

Abstract
The hybrid oncolytic vesicular stomatitis virus (VSV-FH) deleted for its G glycoprotein and displaying the measles virus (MV) envelope glycoproteins (hemagglutinin H and fusion F) is fusogenic, infects cells via any of the three MV receptors and has potent oncolytic activity against subcutaneous and disseminated myeloma tumors. To tailor VSV-FH as an oncolytic virus for ovarian cancer, we ablated its natural tropism and retargeted the virus by display of a single-chain antibody (scFv) with specificity to the HER-2/neu receptor. A panel of six VSVFH-αHER2 viruses displaying anti-HER2 scFv that bind to the same HER2 epitope but with different Kd (10−6 to 10−11 M, VSVFH-αHER2#6 to #11, respectively) were rescued and characterized. A Kd of at least 10−8 M is required for infection of HER-2 positive SKOV3ip.1 cells. The higher affinity viruses (>10−8 M) were able to infect and fuse SKOV3ip.1 cells more efficiently, inducing more extensive cytopathic effects. We next compared the antitumor potency of the viruses against SKOV3ip.1 tumor xenografts. In contrast to the saline-treated animals, one intratumoral injection of VSVFH-αHER2#9, #10, or #11 resulted in efficient tumor control. There was no significant difference between viruses with an affinity higher than 10−9 M in terms of oncolytic potency. VSVFH-αHER2 virus may be a promising agent for the treatment of HER-2 positive malignancies.
Introduction
The human epidermal growth factor receptor-2 (HER-2) is a transmembrane protein member of the epidermal growth factor tyrosine kinases family. Expressed at low levels in healthy tissues, it can form dimers with other members of the ErbB family resulitng in the activation of different cellular pathways mediating cell development, survival, and angiogenesis.1 HER-2 mutation or overexpression has been observed in a number of different malignancies including breast, esophageal, lung, cervical, and endometrial cancer. In particular, HER2 is overexpressed in 9–32% of the cases of ovarian cancer.1–3 Due to its importance in tumorigenesis and metastasis, HER-2 is a target for different therapeutic approaches such as monoclonal antibodies and HER-2 tyrosine kinase inhibitors.4 The use of oncolytic viruses as an anticancer therapy is being extensively investigated in clinical trials with promising results.5,6 Nevertheless, one of the main challenges with the use of such viruses is minimizing nonspecific infection of normal cells and achieving potent virus cytotoxicity in malignant cells. One of the approaches to increase the specificity of infection is to retarget virus infection to receptor-positive malignant cells using a single-chain antibody (scFV) displayed on the viral envelope.
VSV is a negative-strand RNA virus, which has shown antitumor activity in preclinical animal models and is currently being tested in a phase 1 clinical trial utilizing intratumoral injection of the virus into hepatocellular carcinoma and tumors metastatic to the liver at the Mayo Clinic, Arizona (Mitesh Borad, PI, Clinical Trials.gov ID NCT01628640). As a way to increase its safety for systemic therapy, we previously evaluated the feasibility of engineering the VSV glycoprotein (VSV-G) to display a targeting ligand.7 The engineered VSV was able to grow in cultured cells and incorporate the engineered VSV-G displaying a targeting ligand. However, its tropism for cells expressing the VSV natural receptor(s) has not yet been accomplished.7 Therefore, this engineered VSV can still infect, for instance, cells expressing the low-density lipoprotein receptor, which is a known VSV cellular receptor.8
As an alternative method to restrict VSV tropism, our group recently developed a hybrid virus, named VSV-FH. This virus is an engineered replication-competent VSV where the VSV glycoprotein (VSV-G) gene was deleted and replaced with the Measles Virus (MV) hemagglutinin and fusion genes (MV-H and MV-F, respectively). This virus showed a tropism similar to that of MV and demonstrated promising antitumor efficacy against solid plasmacytomas and disseminated human multiple myeloma xenografts.9 In contrast to parental VSV, neurovirulence was significantly diminished after direct intracerebral injection of this virus into the brains of measles receptor positive CD46 transgenic mice.9 However, both VSV and VSV-FH viruses were neurotoxic when given intravenously to CD46 transgenic mice lacking a functional type I IFN receptor (Ifnartm-CD46Gemice).9–11 To reduce the neurotoxicity observed in IfnartmCD46Ge mice, we first evaluated the feasibility of generating retargeted VSV-FH vectors displaying a scFV with specificity to tumor-associated receptors, including CD38, EGFR, α-folate receptor, and prostate-specific membrane antigen (PSMA).12,13 These retargeted nonreplicating vectors were able to efficiently enter cells via the targeted receptors and could no longer infect neuronal cells.13,14
