

Most of the antisense transcripts in bacteria are the product of transcriptional noise derived from spurious promoters.
Keywords: RNA, bacterial antisense RNAs
Abstract
cis-Encoded antisense RNAs (asRNAs) are widespread along bacterial transcriptomes. However, the role of most of these RNAs remains unknown, and there is an ongoing discussion as to what extent these transcripts are the result of transcriptional noise. We show, by comparative transcriptomics of 20 bacterial species and one chloroplast, that the number of asRNAs is exponentially dependent on the genomic AT content and that expression of asRNA at low levels exerts little impact in terms of energy consumption. A transcription model simulating mRNA and asRNA production indicates that the asRNA regulatory effect is only observed above certain expression thresholds, substantially higher than physiological transcript levels. These predictions were verified experimentally by overexpressing nine different asRNAs in Mycoplasma pneumoniae. Our results suggest that most of the antisense transcripts found in bacteria are the consequence of transcriptional noise, arising at spurious promoters throughout the genome.
INTRODUCTION
The catalog of bacteria-encoded RNAs has recently undergone a vast expansion. The canonical mRNAs and known noncoding RNAs [ribosomal RNAs (rRNAs), transfer RNA (tRNAs), transfer mRNA (tmRNA), and others] are now accompanied by a handful of new transcript categories. Small, non–protein-coding RNAs or sRNAs are one of these new categories. The numbers of initially reported sRNAs ranged from dozens to hundreds in different species (1, 2). These include cis-encoded sRNAs, which overlap functionally defined genes, either in sense or antisense (thus named asRNAs), and trans-encoded sRNAs, which are separated from their target genes. These sRNAs span a wide range of lengths: from dozens of to a few thousand base pairs (2). However, recent improvements in techniques for analysis of transcription have revealed that noncoding transcription in prokaryotes is pervasive through the genome (3–5). Still, only few sRNAs have been functionally characterized (6–8), most of which correspond to the category of trans-encoded sRNAs. Examples of these are the ones associated with bacterial virulence (9–11). The most common mechanism of action of sRNAs is via complementary base pairing with coding sequences (fig. S1A). RNA duplex formation between sRNA and mRNA can change mRNA stability, inducing degradation or stabilization of the duplex. This duplex may as well induce or repress mRNA translation by affecting the ribosome binding site (2, 12). Another asRNA regulatory mechanism is transcriptional interference, occurring if two RNA polymerases transcribing in convergent directions collide (13). Other types of RNA having a regulatory role by “nonstandard” mechanisms should not be disregarded. For instance, if there was a Dicer-like mechanism in bacteria as it occurs in eukaryotes (14), low abundant RNAs could exert a strong influence on complementary, more abundant, mRNAs. In this respect, we have the CRISPR (clustered regularly interspaced short palindromic repeats)/Cas system in bacteria, where crRNAs (CRISPR RNAs), even if not abundant, target the enzyme against foreign DNA (15) and/or RNA sequences (16).
There is an ongoing discussion in both eukaryotes and prokaryotes as to what extent this plethora of sRNAs provides a crucial layer of transcriptional and translational regulation, or if a large part of them are the result of transcriptional noise, arising from spurious promoters (17, 18). Bacterial promoters are characterized by low information content, and their major landmark is the Pribnow motif that has the consensus sequence “5′-TANAAT-3′” (19). Other features include (i) the −35 box, although this has been shown not to be essential (especially in Firmicutes) and can be replaced by other elements (20), and (ii) low melting energies, which ultimately depend on the AT composition of the promoter region. Such low information content implies that promoters could easily arise by random mutations in bacterial genomes, especially given the presumptive bias toward G/C nucleotides mutating to A/T (21). If sRNAs are the product of transcriptional noise due to spurious 5′-TANAAT-3′ boxes, we predict that the number of sRNAs in bacteria will strongly correlate to the AT content of their genomes in an exponential manner (fig. S2A). Because of the stochastic nature of transcription and the short half-life of RNAs in bacteria, low levels of random production of asRNA from these spurious Pribnow boxes would not affect the levels of the sense mRNA (fig. S1B).
RESULTS
To investigate these hypotheses, we annotated sRNAs de novo in the genomes of Buchnera aphidicola, Mycoplasma hyopneumoniae, and Mycoplasma mycoides subspecies capri (tables S1 to S3 and fig. S3) in a similar way as we did with Mycoplasma pneumoniae (22). We also considered the sRNAs annotated using deep sequencing data in 17 other bacterial genomes and a chloroplast genome (table S4). These 21 genomes span an AT content ranging from 28 to 80%, and their genome sizes range from 416 kb (B. aphidicola Cc) to 9.02 Mb (Streptomyces avermitilis). Investigating the number of canonical Pribnow boxes in these genomes, we found an exponential dependency of the number of boxes on the AT content, qualitatively similar to our theoretical expectations (fig. S2A). Moreover, comparison of the number of these boxes upstream of open reading frames (ORFs) and sRNAs showed that the proportion of sRNAs with Pribnow boxes is similar to or higher than the proportion of ORFs having them (fig. S2B). This supports the hypothesis that an increase in AT content also results in an increase in spurious Pribnow boxes.
