

Abstract
Oncolytic viruses selectively replicate in cancer cells by exploiting biochemical differences between normal and tumor cells. Treatment with epigenetic modifiers such as 5-Azacytidine, a DNA methyltransferase inhibitor, increases the replication and cytotoxicity of oncolytic viruses in vivo and in vitro. The cotton rat is an attractive animal to study oncolytic viruses, as syngeneic models of breast adenocarcinoma and osteosarcoma are well established, and many features of primary and secondary tumor growth recapitulate human disease. Treatment of LCRT breast cancer cells with 5-Azacytidine increases bovine herpesvirus type 1 (BHV-1)-mediated cytotoxicity in vitro, with Chou-Talalay analysis indicating a very strong synergy. In vivo, BHV-1 monotherapy delayed tumor growth but did not improve survival of cotton rats with subcutaneous breast adenocarcinomas. However, combination therapy significantly decreased the incidence of secondary lesions, with enhanced tumor cell clearance and evidence of immune cell infiltration compared to BHV-1 monotherapy. Together, these results warrant further investigation of BHV-1 combination therapy with epigenetic modifiers for the treatment of breast cancer, particularly in the context of the prevention and treatment of secondary lesions.
Introduction
Oncolytic viruses (OVs) selectively replicate in tumor cells by exploiting the biochemical differences between normal and tumor cells.1,2 OVs can elicit tumor cell death directly through lysis or by stimulating an antitumor immune response. The efficacy of oncolytic herpes simplex virus type 1 (oHSV-1) has been well characterized in preclinical and clinical studies.3 Although the induction of antitumor immunity has been demonstrated following intratumoral (i.t) administration of oHSV-1,4,5 systemic delivery will be required for treating metastatic lesions. However, the high incidence of pre-existing immunity to HSV-1 may limit systemic delivery. Such obstacles warrant the development of nonhuman viruses for OV therapy (OVT).
Bovine herpesvirus type 1 (BHV-1) is a member of the Herpesviridae family that initiates bovine respiratory disease complex in cattle through transient immunosuppression.6 Despite the prevalence of BHV-1 in cattle, no human infections or seroconversions have been reported.6 Normal human cells are susceptible to BHV-1 binding and entry, but are not permissive for BHV-1 replication.7 In contrast, human immortalized, transformed and tumor-initiating cells are permissive to infection.7,8 Interestingly, the ability of BHV-1 to kill human bulk and tumor-initiating breast cancer cells does not depend on the initiation of virus replication or the production of a viral burst.8 Unfortunately, BHV-1 does not efficiently bind and enter murine cells,9 precluding the use of conventional mouse models.
The cotton rat (CR; Sigmodon hispidus) is commonly used in anti-BHV-1 vaccination research, as viral-induced pathology resembles that seen in cattle.10 BHV-1 infection of CRs is immunogenic, inducing mucosal and systemic immune responses, particularly following intranasal inoculation.10 The CR is an attractive syngeneic, immune competent model to study OVs, as cell lines derived from spontaneous fibrosarcomas of the breast (LCRT) and osteosarcomas of the bone (CCRT and VCRT) have been developed.11 CRs are used to evaluate the antitumor efficacy of oncolytic adenoviruses, including neutralization studies to predict human responses to intravenous (i.v) injection.12
Antitumor responses from OVT differ significantly when comparing tolerized and nontolerized tumor-associated antigen (TAA) models. Central and peripheral tolerance can dampen TAA-specific cytotoxic lymphocyte responses, resulting in poor therapeutic outcomes.13,14 TAA-specific CD8+ T cells, essential for mediating tumor regression, are produced in nontolerized syngeneic tumor models, but not in tolerized tumor models.14,15 These observations highlight the importance of evaluating OVs using tolerized animal models, which better recapitulate the human immune landscape, allowing for enhanced understanding of the features that determine therapeutic success.
Aberrant DNA methylation events frequently occur in cancer and include global DNA hypomethylation and CpG island hypermethylation, leading to the silencing of tumor-suppressor genes or the expression of oncogenes.16 Recent studies used DNA methyltransferase inhibitors (DNMTi) such as 5-Azacytidine (5-Aza) to study the role of methylation in the development and prognosis of breast cancer.17,18 While treatment of breast cancer cells with 5-Aza induced differential expression of tumor suppressor genes and oncogenes, it was unable to induce tumor cell death on its own.19,20 OVs have been combined with chemo and radiotherapy to exploit differences in the mechanism of tumor cell death elicited by each treatment, resulting in enhanced antitumor responses.3,21,22 Therefore, combining 5-Aza with agents that increase tumor cell death by counteracting the pro-survival effects of oncogenes may be efficacious.20,23
Results
BHV-1 replication and cytotoxicity in LCRT cells
BHV-1 is able to initiate replication and/or induce cellular cytotoxicity in human breast cancer cells from a variety of subtypes.8 The cytotoxicity of BHV-1 on the CR breast adenocarcinoma cell line LCRT was evaluated. Cells were infected with BHV-1 at a range of multiplicity of infection (MOI) and the initiation of replication, as a measure of green fluorescent protein (GFP) fluorescence, and cellular viability were analyzed 2 days postinfection (pi). While GFP fluorescence is an indicator of the initiation of virus replication only, for simplicity we will refer to this as virus replication. Induction of BHV-1 replication in LCRT cells was observed at MOIs greater than 2.5 (Figure 1a). However, a significant decrease in cellular viability, defined as a decrease greater than 20%, did not occur at any of the MOIs examined (Figure 1b).
Figure 1.
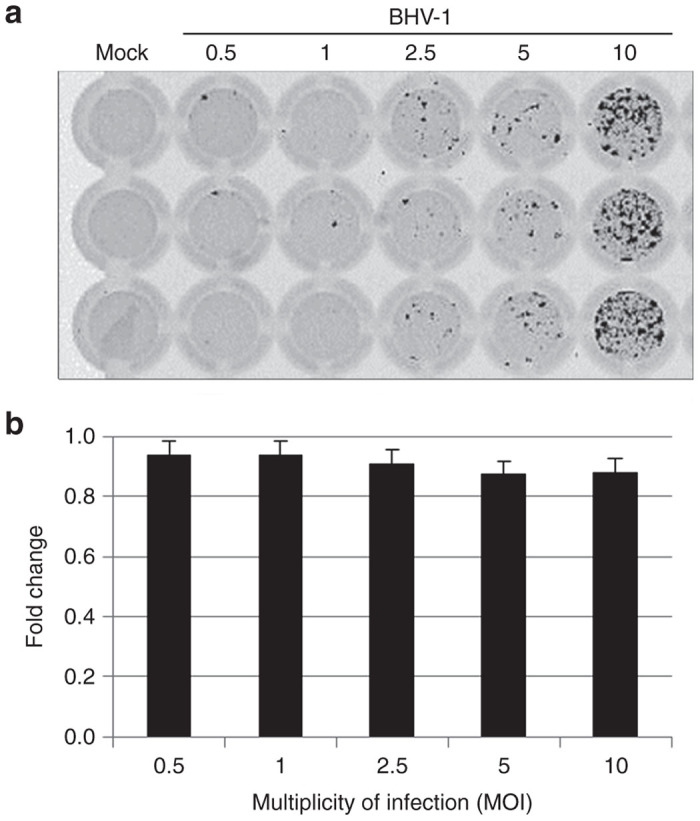
Bovine herpesvirus type 1 (BHV-1) replication and cytotoxicity on LCRT. LCRT cells in 96-well plates were mock or infected with BHV-1 at the indicated MOIs for 1 hour at 37 °C. (a) GFP expression, as a marker of virus replication, was detected using a Typhoon Bioanalyzer (Amersham Biosciences, Piscataway, NJ) at 2 days pi. (b) Cell metabolism, as a measure of cell viability, was assessed using MTT at 2 days pi. Fluorescence was detected using a SpectraMax i3 Multi-Mode Microplate Reader (Molecular Devices, Sunnyvale, CA) and the fold change in fluorescence relative to untreated, uninfected controls was calculated. Data were collected in triplicate and are represented as the mean.
