

Abstract
The use of engineered T cells in adoptive transfer therapies has shown significant promise in treating hematological cancers. However, successes treating solid tumors are much less prevalent. Oncolytic viruses (OVs) have the capacity to induce specific lysis of tumor cells and indirectly impact tumor growth via vascular shutdown. These viruses bear natural abilities to associate with lymphocytes upon systemic administration, but therapeutic doses must be very high in order to evade antibodies and other components of the immune system. As T cells readily circulate through the body, using these cells to deliver OVs directly to tumors may provide an ideal combination. Our studies demonstrate that loading chimeric antigen receptor–engineered T cells with low doses of virus does not impact receptor expression or function in either murine or human T cells. Engineered T cells can deposit virus onto a variety of tumor targets, which can enhance the tumoricidal activity of the combination treatment. This concept appears to be broadly applicable, as we observed similar results using murine or human T cells, loaded with either RNA or DNA viruses. Overall, loading of engineered T cells with OVs represents a novel combination therapy that may increase the efficacy of both treatments.
Introduction
Oncolytic viruses (OVs) are capable of selectively infecting, replicating in, and killing tumor cells, while avoiding healthy tissues.1 In addition, these viruses have been shown to induce robust immune responses, potentiating the antitumor response within a host.2,3 Vesicular stomatitis virus (VSV) has been found to bear these properties.2,4 Mutations in the M protein (VSVΔM51) enhance the interferon sensitivity of this virus, significantly increasing both its safety and its tumor tropism.2,4,5 Vaccinia virus (VV) has also been tested extensively in preclinical models and clinical trials in which systemic treatment with the virus was shown to be safe.6,7 We are particularly interested in a recombinant VV containing deletions of the thymidine kinase and viral growth factor genes, resulting in a “double-deleted” vaccina virus (vvDD).8 This recombinant virus shows enhanced tumor tropism, with limited replication within resting cells.8
To this point, clinical trials of systemic VV have employed high doses of virus, ranging from 1 × 105 to 3 × 107 PFU/kg per patient.1,7 The use of VSV in clinical trials has been limited thus far, though animal studies typically employ doses greater than 5 × 108 PFU per mouse, suggesting human dosages would also be quite high.2,4,9,10 It is speculated that such high doses are required when delivering the virus intravenously because multiple blood-borne defense mechanisms can eliminate the virus, such as complement, antibodies, and immune cells, so the dose must saturate these defense mechanisms to enable delivery of virus to the tumor.11
Adoptive cell transfer (ACT) therapies have emerged as effective treatments for certain types of cancer, including the use of tumor-infiltrating lymphocytes for melanoma and engineered T cells for hematological malignancies.12–16 As evidenced by the successes in ACT studies, adoptive transfer of T cells results in T cells migrating to the tumor site in order to perform their antitumor functions. Interestingly, OVs have been found to naturally associate with circulating lymphocytes such as B cells.17 It is therefore attractive to consider loading lymphocytes with OVs prior to adoptive cell therapy. In this way, the adoptively transferred T cells loaded with OVs should be capable of delivering the OV to the tumor site. Indeed, previous reports have shown that transgenic murine T cells can be used to deliver OVs to established tumors and that this combination can result in tumor rejection.18,19 Loading VSV onto T cells protects the virus from neutralizing antibodies, while retaining its antitumor efficacy.20,21 Similarly, VV can be effectively carried and deposited within tumors using cytokine-induced killer cells, leading again to antitumor efficacy.22,23 With the promising results observed in clinical trials of adoptive transfer of T cells engineered with chimeric antigen receptors (CARs), we were interested in determining whether CAR-engineered T cells could be loaded with OV and maintain their antitumor function, effectively creating dual-pronged antitumor agent. In this article, we demonstrate that both VSVΔM51 and vvDD can be successfully loaded onto murine and human CAR-T cells without affecting CAR expression, viability, or functionality. Our data further show that OV-loaded CAR-T cells are capable of depositing virus onto tumor targets and that this combination has the potential to enhance the efficacy of each of the two approaches. These data provide the basis for combining these two therapies for future therapeutic applications.
Results
OV loading of CAR-T cells does not impact CAR expression
We first sought to determine the feasibility of combining CAR-T cells with OV loading as well as determine the optimal viral multiplicity of infection (MOI) for use in our studies. Murine T cells engineered with a CAR-’ve control retrovirus (to avoid potential effects of the CAR) were loaded with either VSVΔM51-GFP or vvDD-GFP at MOI values of 0.3, 1, and 3 (Figure 1a). Our preliminary experiments found that loading engineered T cells with an MOI of 3 resulted in the highest level of both VSVΔM51-GFP and vvDD-GFP deposition on tumor targets over the lower MOI described above, as well as increased reproducibility between replicates (Figure 1a). We observed the same outcome when testing human T cells engineered with a CAR-’ve lentivirus, with MOI of 3 showing the highest virus deposition (Figure 1b). Based on these results, all subsequent experiments utilized this MOI for all T cells and viruses.
Figure 1.
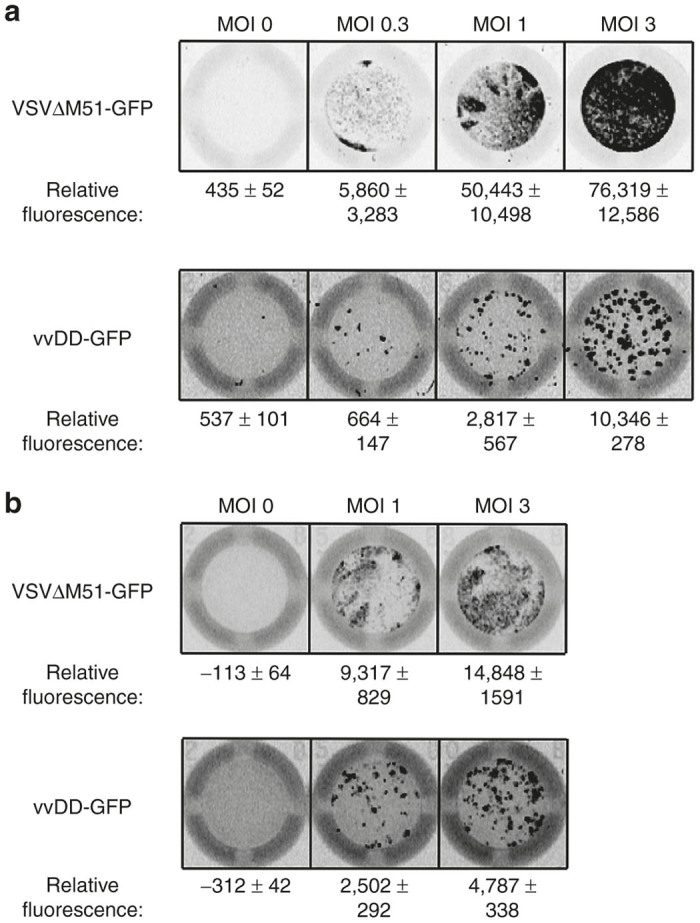
Dose titration of OV-loading. (a) Murine T cells or (b) human T cells engineered with CAR-negative (CAR-’ve) vectors were loaded with the indicated MOI of either VSVΔM51-GFP or vvDD-GFP, washed, and incubated with D2F2 tumor cells to test OV deposition. Relative fluorescence determined by Image Quant software and is presented as mean ± SEM from at least 2–3 replicates, normalized to tumor-only wells. CAR, chimeric antigen receptor; MOI, multiplicity of infection; OV, oncolytic virus; VSV, vesicular stomatitis virus; vvDD, “double-deleted” vaccina virus.