We found that the number of sRNAs normalized by genome size versus the AT content in the studied bacterial species has a clear exponential dependency (Fig. 1A), similar to that of the number of TANAAT motifs randomly expected given a certain AT% (fig. S2A). The exponential trend observed for the sRNAs is conserved, omitting the species whose sRNAs were de novo annotated (R2 = 0.814), indicating that it is not an artifact of the method used to identify them (see fig. S3 and Materials and Methods). In contrast to the observed sRNA trend, the number of coding genes normalized by genome size shows no dependency on AT content, and this trend is invariant with respect to genome size (Fig. 1B). We tested whether the AT dependency held true for both asRNAs and trans-encoded sRNAs. asRNAs follow an exponential dependency on the AT content (fig. S4A), whereas trans-encoded sRNAs behave similarly to coding genes and are uncorrelated to the AT content of the intergenic regions (even when considering a minimal size larger than that of an average asRNA; fig. S4B). These results support the transcriptional noise hypothesis, and that random mutations in coding genes could result in spurious antisense 5′-TANAAT-3′ boxes, in a manner related to the genome AT content, which could drive the expression of asRNAs.
Fig. 1. Different genomic features show distinct dependency on the genomic AT content.
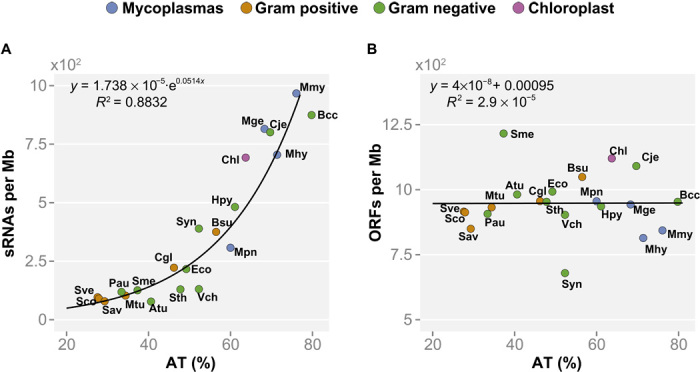
The number of features was divided by the genome size for normalization and represented versus the genomic AT content. The following genomes are represented: Atu, Agrobacterium tumefaciens; Bcc, Buchnera aphidicola (str Cc); Bsu, Bacillus subtilis; Cgl, Corynebacterium glutamicum; Chl, chloroplast (Arabidopsis thaliana); Cje, Campylobacter jejuni; Eco, Escherichia coli; Hpy, Helicobacter pylori; Mge, Mycoplasma genitalium; Mhy, Mycoplasma hyopneumoniae; Mmy, Mycoplasma mycoides; Mpn, Mycoplasma pneumoniae; Mtu, Mycobacterium tuberculosis; Pau, Pseudomonas aeruginosa; Sav, Streptomyces avermitilis; Sco, Streptomyces coelicolor; Sme, Sinorhizobium meliloti; Sth, Salmonella typhimurium; Sve, Streptomyces venezuelae; Syn, Synechocystis spp., Vch, Vibrio cholerae. (A) Number of total sRNAs in different bacteria. Total sRNAs have an exponential dependency on the AT content (R2 = 0.88) and do not correlate with genome size. (B) Genome compaction (that is, number of ORFs normalized by genome size) versus AT content. Genome compaction in the different bacterial genomes analyzed shows no dependency on the AT content. Instead, the number of ORFs in bacterial genomes correlates with the genome size (R = 0.99).
Regarding expression levels, it has been shown that essential ORFs show higher mRNA levels, suggesting that elements with essential roles are more transcribed (23). Therefore, we compared transcript levels of ORFs and asRNAs in eight of the bacteria in our study. In all cases, average asRNA levels were lower than average mRNA levels (fig. S5A). This could indicate that at least a majority of the asRNAs could be nonessential. Indeed, a recent study on the essentiality of the M. pneumoniae genome revealed that only 5% of all sRNAs are essential (23). We also compared the expression of each asRNA to its overlapping mRNA. asRNA-mRNA expression ratios are presented in fig. S5B. These ratios are below 1 in most of the cases (fig. S5B). For three of the species in our study (M. pneumoniae, M. mycoides, and Bacillus subtilis), we compared asRNA levels at exponential and stationary growth phases (fig. S5C). Most of the asRNAs remain unchanged, excluding the effect of the growth phase at where the bacteria were analyzed. Additionally, asRNA and trans-encoded sRNA levels were compared in five species (B. aphidicola, Mycoplasma genitalium, M. pneumoniae, M. mycoides, and M. hyopneumoniae), and we found that asRNA expression is significantly lower than trans-encoded sRNA levels in all cases (Welch’s two-sample t test, P < 0.05).