5-Aza does not alter the permissivity of normal primary cells to BHV-1 infection
While BHV-1 is unable to replicate or induce cytotoxicity in normal human cells,7 epigenetic modifiers may alter cellular sensitivity to the virus. As normal primary CR cell lines do not exist, normal primary human cell lines were used. The IC50 values of 5-Aza were 18 and 3.3 µmol/l on normal human Ventressca and human embryonic lung (HEL) fibroblasts, respectively (data not shown). Treatment with 3 or 10 µmol/l 5-Aza did not increase BHV-1 replication on normal human Ventressca or HEL fibroblasts (data not shown). However, a significant increase in cytotoxicity in Ventressca cells was observed when 5-Aza (10 µmol/l) was combined with BHV-1 (MOI 3 or 5) in comparison to BHV-1 only infected samples (Figure 2a). This effect was also detected in HEL cells when 5-Aza (10 µmol/l) was combined with BHV-1 (MOI 1 and 5) (Figure 2b). However, the combination of 5-Aza with BHV-1 did not increase cytotoxicity over that seen with 5-Aza alone in either cell line (Figure 2), indicating that 5-Aza does not enhance BHV-1-mediated toxicity in normal cells. Therefore, only concentrations below the IC50 value for HEL were used in further experiments.
Figure 2.

Bovine herpesvirus type 1 (BHV-1)-mediated cytotoxicity with and without 5-Aza treatment on normal primary human cell lines Ventressca and human embryonic lung (HEL) cells. (a) Ventressca or (b) HEL cells were seeded into 96-well plates and were mock or infected with BHV-1 at the indicated MOIs for 1 hour at 37 °C. Cell metabolism, as a measure of cell viability, was assessed using MTT at 2 days pi. Fluorescence was detected using a SpectraMax i3 Multi-Mode Microplate Reader (Molecular Devices) and the fold change in fluorescence relative to untreated, uninfected controls was calculated. Data were collected in triplicate and are represented as the mean, n = 2. *P = 0.05
BHV-1 and 5-Aza act synergistically to kill LCRT cells
Studies combining OVs with epigenetic modifiers such as 5-Aza have shown synergistic or additive effects that enhance tumor cell death.23–25 To ensure that 5-Aza was functional in LCRT cells, the expression of Dnmt1 was evaluated as a control. When incorporated into DNA, 5-Aza forms a covalent bond with DNMT such as Dnmt1.26 This bond is irreversible and results in degradation of Dnmt1, reducing cellular levels of the enzyme.26 Western blot analysis indicated that 14-hour treatment with 1 and 3 μmol/l of 5-Aza is sufficient to reduce Dnmt1 expression in LCRT cells (Figure 3a). These data verify 5-Aza activity in LCRT cells. We then determined whether 5-Aza was able to enhance the replication or cytotoxicity of BHV-1. LCRT cells were treated with 5-Aza at 0.5, 1, or 3 µmol/l for 14 hours and subsequently infected with BHV-1 at MOI 3 or 5. Regardless of the concentration, treatment with 5-Aza increased BHV-1 replication (Figure 3b). Likewise, combination treatment was more effective in reducing cellular viability, with 3 µmol/l 5-Aza significantly reducing cellular viability to 60 and 45% of untreated cells following infection with BHV-1 at MOI 3 or 5, respectively (Figure 3c). For reference, we routinely observed a decrease to 20% cellular viability in Madin-Darby bovine kidney (MDBK) cells (BHV-1 MOI 3), which are fully permissive to BHV-1 infection (data not shown).
Figure 3.

5-Aza enhances bovine herpesvirus type 1 (BHV-1) replication and cytotoxicity on LCRT. (a) LCRT cells were treated with 5-Aza at 1 or 3 μmol/l. After 14 hours, whole cell lysates were harvested for western blot analysis with Dnmt1 primary antibody. Actin served as a loading control. Positive (+ve) control HeLa whole cell lysate. LCRT cells in 96-well plates were treated with 5-Aza at 0.5, 1, or 3 μmol/l for 14 hours, then mock or infected with BHV-1 at MOI 3 or 5 for 1 hour at 37 °C. (b) GFP expression, as a marker of virus replication, was detected using a Typhoon Bioanalyzer (Amersham Biosciences) 2 days pi and (c) cell metabolism, as a measure of cell viability, was assessed using MTT at 2 days pi. Fluorescence was detected using a SpectraMax i3 Multi-Mode Microplate Reader (Molecular Devices) and the fold change in fluorescence relative to untreated, uninfected controls was calculated. Error bars represent mean + SEM, n = 3. *P = 0.05
To evaluate whether the interaction between BHV-1 and 5-Aza is synergistic or additive, we generated Chou-Talalay plots using CompuSyn software (data not shown).27 The combination index (CI) for each treatment was calculated and the dose–effect combinations (cellular viability) of 5-Aza and BHV-1 on LCRT were determined. Table 1 shows that there is very strong synergy (CI < 1) between 5-Aza and BHV-1 regardless of the concentration of 5-Aza or MOI of virus used. These results suggest that BHV-1 and 5-Aza synergistically kill LCRT cells.