We next looked to test whether OV loading had any effect on the T cells. Engineered T cells were loaded with an MOI of 3 of either VSVΔM51-GFP or vvDD-GFP. Following washing, cells were incubated overnight and analyzed for virus replication, or changes in CAR expression or functionality. A schematic of the HER2-CAR used in these studies can be found in Supplementary Figure S1. We examined cells for virus infection via GFP production by flow cytometry and found that a small fraction of murine T cells were actually infected with either VSVΔM51-GFP or vvDD-GFP (Figure 2a,b; Supplementary Figure S2a) with similar levels of GFP expression in both CD4+ and CD8+ populations (Supplementary Figure S2b). We went on to characterize the impact of OV loading on CAR expression and found there to be no difference in the level of CAR surface expression after loading with either OV (Figure 2c; Supplementary Figure S1b).
Figure 2.

OV-loading does not impact CAR expression. (a–c) Murine T cells or (d–f) human T cells engineered with either CAR-’ve or HER2-CAR vectors were loaded with VSVΔM51-GFP or vvDD-GFP, washed, and incubated overnight before analysis. (a–b) Murine T cells show minimal VSVΔM51-GFP or vvDD-GFP replication after loading, as measured by GFP+ flow cytometry signal. Data are presented as representative plots or ± SEM from three independent experiments. *P < 0.05, n.s. = not significant. (c) CAR expression was evaluated after OV loading via staining with HER2-Fc Chimera and visualized by flow cytometry (gated on CD8+ cells). Results are presented as mean ± SEM from three independent experiments. (d,e) Human T cells show low levels of VSVΔM51-GFP or vvDD-GFP replication after loading. Data are presented as representative plots or ± SEM from two independent experiments. *P < 0.05; n.s. = not significant. (f) CAR expression was unaffected by OV loading, evaluated as described above (gated on CD8+ cells). Results are presented as mean ± SEM from two independent experiments. CAR, chimeric antigen receptor; MOI, multiplicity of infection; OV, oncolytic virus; VSV, vesicular stomatitis virus; vvDD, “double-deleted” vaccina virus.
We next tested the ability of human CAR-T cells to be loaded with OV. We engineered human T cells with lentiviruses containing the human HER2-CAR (or a CAR-’ve control) and loaded them with VSVΔM51-GFP or vvDD-GFP. Interestingly, we observed significantly higher levels of vvDD-GFP replication in both CAR-’ve and HER2-CAR-T cells, reaching around 12% GFP+ cells at 72 hours after loading (Figures 2d,e and 3b; Supplementary Figure S2b). In contrast, VSVΔM51-GFP-loaded cells never exceeded 2% GFP+ cells over 7 days post-loading (Figures 2d,e and 3b). The increase in virus replication in human CAR-T cells did not result in any changes in T-cell viability for at least 5 days post-loading with OV; the only significant decrease in viability was observed 7 days after vvDD-GFP loading (Figure 3a). In addition, akin to the murine T cells, OV loading did not cause any changes in CAR expression on human T cells (Figure 2f; Supplementary Figure S1b,c). CAR expression was maintained on both CD4+ or CD8+ T cells for at least 7 days post-loading (Supplementary Figure S1; Figure 3c,d). Virus infection (as indicated by GFP+ signal) was not restricted to either CAR+ or CAR− subsets, as we could detect equal (if not higher on CAR+) levels of GFP on both populations of cells, indicating that cells were indiscriminately loaded with OV (Supplementary Table S1). GFP expression was measurable in both CD4+ and CD8+ T cells (Supplementary Figure S2d). To confirm that the OVs were indeed being carried by or loaded onto the CAR+ T-cell population, we loaded HER2-CAR–engineered human T cells with each OV and purified CD3+ CAR+ cells using flow cytometry (Supplementary Figure S4). Virus titration assays confirmed that the CD3+ CAR+ T cells were loaded with replication-competent virus (Supplementary Table S3). Overall, this data suggests that OV loading of CAR+ T cells does not impact CAR surface expression on engineered T cells.
Figure 3.

OV-loading of CAR-T cells shows minimal changes in T cell viability, CAR expression, or functionality over time. Human HER2-CAR-T cells or CAR-’ve controls were loaded with an MOI of 3 of either VSVΔM51-GFP or vvDD-GFP and monitored for 7 days post-loading for (a) viability, (b) virus replication, (c) CAR expression on CD8+ T cells, (d) CAR expression on CD4+ T cells, or cytokine production (IFNγ and/or TNFα positive cells) by (e) CD4+ or (f) CD8+ T cells via flow cytometry on the days indicated. Data are presented as mean ± SEM from two independent T-cell donors. *P < 0.05, **P < 0.01, ***P < 0.001, n.s. = not significant. CAR, chimeric antigen receptor; MOI, multiplicity of infection; OV, oncolytic virus; VSV, vesicular stomatitis virus; vvDD, “double-deleted” vaccina virus.
CAR-T cells show no functional impairments following OV loading
We next sought to determine whether CAR-T-cell functionality was affected by loading with OV. We loaded murine CAR-T cells with OV as described and stimulated with HER2-Fc for 4 hours, followed by flow cytometry for cytokine production. Loading of either VSV or vvDD onto murine T cells resulted in minimal decreases in overall cytokine production that was not significant (Figure 4a). Responding T cells were capable of producing both interferon γ (IFNγ) and tumor necrosis factor α (TNFα) following CAR stimulation (Figure 4a). To evaluate the ability of CAR-T cells to kill target cells with and without OV loading, varying ratios of T cells to tumor targets were co-cultured for 6 hours, minimizing the potential for virus-mediated killing within the short incubation period. Indeed, OV loading did not alter the CAR-T cell’s ability to selectively kill their HER2+ tumor targets (D2F2/E2) while sparing the HER2-’ve D2F2 cells (Figure 4b).
Figure 4.

OV-loading does not impact CAR-T cell function. (a,b) Murine T cells or (c,d) human T cells engineered with CAR-’ve or HER2-CAR vectors were loaded with VSVΔM51-GFP or vvDD-GFP, washed, and incubated overnight before functional testing. (a,c) OV-loaded T cells were stimulated for 4 hours with plate-bound HER2-Fc in the presence of brefeldin A. Cytokine production was equivalent between mock or OV-loaded T cells (gated on CD8+ cells). (a) Data are representative flow plots of two independent experiments, or (c) pooled from at least two independent experiments. (b,d) Mock or OV-loaded CAR-T cells were co-cultured with D2F2 or D2F2/E2 tumor cells for 6 hours. After washing off T cells, tumor cell viability was assessed via alamarBlue assay. Data are representative of two independent experiments for each murine or human T cells. CAR, chimeric antigen receptor; E:T, effector to target ratio; MOI, multiplicity of infection; n.s., not significant; OV, oncolytic virus; VSV, vesicular stomatitis virus; vvDD, “double-deleted” vaccina virus.
For the most part, OV loading of human CAR-T cells did not impact their ability to produce cytokine in response to CAR stimulation (Figures 3e and 4c). Importantly, OV-loaded CAR-T cells retained their ability to specifically kill HER2+ tumor targets, which suggests that any cytokine impairment may not be critical to their cytolytic functions (Figure 4d). Cytokine production from CD4+ T cells was only transiently decreased following loading with VSVΔM51-GFP and recovered within 48 hours of OV loading (Figure 3e). The decrease in CD4+ cytokine production at these early time points was mostly attributed to altered TNFα single producers, with minimal change in IFNγ production (Supplementary Figure S3a,b). Loading with VSVΔM51-GFP did not impact cytokine production from CD8+ T cells (Figure 3f; Supplementary Figure S3c). As there were so few GFP+ cells following VSV loading, we were unable to evaluate the impact of VSV infection directly on the T-cell functionality. Virus infection (as indicated by GFP+) did impact the level of cytokine production from both CD4+ and CD8+ vvDD-loaded HER2-CAR T cells; however, cells were still able to respond to target antigen through the CAR and produce cytokines (Supplementary Table S2 and Figure S3b,c). CD8+ T-cell–mediated production of IFNγ and TNFα was significantly decreased in the days following loading with vvDD-GFP (Figure 3f). This coincided with increasing virus replication within the CAR-T cells (Figure 3b) and was attributed to a decreased functionality within the GFP+ population (Supplementary Table S2). Taken together, our data suggests that engineered T cells can be loaded with OV without causing functional impairment to the CAR-T cells.