In the current study, we sought to determine if it is feasible to generate retargeted replication-competent VSV-FH viruses stably displaying scFVs and if the scFV would be stably retained in the genome of these replication-competent viruses. With ovarian cancer as our target, we engineered the VSV-FH to display a scFv with specificity to HER-2 in the H protein. We also sought to study the effect of scFv affinity to its target. Thus, a panel of anti-HER2 scFV with dissociation constants (Kd) ranging from 10−6 to 10−11 M was used to generate a panel of six HER-2-retargeted VSV-FH (VSVFH-αHER2) viruses.15 This panel of anti-HER2 scFV has been used previously to successfully retarget oncolytic measles viruses (MV) to HER-2. Animal studies done with these HER-2-targeted MV showed that all these engineered MV were equally efficacious against ovarian cancer xenografts regardless of the receptor affinity of the displayed anti-HER-2 scFV.16 In lieu of our previous study showing that the hybrid VSV-FH virus was more potent than MV due to its higher viral yield, faster replication kinetics and more potent fusogenic capabilities, we are keen to investigate the oncolytic potential of HER-2-targeted VSV-FH for ovarian cancer therapy.17
Results
Rescue of VSV-FH bearing scFV against HER2 with different receptor affinities
To produce VSVFH-αHER2, the MV-H gene was replaced with an engineered MV-H fused at the C terminal to a single-chain antibody directed to human HER-2, followed by a six-histidine peptide. MV-H interactions with the MV cellular receptors CD46 and SLAM were ablated by point mutations in amino acids 481 and 533, respectively. The schematic representation of the full-length genome of VSVFH-αHER2 is shown (Figure 1a). Using the Vaccinia-T7 rescue system, we obtained six replication-competent viruses bearing scFV binding to the same HER-2 epitope but with different dissociation constants (Table 1). The viruses were designated from #6 to #11 to allow rapid identification of the approximate dissociation constant (Kd) of the displayed scFV (e.g., VSVFH-αHER2#8 presents a virus displaying scFv with Kd for HER-2 of 10−8 M).
Figure 1.

Rescue of replication-competent VSVFH-αHER2. (a) Schematic representation of the viral genome. Magnified region shows the amino acid numbers (above MV-H) mutated to ablate the interaction between the MV-H protein and the cellular receptors indicated below. (a) A histidine tail (H6) was included at the end of MV-H to allow virus growth in VeroαHis cells. (b) Western blot to detect MV-H incorporation in virions. Supernatants of infected cells were fractionated by polyacrylamide gel electrophoresis (PAGE) and transferred to a polyvinylidene difluoride (PVDF) membrane. MV-H and VSV-M were detected using specific antibodies and a peroxidase-conjugated secondary antibody. (c) Kinetics of viral production. VeroαHis cells were infected at a multiplicity of infection (MOI) of 0.0001 with VSVFH-αHER2#6-11 (abbreviated as αHER2#6-11) or parental VSV-FH (FH). The number of infectious particles produced at the indicated times postinfection (hpi) supernatant was calculated by titration in VeroαHis cells. Bars represent average of three experiments (mean ± SD). (d) Cytotoxicity of viruses. VeroαHis cells were infected at a MOI of 0.1 and at 48 hours postinfection cells were fixed with 5% glutaraldehyde and stained with 0.1% crystal violet. Bar = 800 µm.
Table 1. Constant of dissociation (Kd) of the scFV-αHER-2.
| scFv | Kd (M) of scFv | Recombinant virus |
|---|---|---|
| C6.5Y100kA | 1.6 × 10−6 | VSVFH-αHER2#6 |
| C6.5G98A | 3.2 × 10−7 | VSVFH-αHER2#7 |
| C6.5 | 1.6 × 10−8 | VSVFH-αHER2#8 |
| C6ML3-9 | 1.0 × 10−9 | VSVFH-αHER2#9 |
| C6MH3B1 | 1.2 × 10−10 | VSVFH-αHER2#10 |
| C6B1D2 | 1.5 × 10−11 | VSVFH-αHER2#11 |
By western blot analysis of purified virions, it was observed that all the rescued HER-2-retargeted viruses expressed and incorporated into their virions a larger H (due to the size of the scFV) than the parental MV-H (Figure 1b). Incorporation of MV-H-scFV into virions was comparable between the viruses. Importantly, the inclusion of a scFV did not prevent virus replication, as all the viruses were capable of producing infectious particles in permissive VeroαHis cells (Figure 1c). In these permissive cells, a potent cytopathic effect (CPE), comparable to that of parental VSV-FH, was observed (Figure 1d).