Table 1. Combination index for BHV-1 with 5-Aza on LCRT cells.
| Dose 5-Aza (µmol/l) | Dose BHV-1 (MOI) | Effect | CI |
|---|---|---|---|
| 0.5 | 1.0 | 0.87685 | 0.17848 |
| 0.5 | 3.0 | 0.80107 | 0.06363 |
| 0.5 | 5.0 | 0.56613 | 0.00338 |
| 1.0 | 1.0 | 0.74678 | 0.02872 |
| 1.0 | 3.0 | 0.79427 | 0.07894 |
| 1.0 | 5.0 | 0.56373 | 0.00503 |
| 3.0 | 1.0 | 0.59519 | 0.01460 |
| 3.0 | 3.0 | 0.60882 | 0.01785 |
| 3.0 | 5.0 | 0.43979 | 0.00343 |
Data were collected in triplicate and expressed as means, n = 3. Effect represents cellular viability relative to untreated controls 2 days pi. CI values were calculated using CompuSyn software (Version 1; Cambridge, MA). Synergism (CI < 1), antagonism (CI = 1), additive effect (CI > 1).
5-Aza, 5-Azacytidine; CI, combination index; MOI, multiplicity of infection.
5-Aza increases de novo production of BHV-1 in LCRT cells
To determine whether increased GFP expression is indicative of productive virus replication, the viral burst was determined. LCRT cells were treated with 0, 1, or 3 µmol/l 5-Aza and subsequently infected with BHV-1 at MOI 5. Cells and supernatant were collected 1, 2, and 3 days pi and virus titrated on naive MDBK cells. A minimal viral burst was detected in untreated samples at the time points examined (Figure 4). However, a statistically significant increase in viral titers was detected 2 days pi between untreated cells and cells treated with 1 and 3 µmol/l 5-Aza (Figure 4). Although the increase in virus output with 5-Aza treatment was statistically significant, this increase is not considered biologically significant relative to virus input. For reference, we routinely observe a viral burst of 400 pfu per cell 2 days pi in MDBK cells (BHV-1 MOI 3), which are fully permissive to BHV-1 infection. Additionally, the apparent drop in virus output with 3 µmol/l 5-Aza at 3 dpi is likely due to cellular cytotoxicity induced by the combination therapy at these concentrations (Figure 3c).
Figure 4.
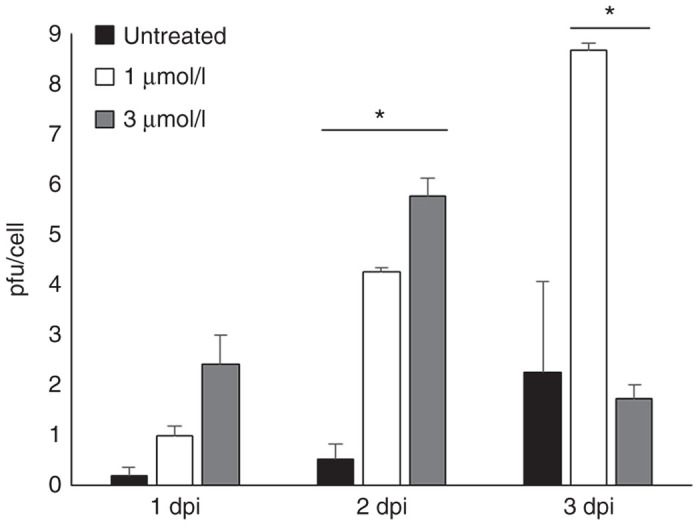
Bovine herpesvirus type 1 (BHV-1) viral burst increases with 5-Aza treatment in LCRT cells. Cells were infected with BHV-1 at MOI 5 for 1 hour at 37 °C. Cell-associated virus particles were collected 1, 2, and 3 days pi and titrated on naive MDBK monolayers. Error bars represent mean + SEM, n = 3. *P = 0.05.
BHV-1 monotherapy does not increase the survival of CR bearing subcutaneous LCRT tumors
Recent studies indicate that in vitro assays do not always predict in vivo outcomes.15,28 Tumors are complex entities that employ a multitude of mechanisms to influence tumor cell survival, proliferation, and spread. These mechanisms impact the success of OVT by affecting virus replication, spread and recruitment of the immune system to the tumor microenvironment.29,30 As a major barrier to effective OVT is central and peripheral tolerance, we evaluated whether BHV-1 possesses antitumor capabilities in a tolerized CR model of breast carcinoma. The CR LCRT model is extremely aggressive; phosphate-buffered saline (PBS)-treated tumors reached endpoint within 10 days on average (Figure 5a). Tumor growth was highly variable with increases in volume from the beginning of treatment to endpoint varying between 11- and 30-fold (PBS controls).
Figure 5.

Kaplan-Meier survival and tumor volumes for cotton rats treated with 5 × 106 pfu BHV-1. When tumors reached treatable size they were treated with 5 × 106 pfu BHV-1 intratumoral (i.t.) daily for 5 days. Tumors were measured every 2 days until animals reached end point. (a) Kaplan-Meier estimates of survival and (b) tumor volumes of cotton rats treated with PBS and (c) 5 × 106 pfu i.t. BHV-1 are shown. BHV-1, bovine herpesvirus type 1.
Preliminary dose-escalation studies were performed to investigate the safety and efficacy of BHV-1 in CRs bearing subcutaneous LCRT tumors. Tumors were treated i.t. with 5 × 106 or 5 × 107 plaque-forming units (pfu) BHV-1 once daily for 5 days and monitored for tumor growth and survival. No survival advantage or tumor regression was observed in animals treated with 5 × 106 pfu BHV-1 (Figure 5). The 5 × 106 pfu BHV-1 dose was well tolerated with no adverse effects observed. Hemorrhagic centers that turned necrotic appeared on large tumors several days post-treatment (Supplementary Figure S1b); however, this was not exclusive to the BHV-1 group, suggesting that this phenomenon may be associated with tumor size.
Animals treated with 5 × 107 pfu BHV-1 displayed significantly increased survival (Supplementary Figure S2); however, all animals reached endpoint due to respiratory distress or tumor burden. Histologically, the lungs contained multiple high-grade tumors that were mostly found around the bronchioles and in the pleura (Supplementary Figure S3c,d). Diffuse alveolar damage and pulmonary hemorrhage was also evident. Extensive damage and edema in the lungs, in conjunction with the secondary lesions in the armpit, contributed to respiratory distress. Pathological analysis suggested that the CRs developed lymphangitic carcinomatosis, which is common in breast adenocarcinoma and is caused by dissemination of tumor cells through the lymphatics in the lung.31 The absence of significant immune cell infiltration suggests that lung pathology was not due directly to BHV-1 infection, but was caused by tumor invasion and growth. Furthermore, hemorrhagic centers formed on large tumors, and severe ulceration was common in BHV-1-treated tumors that eventually turned necrotic (Supplementary Figure S1c).