CAR-T cells can successfully transfer OVs to tumor cells
To determine if OV-loaded CAR-T cells could effectively deposit virus on tumor targets, we co-cultured CAR-’ve or HER2-CAR-T cells loaded with either VSVΔM51-GFP or vvDD-GFP at a 1:1 ratio with D2F2 breast tumor targets, which do not carry the target for the CAR (human HER-2) and thus will not be affected by CAR signaling. After 24 hours, virus replication was evaluated as GFP fluorescence using the Typhoon Imager (Figure 5). We observed that both CAR-’ve and HER2-CAR-T cells could successfully transfer VSVΔM51-GFP to tumor targets with the same level of efficiency (Figure 5a,c). This was true for both murine and human CAR-T cells, which displayed similar patterns of replication within the monolayer (Figure 5a,c left panels). These replication patterns indicated that the virus was infecting specific foci and spreading throughout the monolayer (Figure 5). This furthers the notion that the virus itself is replicating within the tumor cells as opposed to simply infecting cells at the 1:1 culture ratio.
Figure 5.
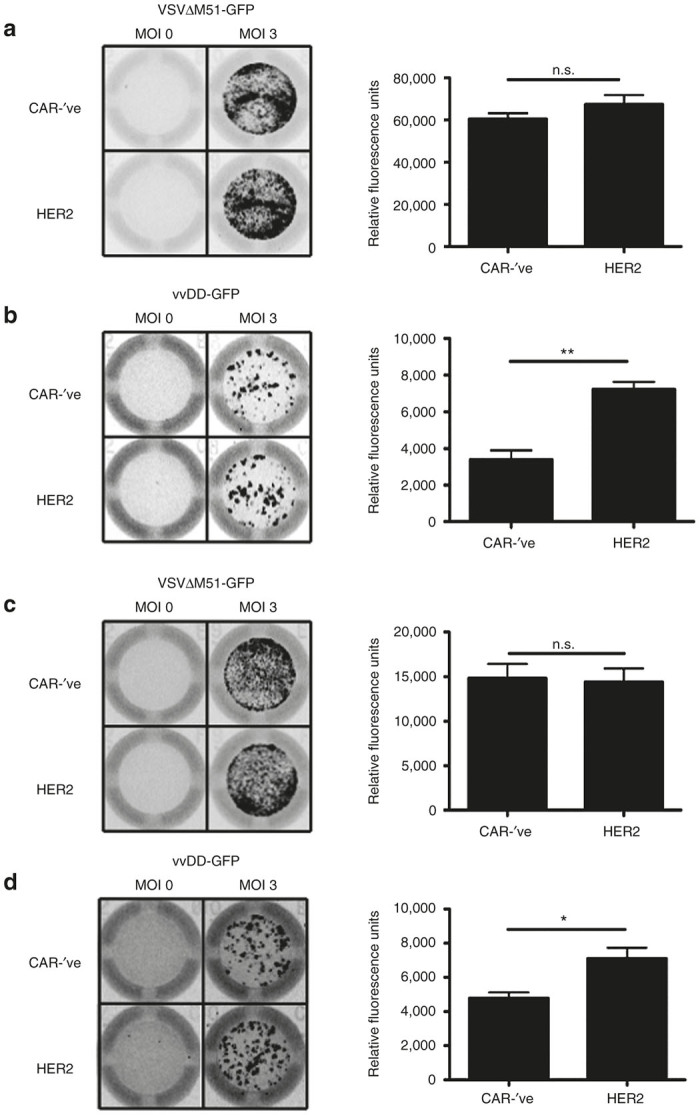
OV-loaded CAR-T cells can deposit OV onto tumor targets. (a,b) Murine or (c,d) human CAR-’ve or HER2-CAR T cells were loaded with MOI of 0 or 3 of (a,c) VSVΔM51-GFP or (b,d) vvDD-GFP and co-cultured with D2F2 tumor targets for 24 hours. Virus replication was visualized using the Typhoon Imager to detect GFP signal (left panels). The level of virus replication was quantified using ImageQuant (right panels). Quantification is expressed as mean ± SEM. *P < 0.05, **P < 0.01, n.s. = not significant. Data is representative of 2–3 independent experiments performed in triplicate for each murine and human cells, normalized to tumor-only wells. CAR, chimeric antigen receptor; MOI, multiplicity of infection; n.s., not significant; OV, oncolytic virus; VSV, vesicular stomatitis virus; vvDD, “double-deleted” vaccina virus.
We also observed successful delivery of vvDD-GFP to tumor targets (Figure 5b,d). vvDD-GFP deposited by either CAR-’ve or HER2-CAR-T cells revealed similar patterns of replication within the wells, again showing specific foci of infection within the cell monolayer (Figure 5b,d, left panels). Similar patterns of infection/replication within the target cells were observed regardless of whether CAR-’ve or CAR+ cells were used, indicating that replication is in general similar between these cells. Overall, our data suggests that CAR-T cells can function as effective vehicles for OV transport.
Cognate interaction between CAR-T cell and tumor does not impact ability to deposit OV
As CAR-T cells produce cytokines such as IFN-γ following CAR ligation, which could impact virus replication, we wanted to determine whether CAR activation would negatively impact OV-deposition onto tumor cells. We co-cultured VSVΔM51-GFP or vvDD-GFP loaded CAR-’ve or HER2-CAR-T cells at a 1:1 ratio with the HER2+ D2F2/E2 cell line for 24 hours and evaluated virus replication by GFP production. We observed no differences in the replication of VSVΔM51-GFP in the presence of cognate interaction between the CAR and the tumor target using either murine or human CAR-T cells (Figure 6a,c). While there appeared to be a trend towards a decreased virus load in the HER2-CAR-T cell groups (Figure 6, right panels), the effect of HER2-CAR-mediated killing of target cells must also be considered, as this reduces the number of target cells available to be infected. With respect to vvDD-GFP replication, there appears to be a slight reduction in the viral replication capacity when comparing the vvDD-GFP replication on HER2-negative targets (Figure 5b,d) to the replication within HER2-positive targets (Figure 6b,d). However, the HER2-CAR T cells may be killing the HER2-positive target cells prior to analysis of OV replication, making it difficult to determine whether this is an impairment of virus infection, or an increase in tumor cell death. Taken together, our data shows that CAR ligation does not significantly impede the ability of OV-loaded T cells to deposit virus or for the virus to replicate in tumor targets.
Figure 6.