VSVFH-αHER2 specifically infects cells expressing HER2
The specificity of VSVFH-αHER2 infection was tested in a panel of Chinese hamster ovary (CHO) cells expressing the known MV receptors CD46, SLAM, or Nectin-4, and human HER-2 protein. As shown in Figure 2, retargeted and parental viruses were unable to infect parental CHO cells, which do not express any of the known MV receptors. Moreover, due to point mutation of the MV-H protein, none of the VSVFH-αHER2 viruses (#6 to #11) were able to infect CHO-CD46 or CHO-SLAM cells. The amino acids of MV-H involved in the interaction with Nectin-4 (a measles receptor) were not modified in the HER2-targeted viruses since it has been shown that Nectin-4 is overexpressed in ovarian cancer.18 Therefore, all the tested viruses were able to produce a CPE in CHO-Nectin-4 cells.
Figure 2.
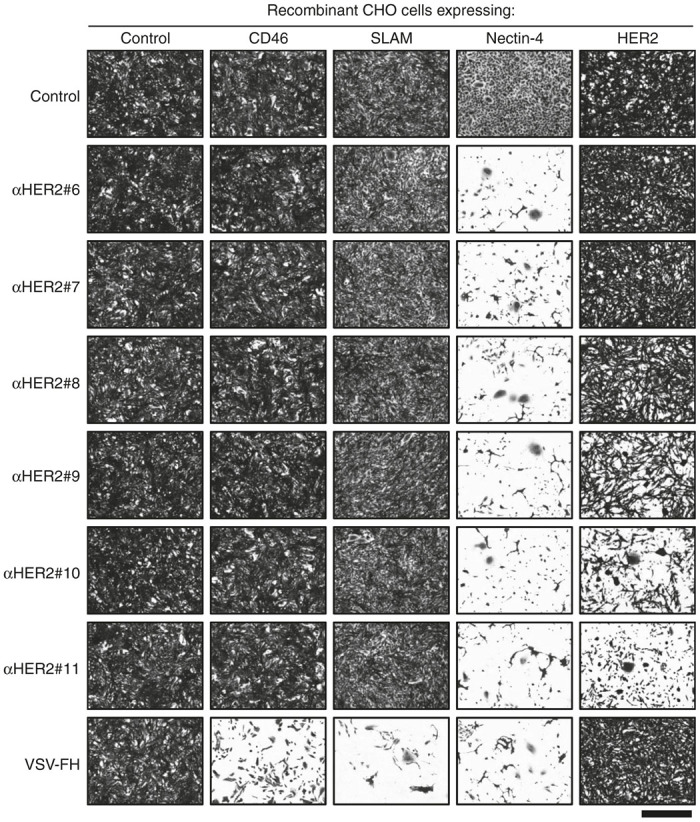
Virus specificity. Recombinant Chinese hamster ovary (CHO) cells expressing the indicated receptor were infected at a multiplicity of infection of 0.1 with VSVFH-αHER2#6-11 (abbreviated as αHER2#6-11) or parental VSV-FH. At 3 days postinfection, cells were fixed with 5% glutaraldehyde and stained with 0.1% crystal violet. Bar = 800 µm.
Lastly, different levels of CPE were observed in the CHO-HER2 cells infected with VSVFH-αHER2. Specifically, no CPE was observed in the CHO-HER2 cells infected with VSV-FH, VSVFH-αHER2#6, #7, or #8, while the destruction of the cell monolayer was visible only in CHO-HER2 cells infected with VSVFH-αHER2#9, #10, or #11, suggesting a threshold in the receptor affinity to allow cell killing.