BHV-1 and 5-Aza combination therapy improved therapeutic efficacy and decreased the incidence of secondary lesions
Although we observed a synergistic effect between 5-Aza and BHV-1 that enhanced cytotoxicity in LCRT cells in vitro, we wanted to determine whether this effect was maintained in vivo. Due to the extensive lung pathology observed in CRs treated with 5 × 107 pfu BHV-1 and that 5-Aza increases BHV-1 replication in vitro, we used 5 × 106 pfu for combination therapy studies. Approximately 2 weeks postsubcutaneous implantation of LCRT cells, CRs were treated with either PBS, 5-Aza (1 dose, 2 mg/kg intraperitoneal (i.p.)), BHV-1 (5 doses, 5 × 106 pfu i.t.) or 5-Aza plus BHV-1. For the combination therapy group, 5-Aza was delivered 1 day prior to commencing daily BHV-1 injections. Combination therapy delayed tumor growth in 38% (3/8) of animals, but did not significantly increase survival compared to BHV-1 treated controls (Figure 6). However, it is important to note that unlike murine models for which tumor growth is relatively uniform, these tumors were highly varied and the conventional method of taking measurements (length and width by caliper measurement) did not provide an accurate means by which to estimate tumor volume. During necropsy procedures, we found that 38% (3/8) of primary tumors harvested from CRs treated with combination therapy (indicated by an * in tumor volume graphs in Figure 6) were mainly comprised of fluid filled space and not solid tumor mass (Supplementary Figure S4).
Figure 6.

Kaplan-Meier survival curve and tumor volumes for cotton rats (CRs) treated with 5 × 106 plaque-forming unit (pfu) bovine herpes virus type 1 (BHV-1), 2 mg/kg 5-Aza, or 5 × 106 pfu BHV-1 and 2 mg/kg 5-Aza combination therapy. 5 × 106 LCRT cells were implanted into CRs by subcutaneous injection. When tumors reached treatable size, they were injected with 5 × 106 pfu BHV-1 intratumoral (i.t.) one dose daily for 5 days, one dose 5-Aza (2 mg/kg) intraperitoneal (i.p.), or pretreated with 5-Aza (2 mg/kg) i.p. 1 day prior to i.t. injection of 5 × 106 pfu BHV-1, one dose daily for 5 days. Tumors were measured every 2 days until animals reached end point. (a) Kaplan-Meier estimates of survival and (b) tumor volumes of CRs treated with 5 × 106 pfu BHV-1 i.t. (c) 2 mg/kg 5-Aza i.p. or, (d) 5 × 106 pfu BHV-1 i.t. and 2 mg/kg 5-Aza i.p. are shown. *Indicates animals for which vacuous cavities were found in primary tumors upon necropsy.
LCRT tumors grow quickly and are very invasive with a high probability of developing lung and lymph lesions.11,32 In this study, secondary lesions were found in the armpit on the lateral side in all CRs in the BHV-1 (3/3) and 5-Aza (4/4) monotherapy groups. The incidence of secondary lesions in the armpit was significantly lower (P = 0.03) in combination therapy treated CRs, in which lesions were detected in only 38% (3/8) of animals. In addition, secondary lesions were also frequently detected posterior to the primary tumor. The incidence of these lesions was 67% in BHV-1 (2/3) and 75% in 5-Aza (3/4) treated CRs, in comparison to only 13% (1/8) in combination therapy treated animals, although these differences were not statistically significant (P = 0.06). Furthermore, lesions found in the lungs of combination treated CRs were fewer and of lower grade when compared to those in the lungs of CRs treated with BHV-1 alone (Supplementary Figure S3e,f). Importantly, we did not detect any respiratory distress in these animals that limited survival.
Combination therapy with BHV-1 and 5-Aza induces immune cell infiltration and tumor cell clearance in subcutaneous LCRT cotton rat tumors
The role of central and peripheral tolerance as a barrier to successful OVT has made the study of OV antitumor efficacy within the context of an immunocompetent host an absolute must.3,14,15,28 Proper engagement of the immune system in OVT achieves prolonged antitumor responses by breaking immune tolerance, long after viral clearance. This outcome circumvents the requirement for extensive OV spread within the tumor, which is thought to play a minor role in the success of HSV OVs.14,15,33 Here, tumors were harvested from PBS, BHV-1, and 5-Aza treatment groups and responders from the combination therapy group at endpoint for histological analysis. A responder was defined as an animal that had slower tumor growth in comparison to monotherapy controls (5-Aza or BHV-1), had increased survival, or for which a fluid filled space was present in the primary tumor at endpoint. It should be noted that the histology for the PBS-treated control was obtained from a previous experiment, but is representative of this group. Histologically, tumors treated with combination therapy had large areas of tumor cell clearance that were more extensive in comparison to the tumors of animals treated with BHV-1 monotherapy (Figure 7a–d). Interestingly, the tumors of CRs treated with combination therapy were densely packed with immune infiltrates (Figure 7e–h). The presence of large granulated cells was observed in combination therapy treated tumors, particularly in cleared areas and around vasculature (Figure 7i,j). Large granulated cells were not observed in PBS or monotherapy-treated tumors. Although the lack of reagents prevents the identification of these infiltrates, large granulated cells in solid tumors have been described and were identified to be NK cells.34 Together, these data suggest that 5-Aza may sensitize LCRT cells to BHV-1-mediated oncolysis and induce an immune response that facilitates tumor destruction and decreases the rate of secondary lesions.
Figure 7.
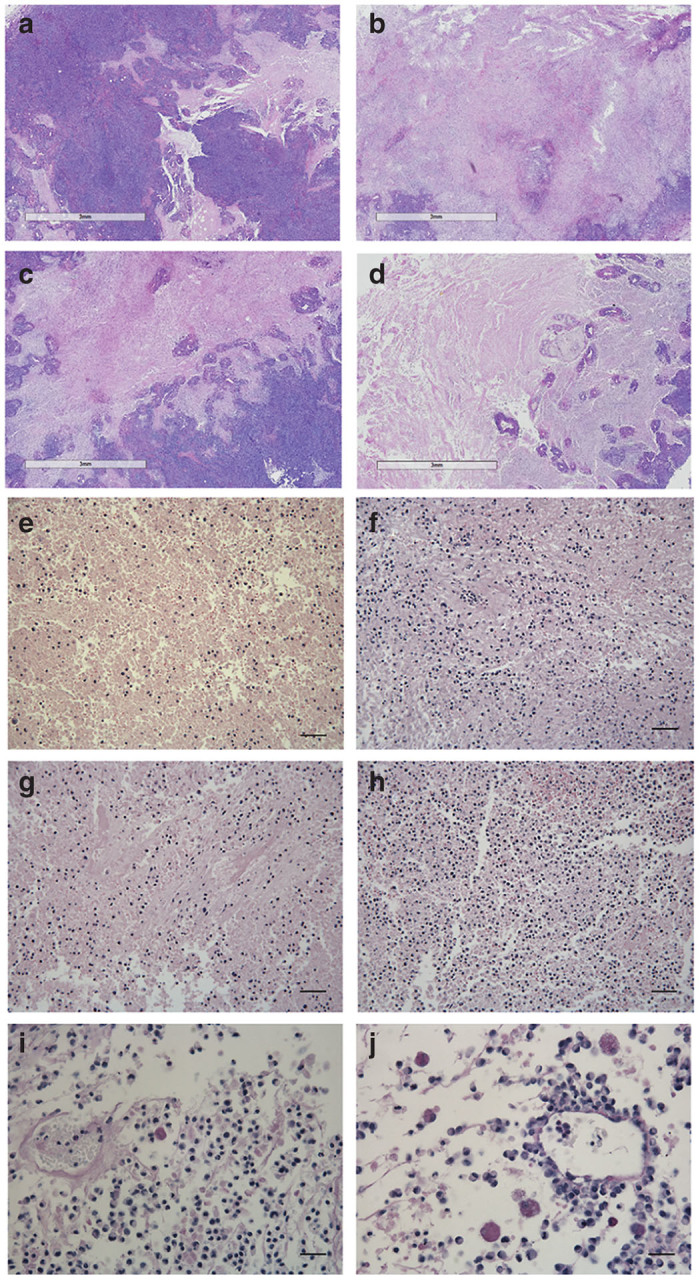
Representative histological analysis of primary tumors from bovine herpesvirus type 1 (BHV-1), 5-Aza and combination therapy treated cotton rats. Tumors were excised from phosphate-buffered saline (a,e), BHV-1 (5 × 106 pfu; (c,g)), 5-Aza (2 mg/kg; (b,f)) and combination treated (d,h–j) animals at endpoint, fixed and H&E or periodic-acid Schiff (PAS) stained for histological analysis. Images were captured at 1× magnification (a–d; tumor cell clearance, H&E) using a Leica Aperio AT Turbo slide scanner or at 20× (e–h; immune cell infiltration, H&E) or 40× (i,j; large granulated lymphocytes, PAS) magnification with a Leica DM IRE2 microscope. Scale bars = 0.3 mm (a–d), 1 mm (e–h), 0.5 mm (i,j).