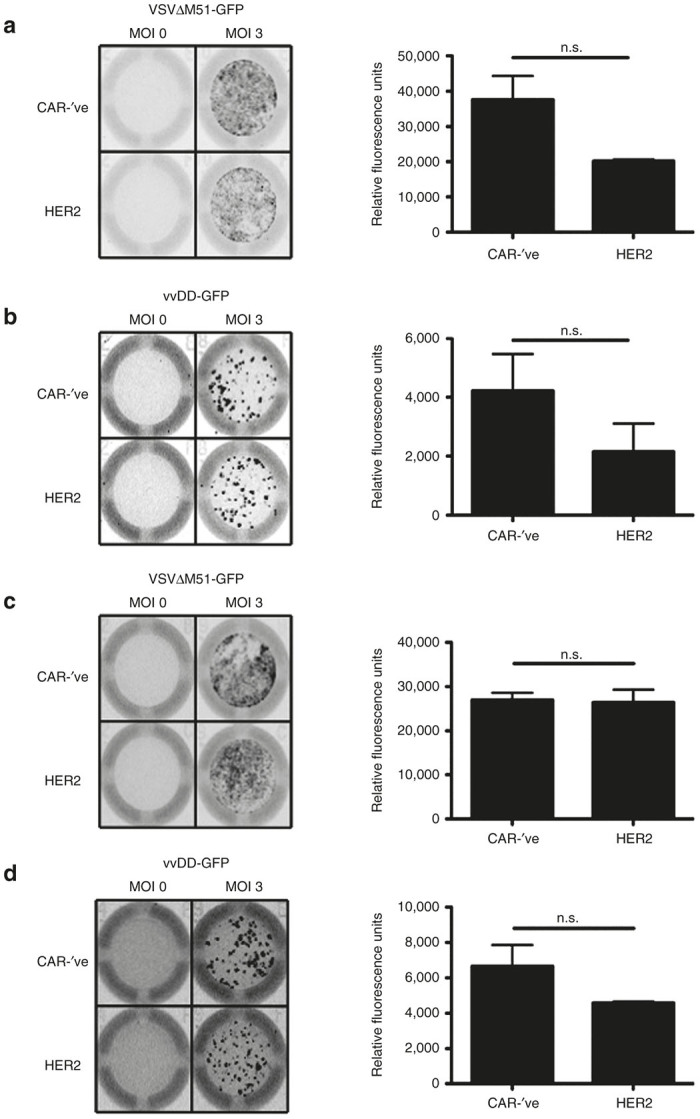
Cognate interaction does not impair CAR-T cell ability to transfer OV. (a,b) Murine or (c,d) human CAR-’ve or HER2-CAR-T cells were loaded with MOI of 0 or 3 of (a,c) VSVΔM51-GFP or (b,d) vvDD-GFP and co-cultured with D2F2/E2 tumor targets for 24 hours. Virus replication was visualized using the Typhoon to detect GFP signal (left panels). Virus replication was quantified using ImageQuant (right panels). Quantification is expressed as mean ± SEM. *P < 0.05, **P < 0.01, n.s. = not significant. Data is representative of 2–3 independent experiments performed in triplicate for each murine and human cells, normalized to tumor-only wells. CAR, chimeric antigen receptor; MOI, multiplicity of infection; OV, oncolytic virus; VSV, vesicular stomatitis virus; vvDD, “double-deleted” vaccina virus.
OV loading of CAR-T cells can enhance tumor cell killing
To evaluate the ability of OV-loaded CAR-T cells to enhance the tumor killing relative to CAR-T cells alone, we evaluated in vitro killing of three different HER-2-positive tumor cell lines. The three lines expressed HER2 to varying degrees (Figure 7a), with A549 showing the lowest level of HER-2 expression, D2F2/E2 displaying the highest level of expression, and T47D revealing an intermediate level of expression. These cell lines are also susceptible to both VSVΔM51-GFP and vvDD-GFP replication, as we could readily detect virus replication in these cells following co-culture with OV-loaded CAR-T cells (Figure 7b). The three cell lines display similar levels of susceptibility to VSVΔM51-GFP infection following deposition by OV-loaded CAR-T cells (Figure 7b, middle column). In contrast, there appeared to be greater difference in susceptibility to infection following deposition of vvDD by the CAR-T cells (Figure 7b, right column), as the A549 cells seem to support greater vvDD replication than the other lines. The 3 cell lines displayed differential sensitivity to killing by CAR-T cells where the A549 cells were relatively resistant to killing, the T47D cells were most sensitive to killing and the D2F2/E2 cells displayed intermediate sensitivity (Figure 7c–e, open circles). The combined differences in sensitivity to CAR-mediated killing and OV infection provide a good spectrum of tumor cells in which to evaluate the efficacy of combining CAR-T cells and OV-loading. The A549 lung adenocarcinoma proved very sensitive to VSV-mediated oncolysis following deposition by T cells but did not reveal any combinatorial effects of the CAR-T cells and the OVs (Figure 7c). Deposition of vvDD-GFP did not affect the viability of the A549 cells despite the observed virus replication (Figure 7b, upper right squares and Figure 7c, triangles). While the T47D line demonstrated sensitivity to killing by both VSV and vvDD deposited by CAR-’ve T cells, the robust killing by the CAR-T cells obscured the benefit of any combinatorial effects (Figure 7d). We observed mild viral oncolysis of D2F2/E2 breast tumor cells following deposition VSVΔM51-GFP by CAR-’ve T cells (Figure 7e, closed squares). As described earlier, the D2F2/E2 line was sensitive to killing by HER2-CAR-T cells and this killing was not affected by loading with vvDD. Interestingly, we did observe a marked combinatorial killing effect of the HER2-CAR-T cells loaded with VSVΔM51-GFP (Figure 7e, open squares). Overall, our data shows that OV-loading of CAR-T cells does not impair the functionality of the CAR-T cells alone and that the addition of the virus enables efficient killing of targets that might otherwise be resistant to CAR-T cell therapy. Moreover, we observed a combinatorial effect on a cell line (D2F2/E2) that was only moderately sensitive to either method alone.
Figure 7.

OV-loading of CAR-T cells can enhance killing of tumor targets. Human HER2- CAR-T cells (or CAR-’ve controls) loaded with an MOI of 3 of either VSVΔM51-GFP or vvDD-GFP were co-cultured at varying effector to target ratios (E:T) with tumor targets. (a) Murine (D2F2 and D2F2/E2) and human (A549, T47D) tumor cell lines were stained for HER2 expression using anti-human HER2, followed by anti-human IgG-PE and visualized by flow cytometry. (b) A549, T47D, and D2F2/E2 show differing susceptibilities to VSV or vvDD replication. Wells were imaged using a Typhoon imager after 24 hours of co-culture at the 1:1 effector to target ratio with OV-loaded CAR-’ve cells as described above. Wells are representative of at least 2–3 independent experiments performed in triplicate. OV-loaded T cells were co-cultured with (c) A549, (d) T47D, or (e) D2F2/E2 tumor cells overnight. After washing off the T cells, tumor cell viability was determined via alamarBlue assay. Data is representative of 2–3 independent experiments, presented as mean ± SEM of triplicate wells. CAR, chimeric antigen receptor; MOI, multiplicity of infection; OV, oncolytic virus; VSV, vesicular stomatitis virus; vvDD, “double-deleted” vaccina virus.
Virus from OV-loaded T cells replicates rapidly upon transfer to tumor targets
To get a better understanding of the degree of virus replication following deposition by T cells, we quantified the virus attached to the input T cells and then measured the virus titers in the supernatant of the infected tumor targets 24 hours following deposition by T cells. Interestingly, we found that VSVΔM51-GFP loading resulted in very low PFU of virus remaining associated with the CAR-T cells, with 20-30 PFU per 1.25 × 105 cells from CAR-’ve or Her-2 CAR-T cells, respectively (Table 1). In contrast, vvDD-GFP loading resulted in significantly higher virus load associated with the T cells, with 0.5−1.2 × 104 PFU per 1.25 × 105 cells (Table 1). To evaluate virus replication after co-culture with tumor targets, we added 1.25 × 105 OV-loaded T cells to 1.25 × 105 D2F2 tumor cells. After 24 hours, we harvested supernatants and titrated the resulting virus. After co-culture of VSV-loaded CAR-T cells, we observed a dramatic amplification of the virus, resulting in up to 2 × 107 PFU (Table 1). This corresponds to a greater than 7.5 × 105-fold increase in viral titer, stemming from an effective MOI of 0.0002. There was no significant difference between cultures where the virus was deposited by CAR-’ve or HER2-CAR-T cells (Table 1). We performed the same experiment with T cells loaded with vvDD achieving MOIs of 0.04 and 0.1 for HER2-CAR and CAR-’ve-T cells, respectively. Virus titers in the culture supernatant reached upwards of 8.88 × 105 PFU24 hours following virus deposition (Table 1). This corresponds to an average 114-fold increase in virus load across T cell types. Again, there was no significant difference between CAR-’ve and HER2-CAR-T cell-derived vvDD-GFP virus titer. Taken together, our data shows that OV-loading of CAR-T cells can be a viable combination, as these T cells can effectively transfer virus to tumor targets, which can serve to enhance the antitumor efficacy of each of these therapies.