Syncytia formation by VSVFH-αHER2
MV-F and MV-H expression in permissive cells results in the fusion of neighboring cells to form syncytia. To confirm that VSVFH-αHER2 is fusogenic and to know to what extent the Kd of the scFV affects the fusogenic activity of MV-F and -H, we measured the syncytial area in two infected HER-2-positive cell lines, TE671 and SKOV3ip.1. It has been reported that TE671 cells express HER-2 at a density of 4.3 × 103 receptor molecules/cell while SKOV3ip.1 cells express higher levels of this protein (1.5 × 105 receptor molecules/cell).15
Cells were infected with VSVFH-αHER2#6-11 and the average area of the syncytia was determined at 48 hours postinfection (Figure 3). Similar to what was observed in the CHO-HER2 cells, there is a threshold in the required receptor affinity of the scFV that allows virus infectivity. No syncytia were observed in cells infected with low-affinity VSVFH-αHER2 (#6 and #7), and only a few small syncytia were observed in SKOV3ip.1 cells infected with VSVFH-αHER2#8. Viruses with a higher receptor affinity formed larger syncytia in both TE671 and SKOV3ip.1 cells, although there was no significant difference between viruses with an affinity of 10−10 and 10−11 M.
Figure 3.

Measurements of VSVFHαHER2-induced syncytia. Confluent TE671 or SKOV3ip.1 cells in 10 cm dishes were infected with VSVFH-αHER2#6-11 (αHER2#6-11). At 3 hours postinfection, the inoculum was removed and replaced with culture media plus methyl cellulose. Pictures of syncytia formed at 48 hpi were taken and syncytia areas were calculated using NIH ImageJ Software. (a) Average area of syncytia plus SD in TE671 cells or (b) in SKOV3ip.1 cells. (c) Representative images of syncytia produced in TE671 (left) or SKOV3ip.1 (right) cells. Nuclei were stained with Hoechst 33342 at a concentration of 0.5 µg/ml. Bar = 200 µm.
Cell killing is dependent on αHER2 receptor affinity
The ability of the chimeric H-scFV protein to trigger syncytia formation might also be correlated with the potential cytotoxic activity of the virus in permissive cells. The viability of TE671 and SKOV3ip.1 cells infected with VSVFH-αHER2#6-11 was measured in vitro (Figure 4). We observed that only viruses with the highest receptor affinity (i.e., VSVFH-αHER2#10 and #11) were capable of killing TE671 cells. On the other hand, the CPE in high-HER-2 expressing SKOV3ip.1 cells started with VSVFH-αHER2#8 (although a higher multiplicity of infection (MOI) of 10 was needed) and continued with the viruses with a higher affinity for HER2.
Figure 4.

Cell killing activity of VSVFH-αHER2. (a) 2 × 105 TE671 (left column) or SKOV3ip.1 (right column) cells were infected with VSVFH-αHER2#6-11 (αHER2#6-11) at a multiplicity of infection (MOI) of 1. At 3 days postinfection, cells were fixed with 5% glutaraldehyde and stained with 0.1% crystal violet. Representative pictures are shown. Bar = 200 µm. (b) 5 × 103 TE671 (left) or SKOV3ip.1 (right) cells per well of a 96-well plate were infected with the same viruses as described above with a MOI of 1 or 10. At 3 days postinfection, cell viability was measured and plotted, considering the cell viability of noninfected cells as 100%. Graphs show the average of three independent experiments plus SD.
VSVFH-αHER2 shows efficacy against ovarian cancer xenografts
To study the oncolytic activity of VSVFH-αHER2 in human ovarian cancer xenografts, SKOV3ip.1 tumors were grown subcutaneously in NCr athymic mice and one single dose of 106 TCID50 units of VSVFH-αHER2#9, #10, or #11 was injected intratumorally. Tumor samples were obtained at intervals thereafter. Infected cells were detected by immunofluorescence staining using a polyclonal antibody against VSV structural proteins, and virus replication in tumors was measured by virus recovery assays and quantitation of infectious viral particles.
At 3 days postinjection, VSV proteins were detected in all the tumor samples from mice treated with VSVFH-αHER2#9, #10, or 11 (Figure 5a). The quantitation of infectious viral particles from the tumor samples (Figure 5b) showed that at day 3 all the viruses were able to replicate and form infectious particles, but viral replication and the number of infectious particles decreased by day 7.
Figure 5.