Discussion
Clinical studies have demonstrated that OVs are an effective and novel cancer therapeutic with unique tumor-targeting mechanisms. However, it has become apparent that combination therapy approaches will be necessary to achieve sustained antitumor responses. Current efforts focused on understanding OV-host interactions have revealed novel agents for combination therapy.35 Epigenetic modifiers enhance OV replication and cytotoxicity by modifying viral gene expression and antiviral immune responses.23–25 In addition to upregulating genes associated with innate and adaptive immunity, 5-Aza increases the sensitivity of tumor cells to T cell mediated cytotoxicity by increasing expression of TAAs.36,37 Enhanced immune activation following 5-Aza treatment may serve to break central and peripheral tolerance to TAAs.36,38 Furthermore, clinical data support the use of epigenetic modifiers, such as 5-Aza, in the treatment of breast cancer as they improve patient responses to therapy.17,18,39
In this study, we show that BHV-1 infection is inefficient in LCRT cells and a significant decrease in cellular viability is not observed. However, when cells were treated with 5-Aza prior to BHV-1 infection, we detected a 55% decrease in cellular viability (Figure 3c). This coincided with an increase in virus replication and bovine infected cell protein 0 (bICP0) expression (Figure 3b and data not shown), but only a modest increase in virus output (Figure 4). We have previously reported that BHV-1 is able to induce cytotoxicity in human breast cancer cells in the absence of a productive infection.8 These data suggest that the presence of a fully permissive cellular environment is not required for BHV-1 to elicit oncolytic activity.
Animal models play an important role in the preclinical evaluation of OVs. However, the relevancy of these models has come under scrutiny as their ability to predict the anticancer efficacy of OVT has been limited.3 Antitumor responses from OVT differ significantly when comparing tolerized and nontolerized TAAs.13,14,28 Although we did not achieve complete responses to BHV-1 mono or combination therapy, by using a tolerized model of breast adenocarcinoma, we were able to evaluate the oncolytic capacity of BHV-1 in the presence of natural local and systemic immunosuppression. This better recapitulates the human immune landscape and is therefore a more accurate predictor of therapeutic efficacy. In contrast to small RNA OVs for which i.t. viral replication and spread are key determinants of therapy success,33 these determinants do not dictate the efficacy of large DNA OVs such as HSV-1, where engagement of the host immune system plays a critical role in antitumor efficacy.14,15,28 We observed an increase in BHV-1 replication in vitro following treatment of LCRT cells with 5-Aza; however, it seems unlikely that combination therapy functions via increased BHV-1 replication in vivo given that combination treatment (5-Aza and BHV-1 5 × 106 pfu) did not result in the extensive pathology seen with 5 × 107 pfu BHV-1, despite the permissivity of CRs to BHV-1 infection.11 These data are consistent with reports that direct virus-mediated effects do not dictate the success of OVT using herpesvirus vectors.14,15,28 Studies from our group have shown that HSV vectors with the greatest oncolytic effect in multiple murine models of breast carcinoma were those which had a lower viral burst and were rapidly cleared from the tumor.15 Here, it appears that combination therapy elicits its in vivo effects by modulating the host antitumor immune response, as observed in other HSV combination therapy approaches.15 In fact, combination therapy elicited tumor cell clearance and induced significant infiltration of immune effectors in comparison to monotherapy treated tumors, including densely granulated immune cells (Figure 7).34 Further characterization of the immune response is not possible due to the lack of reagents in the CR model; however, data from other groups describe a population of large densely granulated immune effectors in solid tumors which were identified as NK cells.34 Furthermore, due to the sensitivity of CRs to anesthetics, we were unable to use in vivo imaging techniques to visualize the biodistribution of BHV-1.
The treatment of secondary lesions remains a major hurdle to successful cancer therapy. The induction of self-perpetuating and sustained systemic antitumoral protection is important in the treatment of nonresectable disease.40–42 LCRT tumors grow quickly and are very invasive with a high probability of developing lung and lymph lesions.11,32 High incidence of secondary lesions to the lymph nodes and lungs is also common in human breast cancer patients.43,44 Treatment of CRs with 5 × 107 pfu BHV-1 caused regression of the primary tumor in 40% (4/10) of animals and significantly increased survival (Supplementary Figure S2). However, this dose was not well tolerated and long-term survival was not achieved as animals reached endpoint due to respiratory distress caused by significant lesions in the lungs. In contrast, CRs treated with combination therapy showed a significant decrease in the number of secondary lesions in the armpit and the number and grade of tumors in the lungs were decreased relative to monotherapy-treated CRs. These data suggest that combination therapy may induce antitumor systemic immune responses to limit the formation of secondary lesions. For cancers such as breast cancer, where primary lesions are often removed surgically, these findings suggest that multiple treatment approaches may offer the best clinical outcome for patients. Moreover, these data highlight the recurring theme that, at least for herpesvirus based OVs, increased and sustained virus replication does not equate with augmented efficacy.15
The disruption of epigenetic processes during virus infection can lead to altered cellular and viral gene expression and function which influence cell status.45,46 Herpesviruses have been shown to induce global cellular epigenetic modification using virus encoded proteins and miRNA species which regulate virus replication and pathogenesis.47,48 Recent studies have described using epigenetic modifiers such as 5-Aza in combination with OVs to improve antitumor responses.23 It is plausible that epigenetic reprogramming by 5-Aza may synergize with that induced by BHV-1 to alter susceptibility of tumor cells to BHV-1 infection and modulate immune mediated antitumor effects. Further studies are required to unravel the relationship between BHV-1 and 5-Aza. How 5-Aza modulates the antiviral response and/or the tumor microenvironment to enhance therapeutic efficacy remains to be elucidated. Understanding the mechanism by which 5-Aza modulates BHV-1 activity can ultimately be exploited to inform further development of novel combination therapy strategies.