Table 1. Virus titrations from loaded CAR-T cellsa.
|
Virus titer (PFU) | ||||
|---|---|---|---|---|
|
VSV |
vvDD |
|||
| Input virus (from 1.25 × 105 T cells) | Virus output (following 24 hours of co-culture) | Input virus (from 1.25 × 105 T cells) | Virus output (following 24 hours of co-culture) | |
| CAR-’ve T cells | 30.02 (±3.46) | 2.03 × 107 (±5.63 × 106) | 1.19 × 104 (±4.83 × 103) | 8.88 × 105 (±1.39 × 105) |
| Her2-CAR-T cells | 20.42 (±3.37) | 1.74 × 107 (±4.21 × 106) | 5.51 × 103 (±4.38 × 102) | 8.38 × 105 (±2.98 × 104) |
T cells were loaded with either VSVΔM51-GFP or vvDD-GFP at an MOI of 3. T cells were collected postwash for “input virus” titration. OV-loaded T cells were co-cultured at a 1:1 ratio with D2F2 tumor cells for 24 hours. Supernatants were collected and virus titrated as “virus output.” VSVΔM51-GFP was titrated using agarose overlays on Vero cells, while vvDD-GFP was titrated using CV-1 cells and visualized with crystal violet staining. Error is expressed as ± SEM. CAR, chimeric antigen receptor; MOI, multiplicity of infection; OV, oncolytic virus; VSV, vesicular stomatitis virus; vvDD, “double-deleted” vaccina virus.
Discussion
Cancer immunotherapy is a rapidly expanding field, with significant advances using T cells, viruses, antibodies, alone or in combination, as therapeutics. While ACT has shown significant promise in treating hematological malignancies, solid tumors continue to show lower levels of response.24 The use of OVs as a standalone therapy has emerged as an additional promising treatment, with examples such as T-VEC showing durable complete or partial clinical responses in a recent phase 3 study.1 However, many of these OV therapies rely on either intratumoral administration, which is difficult in the case of metastatic disease, or very high virus titers for systemic administration, which increases the risk of off-target effects.1,7,25–27 As such, there is considerable room for improvement of each of these therapies. The concept of combining T cells with OV loading has been addressed in a limited number of reports employing either transgenic murine T cells or human cytokine-induced killer cells.18,19,22,28 These studies showed that activated murine T cells were capable of carrying and depositing VSV onto tumor targets, showing an enhanced efficacy over either used as a monotherapy.18 In addition, both mouse and human cytokine-induced killer cells provided successful transport of vvDD, resulting in improved antitumor efficacy.22,28 Engineering T cells to recognize surface-expressed tumor antigens using CARs avoids the MHC restriction encountered by TCR-activated T cells.29 This engineering process allows for precise targeting of known target antigens, and facilitates re-targeting of bulk T cell populations.29 Our study demonstrates the feasibility of loading CAR-engineered T cells with OV for use as a combination therapy.
Loading CAR-T cells with either VSV or vvDD did not result in any phenotypic or functional changes to human or murine CAR-T cells, indicating that the loading of these viruses is a relatively innocuous process. Additionally, the ability of OV-loaded CAR-T cells to transfer their virus load to tumor targets was not impaired by recognition of the tumor by the CAR. This is an important consideration, as combination therapies must not interfere with the effects of each treatment used on its own. The combination of OV with CAR-T cells can provide complementary benefits to the killing capacity of each component. Virus replication within the tumor and lysis of tumor cells induces inflammation, which serves to drive endogenous antitumor immunity.30 In a mouse model of ACT, combining local VSV delivery with tumor-reactive T cells served to maintain the activation status of adoptively transferred cells.30,31 By packaging the OV onto a T cell carrier, the OV is protected from immune recognition in the blood stream, and transported to the tumor.20,21 Thus these therapies possess complementary properties.
One potential concern for combining CAR-T cells with OV is that several OV, including VSV and vvDD, have been shown to induce vascular shut down within the tumor during oncolysis.6,32 This has the potential to prevent CAR-T cells from infiltrating the tumor, which would limit the efficacy of the CAR-T cells. By loading the OV onto CAR-T cells, the virus and T cells would both be present within the tumor together, limiting the likelihood of one restricting access for the other. In order to better drive OV-loaded CAR-T cell trafficking to the tumor, this therapy could be further combined with pre-conditioning irradiation or chemotherapy that have been shown to significantly enhance engraftment of adoptively transferred T cells.12,33,34 These therapies have also been shown to enhance antitumor efficacy and reduce neutralizing antibody generation when using OVs, and so may serve to enhance both arms of this combination.35
Our data supports the use of OV-loaded CAR-T cells to treat a variety of different tumors. In particular, our data shows the effects of treating tumors with varying sensitivities to either OV or CAR-T cell mediated killing. CAR-T cells alone had minimal effect on A549 lung carcinoma cells, however T cell-transported VSVΔM51 proved effective at eliminating these cells (Figure 6c). Treatment of T47D cells showed maximal killing with HER2-CAR-T cells, without a measurable effect of the OVs (Figure 6d). However, as these viruses can disrupt tumor vasculature as well as drive endogenous immune responses in addition to oncolysis, these could still be of use for clinical treatment of tumors with varying sensitivity to CAR-T cells. Finally, when utilizing OV-loaded CAR-T cells to treat D2F2/E2 cells, we observed a significant enhancement of tumor cell killing when combining VSV and HER2-CAR-T cells over each treatment independently (Figure 6e). These data are particularly promising, as they suggest that OV-loaded CAR-T cells may be used to treat heterogeneous tumors. Tumor cells that are resistant to CAR-T-cell–mediated killing may be effectively killed by oncolysis, while those with some degree of susceptibility to each treatment independently can be killed readily by the combination treatment. Additionally, neither treatment appears to impair the antitumor functionality of the other, showing that these two therapies are indeed compatible. Combining these treatments could provide protection against antigen-loss variants, as the OV are capable of driving endogenous immune responses to additional tumor antigens, extending the antitumor response.36 As the effective MOI of viruses derived from OV-loaded CAR-T cells is very low, our data suggests that pre-loading onto tumor-reactive T cells can significantly enhance OV delivery to the tumor. This therefore presents a multi-pronged antitumor approach that allows for significantly lower OV dosages than if the two therapies were used concurrently.
We failed to distinguish an advantage to using vvDD-loaded CAR-T cells over the use of CAR-T cells alone in our studies. We speculate this corresponds to limitations of in vitro assays, where the CAR-T cells are capable of killing tumor targets faster than vvDD can. Our data showed that even in the absence of observable cytolysis, vvDD-GFP replicated readily in all three tumor lines, albeit to varying degrees (Figure 6b). The combination of vvDD loaded onto CAR-T cells may prove a more effective combination in the context of an established tumor. This way, the virus will be able to infect and kill tumor cells unaffected by CAR-T cell treatment, as well target the tumor vasculature, causing tumor destruction through both direct and indirect means.6 Importantly, loading of CAR-T cells with vvDD did not negatively impact the functionality of the CAR-T cells and so could provide benefit as a combination therapy.