Antitumor efficacy of VSV-FH and HER2-targeted viruses. Subcutaneous SKOV3ip.1 tumors were implanted in the right flank of nude NCr mice. Once tumors reached a volume of 50 mm3, 1 × 106 TCID50 units of VSVFH-αHER2 (αHER2) #9-11 were intratumorally administrated in 50 µl of OptiMEM. (a) VSV-positive cells at 3 or 7 days post-treatment were detected by immunofluorescence using an anti-VSV polyclonal antibody. (b) Number of infectious particles present in the tumors, at the same days post-treatment, were determined in the tumors of three mice treated with the HER2-targeted viruses. (c) Tumor growth was monitored three times per week. Average of the tumor size of the mice per group (n = 8) was plotted plus standard error of the mean. (d) Survival of mice treated with the indicated viruses or saline (control). Asterisk (*) indicates the groups that are statistically different from saline control group.
The impact of receptor affinity on the antitumor efficacy of VSVFH-αHER2 was also investigated; for this, tumor size was monitored in animals after one single dose of VSVFH-αHER2, or control saline (Figure 5c). The differences in tumor size between treatments at day 24 were analyzed using JMP version 9 statistics software (2010 SAS Institute, Cary, NC) as previously described.19 The differences in tumor size at this point were significant for the groups treated with all the different viruses (P < 0.0001) with respect to the saline group. Comparison of survival curves showed an increase in mice survival for groups treated with high-receptor affinity VSVFH-αHER2 (i.e., #9, #10, #11) compared to the saline group (P ≤ 0.01). There was no significant difference in antitumor potency between these three viruses.
Discussion
VSV-FH is a recently developed hybrid MV-VSV virus that has been shown to possess potent killing activity against cancer cells and a reduced neurovirulence compared to parental VSV.9 In order to increase its specificity and safety, we decided to investigate if it is possible to retarget VSV-FH by the insertion of a scFV at the C-terminal of MV-H as previously reported for measles virus.12,20 We chose to retarget the virus to HER-2, as this protein is overexpressed in a number of different tumor types, including ovarian tumors.2 Several oncolytic viruses such as Herpes simplex virus,21 measles virus,15 and VSV expressing the Sindbis virus glycoprotein22,23 have also been targeted to HER2 by the inclusion of a scFV in the surface glycoprotein. These viruses have shown to be effective against gliomas,24,25 ovarian cancer,16 and mammary tumors.26,27
A panel of replication-competent HER2-targeted VSV-FH viruses displaying αHER-2 scFV with different affinities for the same epitope was rescued and characterized in vitro and in vivo. None of the rescued VSVFH-αHER2 viruses were able to infect CHO cells genetically engineered to express CD46- or SLAM, as the MV-H gene contained point mutations that specifically blocked interaction with these cellular receptors.28,29 From the in vitro characterization studies, we noted that a threshold level of scFv-receptor affinity was needed for VSVFH-αHER2 to achieve intercellular fusion and killing in HER2-positive cancer cells. Syncytia were seen at a Kd of 10−8 M although the syncytia produced by VSVFH-αHER2#8-infected HER-2-positive cells were small in size, and a high MOI was need to kill the SKOV3ip.1 cells. In low HER-2 expressing TE671 cells, a higher receptor affinity was required to infect and fuse them, and syncytia formation started only with the VSVFH-αHER2#9 virus. We noted similar results for MV retargeted to HER-2 using the same panel of HER-2-specific scFV, where the syncytia formation and cell killing of SKOV3ip.1 cells started with viruses possessing a Kd for HER2 of 10−8 M.15
However, there are important differences in the infection of HER-2 targeted MV versus VSV-FH in vitro. MV-αHER2#6 was able to infect SKOV3ip.1 cells15 while the current study shows that VSVFH-αHER2#6 was not able to do so. This is likely due to the significantly lower amounts of FH incorporated into the VSV virion compared to MV as shown in our previous study.9
Studies undertaken in vivo showed no difference in mice survival among the groups treated with VSVFHαHER2 #9, 10, or 11, indicating that similar to the results observed in vitro, a certain level of affinity for HER-2 is needed to promote cell infection and killing. Affinity above this threshold level did not enhance the oncolytic activity of VSVFH-αHER2. Similar results were observed when mice bearing human ovarian cancer xenografts were treated with HER2-targeted MV. However, in those experiments, all six retargeted MV-αHER2 (i.e., 10−6 to 10−11 M) significantly increased survival of mice when compared to saline-treated mice.16 In contrast, VSVFHαHER2 #6 which could not infect SKOV3ip.1 cells in vitro, and has no antitumor activity against SKOV3ip.1 tumors (Ayala-Breton and Peng, unpublished data).