Materials and Methods
Cell lines
Cell lines were maintained at 37 °C + 5% CO2 in medium supplemented with 2 mmol/l L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. HeLa and HEL cells (American Type Culture Collection, Manassas, VA) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). The CR cell line LCRT was obtained from Dr. Ann Tollefson (St. Louis University) and were cultured in DMEM supplemented with 10% FBS. MDBK cells were obtained from Vikram Misra (University of Saskatchewan) and were maintained in DMEM supplemented with 5% horse serum. Ventressca (primary human adult lung fibroblasts) were obtained from Dr. Jack Gauldie (McMaster University) and were maintained in minimum essential medium/F15 with 15% FBS.
Viruses
BHV-1 expressing GFP was a kind gift from Dr. Günther Keil (Friedrich-Loeffler-Institut, Germany) and was propagated and titrated on MDBK cells. Virus preparations were sucrose cushion purified.8 Purified virus was resuspended in PBS and stored at −80 °C.
Drug preparation
5-Aza (Sigma-Aldrich, St. Louis, MO) stock powder was stored at −20 °C and dissolved in complete DMEM to obtain a working solution. Drug was freshly prepared for each experiment.
Western blot analysis
Cells were treated with 1 or 3 µmol/l 5-Aza for 14 hours24 and whole cell lysates were immediately collected (Dnmt1 blots). Whole cell lysates were collected in whole cell extract buffer (20 mmol/l hydroxyethyl piperazineethanesulfonic acid, pH 7.4, 100 mmol/l NaCl, 10 mmol/l β-glycerophosphate, 0.2% Triton X-100, 1 mmol/l sodium orthovanadate, 50 mmol/l sodium fluoride, 1 mmol/l phenylmethylsulfonyl fluoride, 2 mmol/l dithiothreitol, 1× protease inhibitor cocktail (Sigma)) and lysed on ice for 30 minutes. Lysates were centrifuged at 1,000 rpm for 10 minutes at 4 °C and the supernatants were collected. Protein was quantified using a Bradford assay kit (Bio-Rad Laboratories, Mississauga, ON). Whole cell extracts were boiled in sample buffer containing sodium dodecyl sulfate and β-mercaptoethanol and run on a 7.5% for Dnmt1 expression analysis. Gels were transferred onto polyvinylidene difluoride membranes (Millipore, Billerica, MA) with a wet transfer apparatus at 100 V for 1 hour. All blots were blocked in 5% nonfat milk in Tris-buffered saline (TBS) at room temperature for 2 hours. Blots were probed with primary antibodies specific for Dnmt1 ((C-17); 1:400, Santa Cruz Biotechnology, Dallas, TX) diluted in TBS-Tween (0.1%), overnight at 4 °C. Blots were probed with anti-goat secondary antibodies conjugated to horseradish peroxidase (Sigma) diluted 1:2,000 in 5% nonfat milk in 0.1% TBS-Tween. Blots were visualized by chemiluminescence.
Measurement of virus replication and cellular viability
LCRT cells (1 × 105 cells/ml) were seeded into 96-well plates and treated for 14 hours24 with 1 or 3 µmol/l 5-Aza prior to infection with BHV-1 at MOI 3 or 5 for 1 hour at 37 °C. Two days pi, plates were scanned on a Typhoon BioAnalyzer (GE Healthcare, Piscataway, NJ) to visualize virus replication as a function of GFP fluorescence. Two days pi, cellular viability was assessed using the SpectraMax i3 Multi-Mode Microplate Reader (Molecular Devices, Sunnyvale, CA). Measures of cellular metabolism were assessed using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma-Aldrich). Cells were incubated with MTT (10% v/v) for 4 hours at 37 °C, after which fluorescence was read. Samples were analyzed in triplicate with a total of three independent experiments performed. Data were analyzed relative to uninfected controls and corrected for background fluorescence. The CI for each concentration of 5-Aza with each MOI of BHV-1 was calculated using CompuSyn software (ComboSyn, Paramus, NJ) and used to evaluate pharmacological synergy.
Determination of IC50 values on normal primary cells
Human normal primary cells (1 × 105 cells/ml) were seeded into 96-well plates and treated for 14 hours24 with 0, 0.25, 0.5, 1, 3, 5, 7, or 10 µmol/l 5-Aza. Two days pi, cellular viability was assessed using the SpectraMax i3 Multi-Mode Microplate Reader (Molecular Devices). For both cell lines, survival was measured relative to untreated controls. Median-effect plots were generated using CompuSyn software (ComboSyn) to determine the IC50 values of 5-Aza.
Viral burst
LCRT cells were seeded into six-well plates and treated for 14 hours24 with 1 or 3 µmol/l 5-Aza prior to infection with BHV-1 at MOI 3 or 5 for 1 hour at 37 °C. One, 2, and 3 days pi, viral supernatants and infected cells were collected. Samples were freeze/thawed three times and sonicated for 1 minute prior to centrifugation at 1,000 rpm for 10 minutes at 4 °C. Supernatant was collected and titrated by serial dilution in serum-free DMEM. Dilutions were applied to MDBK cells for 1 hour at 37 °C. MDBK monolayers were maintained in DMEM supplemented with 0.5% horse serum in 1% methylcellulose. At 2 days pi, cells were scanned on a Typhoon BioAnalyzer (GE Healthcare) and pfu were counted and viral burst calculated.
In vivo CR experiments
CRs were maintained at the McMaster University Central Animal Facility and all the procedures were performed in full compliance with the Canadian Council on Animal Care and approved by the Animal Research Ethics Board of McMaster University. Six to seven week old CRs were subcutaneously implanted with 5 × 105 LCRT cells. Tumors reached treatable size within 2 weeks postinjection. Tumors were treated by injecting 5 × 106 or 5 × 107 total pfu BHV-114,28 i.t. (50 µl total) once daily for 5 consecutive days, or 5 × 106 total pfu BHV-1 i.t. (50 µl total) for 5 consecutive days 1 day following a single i.p. injection of 5-Aza (2 mg/kg).49 Tumors were measured every 2 days and fold changes in tumor volume were calculated relative to the volume at the start of treatment (d = 0). Animals were considered to be at endpoint when tumors reached 10% of their total body weight, tumor ulceration occurred in non-BHV-1-treated animals or when breathing difficulties were observed due to metastases.