Additional advantages of packaging OV onto CAR-T cells are that OVs have the unique capabilities of encoding additional genes within them that can be used to enhance the activity of the T cells. The functionality and survival of CAR-T cells can be enhanced through provision of cytokines such as IL-15 or IL-12 within the OV, allowing for production of these cytokines within the tumor microenvironment.9,37 The OV could encode a target antigen recognized by the endogenous T-cell receptor on the CAR-T cell, allowing for boosting of the transferred T cells.38,39 Alternatively, the OV could code for either siRNA or miRNA that could downregulate ligands for immunosuppressive T-cell receptors such as CTLA-4 or PD-L1, as blockade of these pathways has been shown to enhance ACT success.40,41 Thus, there are numerous possibilities for further combination therapies using OV-loaded CAR-T cells.
Overall, our studies combining OV-loading with CAR-T cell transfer provide the proof-of-principle that this combination is both feasible and effective at enhancing antitumor responses. This lays the groundwork to test various other OVs, as well as the modifications of the OV transgenes to drive enhanced T-cell function. Importantly, our data aids in developing an understanding of the interplay between CAR-T cells and OVs when used as a combination therapy.
Materials and Methods
Cell lines
The murine breast tumor cells D2F2 and D2F2/E2 were maintained in Dulbecco’s modified Eagle’s medium supplemented with 5% fetal bovine serum (FBS; Gibco, Life Technologies, Grand Island, NY), 5% cosmic calf serum (Fisher Scientific, Waltham, MA), 1 mmol/l sodium pyruvate, 2 mmol/l l-glutamine, 0.1 mmol/l nonessential amino acids (Gibco), 10 mmol/l HEPES (Gibco), 55 nmol/l β-mercaptoethanol (Gibco), 100 U/ml penicillin, and 100 µg/ml streptomycin. To ensure stable expression of human HER2 was maintained, D2F2/E2 cells were further supplemented with 800 µg/ml G418 (Sigma, St Louis, MO). The human lung tumor cells A549 and the human breast tumor cells T47D were maintained in RPMI 1640 medium containing 10% FBS, 2 mmol/l l-glutamine, 10 mmol/l HEPES, 100 U/ml penicillin, and 100 µg/ml streptomycin. Vero cells were cultured in α-modified Eagle’s medium with 10% FBS, 2 mmol/l l-glutamine, 100 U/ml penicillin, and 100 µg/ml streptomycin. CV-1 cells were cultured in Dulbecco’s modified Eagle’s medium with 10% FBS, 2 mmol/l l-glutamine, 100 U/ml penicillin, and 100 µg/ml streptomycin. For vaccinia titrations, CV-1 medium used 2% FBS instead. All cells were cultured at 37 °C with 5% CO2.
Generation of CAR retroviral and lentiviral vectors
We generated an in-house variant of a previously-reported human anti-HER2-CAR42 for use in murine T cells, in which the human sequences from the HER2-CAR were replaced by equivalent murine sequences. This HER2-CAR was comprised of a single chain antibody fragment (scFv) specific for human HER2 (ref. 43), a short marker epitope of c-myc, the hinge region from murine CD8, the transmembrane and cytoplasmic portions of murine CD28, and finally the cytoplasmic portion of murine CD3ζ. These components were cloned into the MSCV-based retroviral vector pRV2011 oFL.44 Murine ecotropic retroviruses were packaged by transfecting Platinum-E (PLAT-E) cells with retrovirus vectors and the helper plasmid pCL-Eco using Lipofectamine 2000 (Life Technologies) as described previously.44,45 Retrovirus supernatants were harvested at 48 hours post transfection, concentrated 10× using Amicon Ultra filters, and used immediately to transduce murine T cells. Negative control murine T cells (CAR-’ve) were prepared using the same methods, using viruses were prepared from transfections with the pRV2011 oFL plasmid, which lacks the CAR cDNA.
For human T-cell engineering, the human HER2-CAR was cloned into the lentiviral vector pCCL-ΔNGFR vector.46 Lentiviruses were produced by four-plasmid transfection of HEK 293T cells as described previously.47,48 Virus titers were determined using serial dilutions on 293T cells and evaluated by flow cytometric staining for NGFR. Negative control human T cells (CAR-’ve) were prepared using the same methods, with viruses were prepared from transfections with the pCCL-ΔNGFR plasmid, which lacks the CAR cDNA.
Murine T-cell transduction
Female BALB/c mice were purchased from Charles River Breeding Laboratories (Wilmington, MA). The McMaster University Animal Research Ethics Board has approved all animal usage. Splenocytes were isolated and cultured in T cell growth medium, consisting of RPMI 1640 with 10% fetal bovine serum, 10 mmol/l HEPES, 2 mmol/l l-glutamine, 0.1 mmol/l nonessential amino acids, 1 mmol/l sodium pyruvate, 55 nmol/l β-mercaptoethanol, and 100 µg/ml Normocin (Invivogen, San Diego, CA). T cells were activated using 0.1 µg/ml each hamster anti-mouse CD3 (clone 2C11; BD Biosciences, Franklin Lakes, NJ) and hamster anti-mouse CD28 (clone 37.51; BD Biosciences), and grown in the presence of 60 IU rhIL-2 (Peprotech, Rocky Hill, NJ). After 24 hours, T cells were transduced with 100 µl of concentrated retrovirus, 1.6 µg/ml polybrene (Sigma), and 2 µg/ml lipofectamine via spinfection at 2,000 rpm for 90 minutes. Cells were expanded in T-cell medium containing 60 IU/ml rhIL-2, and loaded with OV three days after transduction.
Human T-cell transduction
Human peripheral blood cells (PBMCs) were isolated from healthy donors. T cells were activated using anti-CD3/CD28 Dynabeads (Gibco) in the T cell media described above, in the presence of 100 IU/ml IL-2 and 10 ng/ml IL-7 (Peprotech). Twenty-four hours after activation, T cells were transduced with an MOI of 1 of the indicated lentiviruses. T cells were expanded in T cell medium with 100 IU/ml IL-2 and 10 ng/ml IL-7, feeding with fresh medium and cytokines every 2–3 days, and loaded with OVs 14 days after activation.
OVs and T-cell loading
We utilized the interferon-sensitive ΔM51 mutant of VSV expressing GFP (VSVΔM51-GFP).9 The VGF-, TK- vvDD expressing GFP has been described previously.8 Murine and human CAR-T cells were loaded with OV following the same protocol. CAR-T cells were incubated with a MOI of either 0 (mock) or 3 of the indicated virus for 3 hours at 37 °C. Cells were washed 4 times in phosphate-buffered saline at 4 °C, resuspended in T-cell growth medium and used in the described experiments.
Intracellular cytokine staining
Both murine and human CAR-T cells were stimulated using plate-bound recombinant human HER2-Fc chimera (1,000 ng/ml in phosphate-buffered saline; R&D Systems, Minneapolis, MN) or vehicle control for 4 hours at 37 °C in the presence of GolgiPlug protein transport inhibitor (BD Pharmingen, San Jose, CA). Following stimulation, cells were resuspended in 5% FBS (in phosphate-buffered saline) and stored at 4 °C overnight.
