Nat Med 6: 1134–1139. [DOI] [PubMed] [Google Scholar]
- Fueyo, J, Gomez-Manzano, C, Alemany, R, Lee, PS, McDonnell, TJ, Mitlianga, P et al. (2000). A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene 19: 2–12. [DOI] [PubMed] [Google Scholar]
- Rodriguez, R, Schuur, ER, Lim, HY, Henderson, GA, Simons, JW and Henderson, DR (1997). Prostate attenuated replication competent adenovirus (ARCA) CN706: a selective cytotoxic for prostate-specific antigen-positive prostate cancer cells. Cancer Res 57: 2559–2563. [PubMed] [Google Scholar]
- Matsubara, S, Wada, Y, Gardner, TA, Egawa, M, Park, MS, Hsieh, CL et al. (2001). A conditional replication-competent adenoviral vector, Ad-OC-E1a, to cotarget prostate cancer and bone stroma in an experimental model of androgen-independent prostate cancer bone metastasis. Cancer Res 61: 6012–6019. [PubMed] [Google Scholar]
- Mathis, JM, Stoff-Khalili, MA and Curiel, DT (2005). Oncolytic adenoviruses - selective retargeting to tumor cells. Oncogene 24: 7775–7791. [DOI] [PubMed] [Google Scholar]
- Stoff-Khalili, MA, Rivera, AA, Nedeljkovic-Kurepa, A, DeBenedetti, A, Li, XL, Odaka, Y et al. (2008). Cancer-specific targeting of a conditionally replicative adenovirus using mRNA translational control. Breast Cancer Res Treat 108: 43–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylösmäki, E, Hakkarainen, T, Hemminki, A, Visakorpi, T, Andino, R and Saksela, K (2008). Generation of a conditionally replicating adenovirus based on targeted destruction of E1A mRNA by a cell type-specific MicroRNA. J Virol 82: 11009–11015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leja, J, Nilsson, B, Yu, D, Gustafson, E, Akerström, G, Oberg, K et al. (2010). Double-detargeted oncolytic adenovirus shows replication arrest in liver cells and retains neuroendocrine cell killing ability. PLoS ONE 5: e8916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noureddini, SC, Krendelshchikov, A, Simonenko, V, Hedley, SJ, Douglas, JT, Curiel, DT et al. (2006). Generation and selection of targeted adenoviruses embodying optimized vector properties. Virus Res 116: 185–195. [DOI] [PubMed] [Google Scholar]
- Magnusson, MK, Hong, SS, Henning, P, Boulanger, P and Lindholm, L (2002). Genetic retargeting of adenovirus vectors: functionality of targeting ligands and their influence on virus viability. J Gene Med 4: 356–370. [DOI] [PubMed] [Google Scholar]
- Goldstein, MJ and Mitchell, EP (2005). Carcinoembryonic antigen in the staging and follow-up of patients with colorectal cancer. Cancer Invest 23: 338–351. [DOI] [PubMed] [Google Scholar]
- Kanetaka, K, Ito, S, Susumu, S, Yoneda, A, Fujita, F, Takatsuki, M et al. (2013). Clinical significance of carcinoembryonic antigen in peritoneal lavage from patients with gastric cancer. Surgery 154: 563–572. [DOI] [PubMed] [Google Scholar]
- Grunnet, M and Sorensen, JB (2012). Carcinoembryonic antigen (CEA) as tumor marker in lung cancer. Lung Cancer 76: 138–143. [DOI] [PubMed] [Google Scholar]
- Ballesta, AM, Molina, R, Filella, X, Jo, J and Giménez, N (1995). Carcinoembryonic antigen in staging and follow-up of patients with solid tumors. Tumour Biol 16: 32–41. [DOI] [PubMed] [Google Scholar]
- Cojoc, M, Peitzsch, C, Trautmann, F, Polishchuk, L, Telegeev, GD and Dubrovska, A (2013). Emerging targets in cancer management: role of the CXCL12/CXCR4 axis. Onco Targets Ther 6: 1347–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitzenfeld, P and Ben-Baruch, A (2014). The chemokine system, and its CCR5 and CXCR4 receptors, as potential targets for personalized therapy in cancer. Cancer Lett 352: 36–53. [DOI] [PubMed] [Google Scholar]
- Thomas, MA, Spencer, JF, La Regina, MC, Dhar, D, Tollefson, AE, Toth, K et al. (2006). Syrian hamster as a permissive immunocompetent animal model for the study of oncolytic adenovirus vectors. Cancer Res 66: 1270–1276. [DOI] [PubMed] [Google Scholar]
- Khare, R, Chen, CY, Weaver, EA and Barry, MA (2011). Advances and future challenges in adenoviral vector pharmacology and targeting. Curr Gene Ther 11: 241–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alba, R, Bradshaw, AC, Parker, AL, Bhella, D, Waddington, SN, Nicklin, SA et al. (2009). Identification of coagulation factor (F)X binding sites on the adenovirus serotype 5 hexon: effect of mutagenesis on FX interactions and gene transfer. Blood 114: 965–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short, JJ, Rivera, AA, Wu, H, Walter, MR, Yamamoto, M, Mathis, JM et al. (2010). Substitution of adenovirus serotype 3 hexon onto a serotype 5 oncolytic adenovirus reduces factor X binding, decreases liver tropism, and improves antitumor efficacy. Mol Cancer Ther 9: 2536–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaliberov, SA, Kaliberova, LN, Hong Lu, Z, Preuss, MA, Barnes, JA, Stockard, CR et al. (2013). Retargeting of gene expression using endothelium specific hexon modified adenoviral vector. Virology 447: 312–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlan, L, Alba, R, Parker, AL, Bradshaw, AC, McNeish, IA, Nicklin, SA et al. (2010). Tropism-modification strategies for targeted gene delivery using adenoviral vectors. Viruses 2: 2290–2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo, NC, van Rooijen, N and Shayakhmetov, DM (2009). Redundant and synergistic mechanisms control the sequestration of blood-born adenovirus in the liver. Mol Ther 17: 675–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belousova, N, Korokhov, N, Krendelshchikova, V, Simonenko, V, Mikheeva, G, Triozzi, PL et al. (2003). Genetically targeted adenovirus vector directed to CD40-expressing cells. J Virol 77: 11367–11377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins, PF, Kantor, JA, Salgaller, M, Hand, PH, Fernsten, PD and Schlom, J (1991). Transduction and expression of the human carcinoembryonic antigen gene in a murine colon carcinoma cell line. Cancer Res 51: 3657–3662. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.