In conclusion, we have created and characterized a panel of HER2-retargeted VSV/Measles hybrid viruses with different binding affinities to HER2 receptor. A receptor affinity of at least 10−9 M is necessary to allow syncytia formation in high HER2 expressing SKOV3ip.1 cells, and to have significant in vivo oncolytic activity against these human ovarian cancer xenografts. Overall, the high affinity VSVFH-αHER2 viruses might be suitable candidates to explore for clinical evaluation in ovarian cancer.
Materials and Methods
Cell culture
Baby Hamster Kidney Cells and VeroαHis cells were cultured in Dulbecco’s modified Eagle medium containing 5% fetal bovine serum and 1% penicillin-streptomycin. TE671 and CHO cells were maintained in Dulbecco’s modified Eagle medium containing 10% (fetal bovine serum) and 1% penicillin-streptomycin. SKOV3ip.1 cells were cultured in α-minimum essential medium containing 20% fetal bovine serum and 1% penicillin-streptomycin. CHO-HER2 and CHO-CD46 were maintained in Dulbecco’s modified Eagle medium containing 10% fetal bovine serum and 1% penicillin-streptomycin. CHO-HER2 cells were maintained as described previously.15
Generation of HER2-retargeted VSV/MV hybrid displaying different affinity mutants
The NotI restriction site in the plasmid MC11-VSV-eGFP30 was removed using the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent technologies, Santa Clara, CA) with the primers NotI-mut-F/R described in Table 2. MluI and AvrII restriction sites were introduced into the beginning and end, respectively, of MVHaa-scFVαPSMA31 by polymerase chain reaction (primers Haa-F/R, Table 2) and cloned into the plasmid pCR-Blunt II-Topo (Invitrogen, Carlsbad, CA). MVHaa-scFVαPSMA was cut and cloned into the MluI-AvrII restriction sites of plasmid MC11-VSV-eGFP. MV-F was amplified from pCGF with added SphI restriction sites at the beginning and end of the gene (primers MVF-F/R, Table 2). Then, MV-F was digested with SphI and cloned into MC11-VSV-∆G-MVHaaαPSMA-eGFP. The new plasmid, containing MV-F and retargeted MV-H, was digested with SfiI and NotI to remove the scFVαPSMA, and replaced with the scFVαHER2 from the library of plasmids containing scFVαHER2 with different receptor affinities (Table 1). Replication-competent viruses were rescued as previously described.9 All the viruses were grown and titered in VeroαHis cells.
Table 2. List of primers used to construct VSVFH-αHER2 plasmids.
| Primer | Sequence (5′–3′) |
|---|---|
| NotI-mut-Fa | CTCGGATGGCTAAGGGAGAGCCAGCtagCGCTTCGAGCAGACATG |
| NotI-mut-Ra | CATGTCTGCTCGAAGCGctaGCTGGCTCTCCCTTAGCCATCCGAG |
| Haa-F | ACGCGTAGATCATCGATAATGTCACCACAACGAGACCGGAT |
| Haa-R | CCTAGGATTGCTGTTAGTTTTTTTCATACCTGCAGGCCTA GTTTTCACTA TCAGTG |
| MVF-F | GCATGCTATGAAAAAAACTAACAGATATCACACCGGGAAT CCCAGAATCA |
| MVF-R | TCGATCAGTG GCTCGAGGCATGCCTACCGATATTGTTCGG CCAGAGGGA |
Mutated nucleotides are indicated in lower case.
Immunoblotting
Viruses from the supernatant of VSVFH- and VSVFHαHER2-infected VeroαHis cells were purified by ultrafiltration using Amicon Ultra-15 Centrifugal Filter Units 100 kDa NMWL (EMD Millipore, Billerica, MA). Equivalent amounts of purified infectious viruses (1.5 × 106 TCID50) were mixed with an equal volume of Laemmli sample buffer (BIO-RAD, Hercules, CA). Samples were incubated for 5 minutes at 80 °C and samples were fractionated by SDS–PAGE through a 10% Tris-HCl gel and blotted to a polyvinylidene difluoride membrane. VSV-M and MV-H proteins were detected as previously described.9
Quantification of syncytia sizes
Confluent TE671 or SKOV3ip.1 cells in 10-cm dishes were infected at a MOI of 0.01. After 3 hours of incubation, the inoculum was removed and replaced with complete medium with 1% methyl cellulose. At 48 hpi photographs of the syncytia were taken and the size determined using the NIH ImageJ software.32
Virus cytotoxicity assays
5 × 103 TE671 or SKOV3ip.1 cells were seeded per well of a 96-well plate. The next day cells were infected with the indicated viruses at a MOI of 1 or 10. At 3 days postinfection, cell viability was determined using CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI) following the manufacturer protocol.