Flow cytometry antibodies and analytical instruments
For murine T cell work, we used the following BD Bioscience antibodies: mouse Fc-Block, anti-CD4 (clone RM4-5), anti-CD8a (clone 53-6.7), anti-IFNγ (clone XMG1.2), and anti-TNFα (clone MP6-XT22). For human T-cell staining, we used the following BD Bioscience antibodies: anti-CD4 (clone RPA-T4), anti-NGFR (clone C40-1457), anti-IFNγ (clone B27), and anti-TNFα (clone MAb11). The anti-CD8a clone OKT8 from eBiosciences was also used. Recombinant human HER2-Fc (R&D Systems) was used to stain for HER2-CAR expression on both murine and human T cells and detected with goat-anti-human IgG (Jackson ImmunoResearch, West Grove, PA). Intracellular staining were performed using the cytofix/cytoperm reagent and associated protocol (BD Biosciences). T-cell viability was evaluated using the Molecular Probes LIVE/DEAD Fixable Near-IR kit (Life Technologies). Results were acquired on a FACSCanto or LSRII (BD Biosciences) and analyzed using FlowJo software (FlowJo, Ashland, OR).
In vitro cytotoxicity assay
Murine D2F2 or D2F2/E2 or human A579 or T47D tumor cells were used for in vitro cytotoxicity assays. These assays were performed by co-culturing varying ratios of mock, VSVΔM51-GFP, or vvDD-GFP loaded T cells with 1.25 × 104 target cells per well in triplicate in 96-well flat-bottom plates in 200 µl. For assays investigating CAR-T-cell–mediated killing after virus loading, plates were incubated for 6 hours. For assays evaluating combined killing between OV and CAR-T cells, plates were incubated for 24 hours, after which T cells were washed off, and 100 µl of 10% alamarBlue (Life Technologies) in T-cell media was added. After 3 hours, fluorescence was quantified by a Safire plate reader (Tecan, Maennendorf, Switzerland), using 530 nm as excitation and 590 nm as emission wavelengths. Tumor cell viability was calculated as the loss of fluorescence in experimental wells compared to untreated target cells.
Virus titrations
All samples taken for virus titrations were frozen at −80 °C prior to titration and thawed only to titrate virus. To determine vaccinia titrations, confluent CV-1 cells were infected and visualized using crystal violet as described previously.49
Virus titers for VSV were determined utilizing confluent Vero cells in 60 mm dishes. Serial virus dilutions were prepared and added to Vero cells in a 100 µl volume for 45 minutes. After allowing for adsorption, 3 ml of prepared agarose overlay was added (1:1 of 1% agarose, 2× F11 medium with 20% FBS). Plaques were counted at 24 and 48 hours later.
Statistical analysis
Student’s t-tests were used to compare data between two groups, while analysis of variance was used for data involving multiple groups or multiple time points. Results were prepared using GraphPad Prism 5. Significant differences were defined as: *P < 0.05, **P < 0.01, ***P < 0.001; n.s. = not significant.
Acknowledgments
This work was supported by funds from the Terry Fox Foundation and the Canadian Institutes for Health Research (CIHR). H.V. was supported by a Canada Graduate Scholarship from CIHR and by the Government of Ontario, and J.L.B. holds the Canada Research Chair in Translational Cancer Immunology. Murine D2F2 and D2F2/E2 cells were provided by Wei-Zen Wei (Barbara Ann Karmanos Cancer Institute, Detroit, MI). The human anti-HER2-CAR was provided by Philip K. Darcy (University of Melbourne, Victoria, Australia). Retroviral vectors were provided by Brian Rabinovich (MD Anderson Cancer Centre, Houston, TX). Lentiviral vectors were provided by Megan Levings (University of British Columbia, Canada). We obtained VSVΔM51-GFP from Brian Lichty (McMaster University, Hamilton, Ontario, Canada), and vvDD-GFP from Andrea McCart (University of Toronto, Ontario, Canada) and John C. Bell (University of Ottawa, Ontario, Canada).
The authors declare no conflict of interest.
References
- Pol, J, Bloy, N, Obrist, F, Eggermont, A, Galon, J, Cremer, I et al. (2014). Trial watch: oncolytic viruses for cancer therapy. Oncoimmunology 3: e28694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojdl, DF, Lichty, BD, tenOever, BR, Paterson, JM, Power, AT, Knowles, S et al. (2003). VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell 4: 263–275. [DOI] [PubMed] [Google Scholar]
- Bridle, BW, Stephenson, KB, Boudreau, JE, Koshy, S, Kazdhan, N, Pullenayegum, E et al. (2010). Potentiating cancer immunotherapy using an oncolytic virus. Mol Ther 18: 1430–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojdl, DF, Lichty, B, Knowles, S, Marius, R, Atkins, H, Sonenberg, N et al. (2000). Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med 6: 821–825. [DOI] [PubMed] [Google Scholar]
- Bell, JC, Lichty, B and Stojdl, D (2003). Getting oncolytic virus therapies off the ground. Cancer Cell 4: 7–11. [DOI] [PubMed] [Google Scholar]
- Breitbach, CJ, Arulanandam, R, De Silva, N, Thorne, SH, Patt, R, Daneshmand, M et al. (2013). Oncolytic vaccinia virus disrupts tumor-associated vasculature in humans. Cancer Res 73: 1265–1275. [DOI] [PubMed] [Google Scholar]
- Breitbach, CJ, Burke, J, Jonker, D, Stephenson, J, Haas, AR, Chow, LQ et al. (2011). Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature 477: 99–102. [DOI] [PubMed] [Google Scholar]
- McCart, JA, Ward, JM, Lee, J, Hu, Y, Alexander, HR, Libutti, SK et al. (2001). Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer Res 61: 8751–8757. [PubMed] [Google Scholar]
- Stephenson, KB, Barra, NG, Davies, E, Ashkar, AA and Lichty, BD (2012). Expressing human interleukin-15 from oncolytic vesicular stomatitis virus improves survival in a murine metastatic colon adenocarcinoma model through the enhancement of anti-tumor immunity. Cancer Gene Ther 19: 238–246. [DOI] [PubMed] [Google Scholar]
- Bridle, BW, Chen, L, Lemay, CG, Diallo, JS, Pol, J, Nguyen, A et al. (2013). HDAC inhibition suppresses primary immune responses, enhances secondary immune responses, and abrogates autoimmunity during tumor immunotherapy. Mol Ther 21: 887–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willmon, C, Harrington, K, Kottke, T, Prestwich, R, Melcher, A and Vile, R (2009). Cell carriers for oncolytic viruses: Fed Ex for cancer therapy. Mol Ther 17: 1667–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinrichs, CS and Rosenberg, SA (2014). Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunol Rev 257: 56–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg, SA and Dudley, ME (2004). Cancer regression in patients with metastatic melanoma after the transfer of autologous antitumor lymphocytes. Proc Natl Acad Sci USA 101 (suppl. 2): 14639–14645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brentjens, RJ, Davila, ML, Riviere, I, Park, J, Wang, X, Cowell, LG et al. (2013). CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med 5: 177ra38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davila, ML, Kloss, CC, Gunset, G and Sadelain, M (2013). CD19 CAR-targeted T cells induce long-term remission and B cell aplasia in an immunocompetent mouse model of B cell acute lymphoblastic leukemia. PLoS ONE 8: e61338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalos, M, Levine, BL, Porter, DL, Katz, S, Grupp, SA, Bagg, A et al. (2011). T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med 3: 95ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L, Bridle, BW, Chen, L, Pol, J, Spaner, D, Boudreau, JE et al. (2013). Delivery of viral-vectored vaccines by B cells represents a novel strategy to accelerate CD8(+) T-cell recall responses. Blood 121: 2432–2439. [DOI] [PubMed] [Google Scholar]