Histological analysis
Tumors were resected from animals at endpoint and fixed in 10% neutral buffered formalin for 2–5 days depending on the size of the tumor (5 days for large tumors) and transferred to 70% ethanol for preservation. Tumor tissue was embedded in paraffin and 4-µm sections were prepared. Sections were stained with hematoxylin and eosin (H&E) or periodic-acid Schiff (PAS) and subsequently analyzed using a Leica DM IRE2 microscope or Leica Aperio AT Turbo slide scanner.
Statistical analysis
One-way analysis of variance was used to analyze the significance of the differences in viral burst with a Bonferroni post hoc test to compare the pairs of data within the distribution. The log-rank Mantel-Cox test was used to determine statistical significance for the difference in Kaplan-Meier survival between treatments. The χ2 test was used to determine the statistical significance of the incidence in secondary lesions between treatments. The null hypothesis was rejected for P values less than 0.05. Survival analysis was carried out using GraphPad Prism (LaJolla, CA) and all the other analyses were performed using Microsoft Excel (Redmond, WA).
Acknowledgments
We thank Vikram Misra (University of Saskatchewan, Canada), Günther Keil (Friedrich-Loeffler-Institut, Germany) and Clinton Jones (University of Nebraska, USA) for reagents and Derek Cummings for technical assistance. We would also like to thank Dr. Jean-Claude Cutz (McMaster University, Canada) for his help with analyzing lung histology samples. Breanne Cuddington was the recipient of a fellowship from the Canadian Breast Cancer Foundation. This work was sponsored by operating grants from the Cancer Research Society and the Canadian Cancer Society Research Institute (formerly the Canadian Breast Cancer Research Alliance). The authors acknowledge there are no financial conflicts of interest related to this research.
The authors declare no conflict of interest.
References
- Cervantes-García, D, Ortiz-López, R, Mayek-Pérez, N and Rojas-Martínez, A (2008). Oncolytic virotherapy. Ann Hepatol 7: 34–45. [PubMed] [Google Scholar]
- Vähä-Koskela, MJ, Heikkilä, JE and Hinkkanen, AE (2007). Oncolytic viruses in cancer therapy. Cancer Lett 254: 178–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell, SJ, Peng, KW and Bell, JC (2012). Oncolytic virotherapy. Nat Biotechnol 30: 658–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman, HL, Kim, DW, DeRaffele, G, Mitcham, J, Coffin, RS and Kim-Schulze, S (2010). Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann Surg Oncol 17: 718–730. [DOI] [PubMed] [Google Scholar]
- Senzer, NN, Kaufman, HL, Amatruda, T, Nemunaitis, M, Reid, T, Daniels, G et al. (2009). Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J Clin Oncol 27: 5763–5771. [DOI] [PubMed] [Google Scholar]
- Jones, C and Chowdhury, S (2010). Bovine herpesvirus type 1 (BHV-1) is an important cofactor in the bovine respiratory disease complex. Vet Clin North Am Food Anim Pract 26: 303–321. [DOI] [PubMed] [Google Scholar]
- Rodrigues, R, Cuddington, B and Mossman, K (2010). Bovine herpesvirus type 1 as a novel oncolytic virus. Cancer Gene Ther 17: 344–355. [DOI] [PubMed] [Google Scholar]
- Cuddington, BP, Dyer, AL, Workenhe, ST and Mossman, KL (2013). Oncolytic bovine herpesvirus type 1 infects and kills breast tumor cells and breast cancer-initiating cells irrespective of tumor subtype. Cancer Gene Ther 20: 282–289. [DOI] [PubMed] [Google Scholar]
- Hushur, O, Takashima, Y, Matsumoto, Y and Otsuka, H (2004). Restriction of bovine herpesvirus 1 (BHV-1) growth in non-permissive cells beyond the expression of immediate early genes. J Vet Med Sci 66: 453–455. [DOI] [PubMed] [Google Scholar]
- Papp, Z, Babiuk, LA and Baca-Estrada, ME (1998). Induction of immunity in the respiratory tract and protection from bovine herpesvirus type 1 infection by different routes of immunization with recombinant adenovirus. Viral Immunol 11: 79–91. [DOI] [PubMed] [Google Scholar]
- Prince, GA (1994). The cotton rat in biomedical research. Animal Welfare Information Center Newsletter 5: 3–5. [Google Scholar]
- Toth, K, Spencer, JF and Wold, WS (2007). Immunocompetent, semi-permissive cotton rat tumor model for the evaluation of oncolytic adenoviruses. Methods Mol Med 130: 157–168. [DOI] [PubMed] [Google Scholar]
- Frey, AB and Monu, N (2006). Effector-phase tolerance: another mechanism of how cancer escapes antitumor immune response. J Leukoc Biol 79: 652–662. [DOI] [PubMed] [Google Scholar]
- Sobol, PT, Boudreau, JE, Stephenson, K, Wan, Y, Lichty, BD and Mossman, KL (2011). Adaptive antiviral immunity is a determinant of the therapeutic success of oncolytic virotherapy. Mol Ther 19: 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workenhe, ST, Simmons, G, Pol, JG, Lichty, BD, Halford, WP and Mossman, KL (2014). Immunogenic HSV-mediated oncolysis shapes the antitumor immune response and contributes to therapeutic efficacy. Mol Ther 22: 123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, PA and Baylin, SB (2002). The fundamental role of epigenetic events in cancer. Nat Rev Genet 3: 415–428. [DOI] [PubMed] [Google Scholar]
- Byler, S, Goldgar, S, Heerboth, S, Leary, M, Housman, G, Moulton, K et al. (2014). Genetic and epigenetic aspects of breast cancer progression and therapy. Anticancer Res 34: 1071–1077. [PubMed] [Google Scholar]
- Yang, X, Ferguson, AT, Nass, SJ, Phillips, DL, Butash, KA, Wang, SM et al. (2000). Transcriptional activation of estrogen receptor alpha in human breast cancer cells by histone deacetylase inhibition. Cancer Res 60: 6890–6894. [PubMed] [Google Scholar]
- Radpour, R, Barekati, Z, Kohler, C, Schumacher, MM, Grussenmeyer, T, Jenoe, P et al. (2011). Integrated epigenetics of human breast cancer: synoptic investigation of targeted genes, microRNAs and proteins upon demethylation treatment. PLoS ONE 6: e27355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirza, S, Sharma, G, Pandya, P and Ralhan, R (2010). Demethylating agent 5-aza-2-deoxycytidine enhances susceptibility of breast cancer cells to anticancer agents. Mol Cell Biochem 342: 101–109. [DOI] [PubMed] [Google Scholar]
- Zeng, WG, Li, JJ, Hu, P, Lei, L, Wang, JN and Liu, RB (2013). An oncolytic herpes simplex virus vector, G47?, synergizes with paclitaxel in the treatment of breast cancer. Oncol Rep 29: 2355–2361. [DOI] [PubMed] [Google Scholar]