Animal studies
5 × 106 SKOV3ip.1 cells were implanted in the right flank of nude NCr mice (Harlan, Indianapolis, IN). When tumors reached a volume of 50 mm3, mice were treated with one intratumoral injection of 50 µl of saline (control) or 1 × 106 TCID50 of the indicated viruses in 50 µl of OptiMEM. For antitumor efficacy, tumor size was measured at the indicated days post-treatment, mice were euthanized when tumor reached a volume of, or bigger than 2,000 mm3. Other sets of mice were euthanized at days 3 and 7 post-treatment, tumors were removed and one part stored in optimal cutting temperature compound for histological analysis and another part was frozen for TCID50 calculations.
Acknowledgments
This work was funded by a grant from the NIH/NCI (R01CA129193). N.W. was supported by a training fellowship grant and the Mahidol University (Thai Royal Golden Jubilee) PhD Scholarship.
References
- Iqbal, N and Iqbal, N (2014). Human Epidermal Growth Factor Receptor 2 (HER2) in Cancers: Overexpression and Therapeutic Implications. Mol Biol Int 2014: 852748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English, DP, Roque, DM and Santin, AD (2013). HER2 expression beyond breast cancer: therapeutic implications for gynecologic malignancies. Mol Diagn Ther 17: 85–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai, W, Mahato, R and Cheng, K (2010). The role of HER2 in cancer therapy and targeted drug delivery. J Control Release 146: 264–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradishar, WJ (2013). Emerging approaches for treating HER2-positive metastatic breast cancer beyond trastuzumab. Ann Oncol 24: 2492–2500. [DOI] [PubMed] [Google Scholar]
- Bell, J and McFadden, G (2014). Viruses for tumor therapy. Cell Host Microbe 15: 260–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell, SJ, Peng, KW and Bell, JC (2012). Oncolytic virotherapy. Nat Biotechnol 30: 658–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammayappan, A, Peng, KW and Russell, SJ (2013). Characteristics of oncolytic vesicular stomatitis virus displaying tumor-targeting ligands. J Virol 87: 13543–13555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelshtein, D, Werman, A, Novick, D, Barak, S and Rubinstein, M (2013). LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc Natl Acad Sci USA 110: 7306–7311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala-Breton, C, Suksanpaisan, L, Mader, EK, Russell, SJ and Peng, KW (2013). Amalgamating oncolytic viruses to enhance their safety, consolidate their killing mechanisms, and accelerate their spread. Mol Ther 21: 1930–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrkic, B, Odermatt, B, Klein, MA, Billeter, MA, Pavlovic, J and Cattaneo, R (2000). Lymphatic dissemination and comparative pathology of recombinant measles viruses in genetically modified mice. J Virol 74: 1364–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roscic-Mrkic, B, Schwendener, RA, Odermatt, B, Zuniga, A, Pavlovic, J, Billeter, MA et al. (2001). Roles of macrophages in measles virus infection of genetically modified mice. J Virol 75: 3343–3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura, T, Peng, KW, Harvey, M, Greiner, S, Lorimer, IA, James, CD et al. (2005). Rescue and propagation of fully retargeted oncolytic measles viruses. Nat Biotechnol 23: 209–214. [DOI] [PubMed] [Google Scholar]
- Ayala-Breton, C, Barber, GN, Russell, SJ and Peng, KW (2012). Retargeting vesicular stomatitis virus using measles virus envelope glycoproteins. Hum Gene Ther 23: 484–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paraskevakou, G, Allen, C, Nakamura, T, Zollman, P, James, CD, Peng, KW et al. (2007). Epidermal growth factor receptor (EGFR)-retargeted measles virus strains effectively target EGFR- or EGFRvIII expressing gliomas. Mol Ther 15: 677–686. [DOI] [PubMed] [Google Scholar]