- Qiao, J, Wang, H, Kottke, T, Diaz, RM, Willmon, C, Hudacek, A et al. (2008). Loading of oncolytic vesicular stomatitis virus onto antigen-specific T cells enhances the efficacy of adoptive T-cell therapy of tumors. Gene Ther 15: 604–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole, C, Qiao, J, Kottke, T, Diaz, RM, Ahmed, A, Sanchez-Perez, L et al. (2005). Tumor-targeted, systemic delivery of therapeutic viral vectors using hitchhiking on antigen-specific T cells. Nat Med 11: 1073–1081. [DOI] [PubMed] [Google Scholar]
- Kottke, T, Diaz, RM, Kaluza, K, Pulido, J, Galivo, F, Wongthida, P et al. (2008). Use of biological therapy to enhance both virotherapy and adoptive T-cell therapy for cancer. Mol Ther 16: 1910–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao, J, Kottke, T, Willmon, C, Galivo, F, Wongthida, P, Diaz, RM et al. (2008). Purging metastases in lymphoid organs using a combination of antigen-nonspecific adoptive T cell therapy, oncolytic virotherapy and immunotherapy. Nat Med 14: 37–44. [DOI] [PubMed] [Google Scholar]
- Thorne, SH, Liang, W, Sampath, P, Schmidt, T, Sikorski, R, Beilhack, A et al. (2010). Targeting localized immune suppression within the tumor through repeat cycles of immune cell-oncolytic virus combination therapy. Mol Ther 18: 1698–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampath, P, Li, J, Hou, W, Chen, H, Bartlett, DL and Thorne, SH (2013). Crosstalk between immune cell and oncolytic vaccinia therapy enhances tumor trafficking and antitumor effects. Mol Ther 21: 620–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranda, F, Vacchelli, E, Obrist, F, Eggermont, A, Galon, J, Hervé Fridman, W et al. (2014). Trial watch: adoptive cell transfer for anticancer immunotherapy. Oncoimmunology 3: e28344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo, J, Reid, T, Ruo, L, Breitbach, CJ, Rose, S, Bloomston, M et al. (2013). Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med 19: 329–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacchelli, E, Eggermont, A, Sautès-Fridman, C, Galon, J, Zitvogel, L, Kroemer, G et al. (2013). Trial watch: oncolytic viruses for cancer therapy. Oncoimmunology 2: e24612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi, L and Lugli, E (2013). Cancer immunotherapy turns viral. Oncoimmunology 2: e24802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorne, SH, Negrin, RS and Contag, CH (2006). Synergistic antitumor effects of immune cell-viral biotherapy. Science 311: 1780–1784. [DOI] [PubMed] [Google Scholar]
- Sadelain, M, Brentjens, R and Rivière, I (2009). The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol 21: 215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottolino-Perry, K, Diallo, JS, Lichty, BD, Bell, JC and McCart, JA (2010). Intelligent design: combination therapy with oncolytic viruses. Mol Ther 18: 251–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz, RM, Galivo, F, Kottke, T, Wongthida, P, Qiao, J, Thompson, J et al. (2007). Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Res 67: 2840–2848. [DOI] [PubMed] [Google Scholar]
- Breitbach, CJ, Paterson, JM, Lemay, CG, Falls, TJ, McGuire, A, Parato, KA et al. (2007). Targeted inflammation during oncolytic virus therapy severely compromises tumor blood flow. Mol Ther 15: 1686–1693. [DOI] [PubMed] [Google Scholar]
- Dudley, ME, Wunderlich, JR, Yang, JC, Sherry, RM, Topalian, SL, Restifo, NP et al. (2005). Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol 23: 2346–2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrzesinski, C, Paulos, CM, Kaiser, A, Muranski, P, Palmer, DC, Gattinoni, L et al. (2010). Increased intensity lymphodepletion enhances tumor treatment efficacy of adoptively transferred tumor-specific T cells. J Immunother 33: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao, J, Wang, H, Kottke, T, White, C, Twigger, K, Diaz, RM et al. (2008). Cyclophosphamide facilitates antitumor efficacy against subcutaneous tumors following intravenous delivery of reovirus. Clin Cancer Res 14: 259–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prestwich, RJ, Errington, F, Diaz, RM, Pandha, HS, Harrington, KJ, Melcher, AA et al. (2009). The case of oncolytic viruses versus the immune system: waiting on the judgment of Solomon. Hum Gene Ther 20: 1119–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chmielewski, M and Abken, H (2012). CAR T cells transform to trucks: chimeric antigen receptor-redirected T cells engineered to deliver inducible IL-12 modulate the tumour stroma to combat cancer. Cancer Immunol Immunother 61: 1269–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossig, C, Bollard, CM, Nuchtern, JG, Rooney, CM and Brenner, MK (2002). Epstein-Barr virus-specific human T lymphocytes expressing antitumor chimeric T-cell receptors: potential for improved immunotherapy. Blood 99: 2009–2016. [DOI] [PubMed] [Google Scholar]
- Nakazawa, Y, Huye, LE, Salsman, VS, Leen, AM, Ahmed, N, Rollins, L et al. (2011). PiggyBac-mediated cancer immunotherapy using EBV-specific cytotoxic T-cells expressing HER2-specific chimeric antigen receptor. Mol Ther 19: 2133–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGray, AJ, Hallett, R, Bernard, D, Swift, SL, Zhu, Z, Teoderascu, F et al. (2014). Immunotherapy-induced CD8+ T cells instigate immune suppression in the tumor. Mol Ther 22: 206–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, JH, Park, HB, Oh, YM, Lim, DP, Lee, JE, Seo, HH et al. (2012). Positive conversion of negative signaling of CTLA4 potentiates antitumor efficacy of adoptive T-cell therapy in murine tumor models. Blood 119: 5678–5687. [DOI] [PubMed] [Google Scholar]
- Moeller, M, Haynes, NM, Trapani, JA, Teng, MW, Jackson, JT, Tanner, JE et al. (2004). A functional role for CD28 costimulation in tumor recognition by single-chain receptor-modified T cells. Cancer Gene Ther 11: 371–379. [DOI] [PubMed] [Google Scholar]
- Wels, W, Harwerth, IM, Zwickl, M, Hardman, N, Groner, B and Hynes, NE (1992). Construction, bacterial expression and characterization of a bifunctional single-chain antibody-phosphatase fusion protein targeted to the human erbB-2 receptor. Biotechnology (NY) 10: 1128–1132. [DOI] [PubMed] [Google Scholar]
- Rabinovich, BA, Ye, Y, Etto, T, Chen, JQ, Levitsky, HI, Overwijk, WW et al. (2008). Visualizing fewer than 10 mouse T cells with an enhanced firefly luciferase in immunocompetent mouse models of cancer. Proc Natl Acad Sci USA 105: 14342–14346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita, S, Kojima, T and Kitamura, T (2000). Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther 7: 1063–1066. [DOI] [PubMed] [Google Scholar]
- McMurchy, AN, Gillies, J, Gizzi, MC, Riba, M, Garcia-Manteiga, JM, Cittaro, D et al. (2013). A novel function for FOXP3 in humans: intrinsic regulation of conventional T cells. Blood 121: 1265–1275. [DOI] [PubMed] [Google Scholar]
- Allan, SE, Alstad, AN, Merindol, N, Crellin, NK, Amendola, M, Bacchetta, R et al. (2008). Generation of potent and stable human CD4+ T regulatory cells by activation-independent expression of FOXP3. Mol Ther 16: 194–202. [DOI] [PubMed] [Google Scholar]
- Amendola, M, Venneri, MA, Biffi, A, Vigna, E and Naldini, L (2005). Coordinate dual-gene transgenesis by lentiviral vectors carrying synthetic bidirectional promoters. Nat Biotechnol 23: 108–116. [DOI] [PubMed] [Google Scholar]
- Dayball, K, Millar, J, Miller, M, Wan, YH and Bramson, J (2003). Electroporation enables plasmid vaccines to elicit CD8+ T cell responses in the absence of CD4+ T cells. J Immunol 171: 3379–3384. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.