- Chung, SM, Advani, SJ, Bradley, JD, Kataoka, Y, Vashistha, K, Yan, SY et al. (2002). The use of a genetically engineered herpes simplex virus (R7020) with ionizing radiation for experimental hepatoma. Gene Ther 9: 75–80. [DOI] [PubMed] [Google Scholar]
- Okemoto, K, Kasai, K, Wagner, B, Haseley, A, Meisen, H, Bolyard, C et al. (2013). DNA demethylating agents synergize with oncolytic HSV1 against malignant gliomas. Clin Cancer Res 19: 5952–5959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsuki, A, Patel, A, Kasai, K, Suzuki, M, Kurozumi, K, Chiocca, EA et al. (2008). Histone deacetylase inhibitors augment antitumor efficacy of herpes-based oncolytic viruses. Mol Ther 16: 1546–1555. [DOI] [PubMed] [Google Scholar]
- Bridle, BW, Chen, L, Lemay, CG, Diallo, JS, Pol, J, Nguyen, A et al. (2013). HDAC inhibition suppresses primary immune responses, enhances secondary immune responses, and abrogates autoimmunity during tumor immunotherapy. Mol Ther 21: 887–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stresemann, C and Lyko, F (2008). Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int J Cancer 123: 8–13. [DOI] [PubMed] [Google Scholar]
- Chou, TC and Talalay, P (1984). Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 22: 27–55. [DOI] [PubMed] [Google Scholar]
- Workenhe, ST, Pol, JG, Lichty, BD, Cummings, DT and Mossman, KL (2013). Combining oncolytic HSV-1 with immunogenic cell death-inducing drug mitoxantrone breaks cancer immune tolerance and improves therapeutic efficacy. Cancer Immunol Res 1: 309–319. [DOI] [PubMed] [Google Scholar]
- Kanai, R, Wakimoto, H, Cheema, T and Rabkin, SD (2010). Oncolytic herpes simplex virus vectors and chemotherapy: are combinatorial strategies more effective for cancer? Future Oncol 6: 619–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojton, J and Kaur, B (2010). Impact of tumor microenvironment on oncolytic viral therapy. Cytokine Growth Factor Rev 21: 127–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold, CJ, Bankier, AA and Fleischmann, D (1996). Lung metastases. Eur Radiol 6: 596–606. [DOI] [PubMed] [Google Scholar]
- Toth, K, Spencer, JF, Tollefson, AE, Kuppuswamy, M, Doronin, K, Lichtenstein, DL et al. (2005). Cotton rat tumor model for the evaluation of oncolytic adenoviruses. Hum Gene Ther 16: 139–146. [DOI] [PubMed] [Google Scholar]
- Ayala-Breton, C, Russell, LO, Russell, SJ and Peng, KW (2014). Faster replication and higher expression levels of viral glycoproteins give the vesicular stomatitis virus/measles virus hybrid VSV-FH a growth advantage over measles virus. J Virol 88: 8332–8339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, RB, Engels, B, Arina, A, Schreiber, K, Hyjek, E, Schietinger, A et al. (2012). Densely granulated murine NK cells eradicate large solid tumors. Cancer Res 72: 1964–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diallo, JS, Le Boeuf, F, Lai, F, Cox, J, Vaha-Koskela, M, Abdelbary, H et al. (2010). A high-throughput pharmacoviral approach identifies novel oncolytic virus sensitizers. Mol Ther 18: 1123–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gang, AO, Frøsig, TM, Brimnes, MK, Lyngaa, R, Treppendahl, MB, Grønbæk, K et al. (2014). 5-Azacytidine treatment sensitizes tumor cells to T-cell mediated cytotoxicity and modulates NK cells in patients with myeloid malignancies. Blood Cancer J 4: e197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H, Chiappinelli, KB, Guzzetta, AA, Easwaran, H, Yen, RW, Vatapalli, R et al. (2014). Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget 5: 587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasi, TB, Magner, WJ and Khan, AN (2006). Epigenetic regulation of immune escape genes in cancer. Cancer Immunol Immunother 55: 1159–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly, RM, Andreopoulou, E, Allred, JB, Jeter, SC, Adam, BM, Espinoza-Delagado, I et al. (2013). A phase 2 study investigating the safety, efficacy, and surrogate biomarkers or response of 5-Azacytidine (5-AZA) and entinostat (MS-275) in patients with advanced breast cancer. Cancer Res 73: 4666. [Google Scholar]
- Yu, F, Wang, X, Guo, ZS, Bartlett, DL, Gottschalk, SM and Song, XT (2014). T-cell engager-armed oncolytic vaccinia virus significantly enhances antitumor therapy. Mol Ther 22: 102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo, J, Reid, T, Ruo, L, Breitbach, CJ, Rose, S, Bloomston, M et al. (2013). Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med 19: 329–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesonen, S, Diaconu, I, Kangasniemi, L, Ranki, T, Kanerva, A, Pesonen, SK et al. (2012). Oncolytic immunotherapy of advanced solid tumors with a CD40L-expressing replicating adenovirus: assessment of safety and immunologic responses in patients. Cancer Res 72: 1621–1631. [DOI] [PubMed] [Google Scholar]
- Berman, AT, Thukral, AD, Hwang, WT, Solin, LJ and Vapiwala, N (2013). Incidence and patterns of distant metastases for patients with early-stage breast cancer after breast conservation treatment. Clin Breast Cancer 13: 88–94. [DOI] [PubMed] [Google Scholar]
- Disibio, G and French, SW (2008). Metastatic patterns of cancers: results from a large autopsy study. Arch Pathol Lab Med 132: 931–939. [DOI] [PubMed] [Google Scholar]
- Sharma, S, Kelly, TK and Jones, PA (2010). Epigenetics in cancer. Carcinogenesis 31: 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez, AF and Esteller, M (2010). Viral epigenomes in human tumorigenesis. Oncogene 29: 1405–1420. [DOI] [PubMed] [Google Scholar]
- Adhya, D and Basu, A (2010). Epigenetic modulation of host: new insights into immune evasion by viruses. J Biosci 35: 647–663. [DOI] [PubMed] [Google Scholar]
- Cullen, BR (2011). Viruses and microRNAs: RISCy interactions with serious consequences. Genes Dev 25: 1881–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belinsky, SA, Grimes, MJ, Picchi, MA, Mitchell, HD, Stidley, CA, Tesfaigzi, Y et al. (2011). Combination therapy with vidaza and entinostat suppresses tumor growth and reprograms the epigenome in an orthotopic lung cancer model. Cancer Res 71: 454–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.