- Hasegawa, K, Hu, C, Nakamura, T, Marks, JD, Russell, SJ and Peng, KW (2007). Affinity thresholds for membrane fusion triggering by viral glycoproteins. J Virol 81: 13149–13157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suksanpaisan, L, Russell, SJ and Peng, KW (2014). High scFv-receptor affinity does not enhance the antitumor activity of HER2-retargeted measles virus. Cancer Gene Ther 21: 256–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala-Breton, C, Russell, LO, Russell, SJ and Peng, KW (2014). Faster replication and higher expression levels of viral glycoproteins give the vesicular stomatitis virus/measles virus hybrid VSV-FH a growth advantage over measles virus. J Virol 88: 8332–8339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derycke, MS, Pambuccian, SE, Gilks, CB, Kalloger, SE, Ghidouche, A, Lopez, M et al. (2010). Nectin 4 overexpression in ovarian cancer tissues and serum: potential role as a serum biomarker. Am J Clin Pathol 134: 835–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, YP, Wang, J, Avanzato, VA, Bakkum-Gamez, JN, Russell, SJ, Bell, JC et al. (2014). Oncolytic vaccinia virotherapy for endometrial cancer. Gynecol Oncol 132: 722–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadac, EM, Peng, KW, Nakamura, T and Russell, SJ (2004). Reengineering paramyxovirus tropism. Virology 329: 217–225. [DOI] [PubMed] [Google Scholar]
- Menotti, L, Cerretani, A and Campadelli-Fiume, G (2006). A herpes simplex virus recombinant that exhibits a single-chain antibody to HER2/neu enters cells through the mammary tumor receptor, independently of the gD receptors. J Virol 80: 5531–5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman, I, Whitaker-Dowling, P, Gao, Y, Griffin, JA and Watkins, SC (2003). Vesicular stomatitis virus expressing a chimeric Sindbis glycoprotein containing an Fc antibody binding domain targets to Her2/neu overexpressing breast cancer cells. Virology 316: 337–347. [DOI] [PubMed] [Google Scholar]
- Bergman, I, Whitaker-Dowling, P, Gao, Y and Griffin, JA (2004). Preferential targeting of vesicular stomatitis virus to breast cancer cells. Virology 330: 24–33. [DOI] [PubMed] [Google Scholar]
- Reisoli, E, Gambini, E, Appolloni, I, Gatta, V, Barilari, M, Menotti, L et al. (2012). Efficacy of HER2 retargeted herpes simplex virus as therapy for high-grade glioma in immunocompetent mice. Cancer Gene Ther 19: 788–795. [DOI] [PubMed] [Google Scholar]
- Gambini, E, Reisoli, E, Appolloni, I, Gatta, V, Campadelli-Fiume, G, Menotti, L et al. (2012). Replication-competent herpes simplex virus retargeted to HER2 as therapy for high-grade glioma. Mol Ther 20: 994–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman, I, Griffin, JA, Gao, Y and Whitaker-Dowling, P (2007). Treatment of implanted mammary tumors with recombinant vesicular stomatitis virus targeted to Her2/neu. Int J Cancer 121: 425–430. [DOI] [PubMed] [Google Scholar]
- Gao, Y, Whitaker-Dowling, P, Griffin, JA, Barmada, MA and Bergman, I (2009). Recombinant vesicular stomatitis virus targeted to Her2/neu combined with anti-CTLA4 antibody eliminates implanted mammary tumors. Cancer Gene Ther 16: 44–52. [DOI] [PubMed] [Google Scholar]
- Bartz, R, Brinckmann, U, Dunster, LM, Rima, B, Ter Meulen, V and Schneider-Schaulies, J (1996). Mapping amino acids of the measles virus hemagglutinin responsible for receptor (CD46) downregulation. Virology 224: 334–337. [DOI] [PubMed] [Google Scholar]
- Vongpunsawad, S, Oezgun, N, Braun, W and Cattaneo, R (2004). Selectively receptor-blind measles viruses: Identification of residues necessary for SLAM- or CD46-induced fusion and their localization on a new hemagglutinin structural model. J Virol 78: 302–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammayappan, A, Nace, R, Peng, KW and Russell, SJ (2013). Neuroattenuation of vesicular stomatitis virus through picornaviral internal ribosome entry sites. J Virol 87: 3217–3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura, T, Peng, KW, Vongpunsawad, S, Harvey, M, Mizuguchi, H, Hayakawa, T et al. (2004). Antibody-targeted cell fusion. Nat Biotechnol 22: 331–336. [DOI] [PubMed] [Google Scholar]
- Schneider, CA, Rasband, WS and Eliceiri, KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]