We estimated the energy consumed by the cells in transcribing these asRNAs in M. pneumoniae, considering the number of noncoding RNAs, their length, and their transcription rate, compared to those of mRNAs, tRNAs, and rRNAs (see Materials and Methods). M. pneumoniae spends ~5000 adenosine triphosphate (ATP) units per cell per second in transcribing mRNAs, tRNAs, and rRNAs (24). This amount is proportional to the transcription rate of these molecules, their length, and their copy number in the cell. Taking into account these parameters for sRNAs, we estimate that M. pneumoniae spends 2.94% of the energy of RNA transcription in synthesizing sRNAs, equivalent to ~147 ATP units per cell per second. This number represents 0.24% of the total ATP generated per cell per second (24). Thus, according to our calculations, the energetic impact of spurious transcription is not high even in bacteria with a large number of asRNAs.
asRNAs have been proposed to play a role in transcription regulation complementing the role of transcription factors (25). Should this be the case, we would expect a negative dependency with the number of transcription factors in the different bacteria analyzed here. The number of transcription factors, as reported in the P2TF database (26), shows a linear trend with genome size as previously described (27) (fig. S6A). However, this trend does not exist for asRNAs (fig. S6B). To determine if there is a negative dependency between transcription factors and asRNAs, we considered groups of genomes with approximately similar AT content and different numbers of transcription factors. We found no negative relationship between the number of transcription factors and the number of asRNAs per genome having similar AT content (>60%) (fig. S6C). For bacteria with high AT content, there is a positive correlation, contrary to what we would expect (R = 0.94). This can be explained by the fact that for this group, larger genomes present both more transcription factors and more asRNAs. Indeed, for bacteria with similar AT content, the number of asRNAs correlates with the number of genes, indicative of genome size (fig. S6D).
As we indicated in fig. S1B, asRNAs expressed at low levels could barely encounter its sense mRNA, given the stochastic nature of transcription. Therefore, no effect on mRNA half-life or translation would be expected. To see if this is the case, we constructed a mathematical model of transcription and translation of a gene in the bacterium M. pneumoniae. We modeled three possible effects of the asRNA: (i) the binding of the asRNA to the mRNA induces degradation of the duplex, (ii) the binding of the asRNA to the mRNA induces degradation of the mRNA, and (iii) the binding of the asRNA to the mRNA is stable but prevents translation (fig. S1A). In all cases, binding of the mRNA to the ribosome prevents degradation of the mRNA. Parameters for this model were determined from experimental data (see Materials and Methods). Other possible effects, such as transcriptional interference, were not considered as the low transcription rates in M. pneumoniae deem the collision of transcribing polymerases to be very unlikely. We scanned the parameter space of the mRNA and the asRNA transcription rates, from typical wild-type levels to ~100-fold overexpression (Fig. 2 and fig. S7). We found that for the three cases modeled, the region with low concentrations of both asRNA and mRNA shows no changes with respect to the control simulations. This can be explained by the fact that in this region, RNA copy numbers are below 1 per cell, and thus the chance of an mRNA and an asRNA to occur simultaneously at the same cell is negligible (fig. S1B). Remarkably, most of the RNAs in different bacteria are present at concentrations that yield no asRNA effect (28), although some exceptions have been described, showing that some asRNAs can have a regulatory role (29–31) (Fig. 2A). This mathematical model can be a valuable resource to identify putative functional asRNAs in a given organism according to their expression levels. By determining the concentrations of all asRNAs in M. pneumoniae, we can determine a list of potential functional asRNA candidates. In this bacterium, asRNAs are insufficiently expressed to trigger an effect in their overlapping mRNAs, according to our simulations. It has to be noted, though, that the values of decay rates used in these simulations represent the average values determined for M. pneumoniae. Individual transcripts with decay rates that differ significantly from the average should be analyzed on a case-by-case basis. With the adequate parameters, the model could be extended to other bacteria, given that the action mechanism of asRNAs is known beforehand.
Fig. 2. Simulation of the effect of the asRNAs, assuming that the asRNA-mRNA pairing causes duplex degradation.
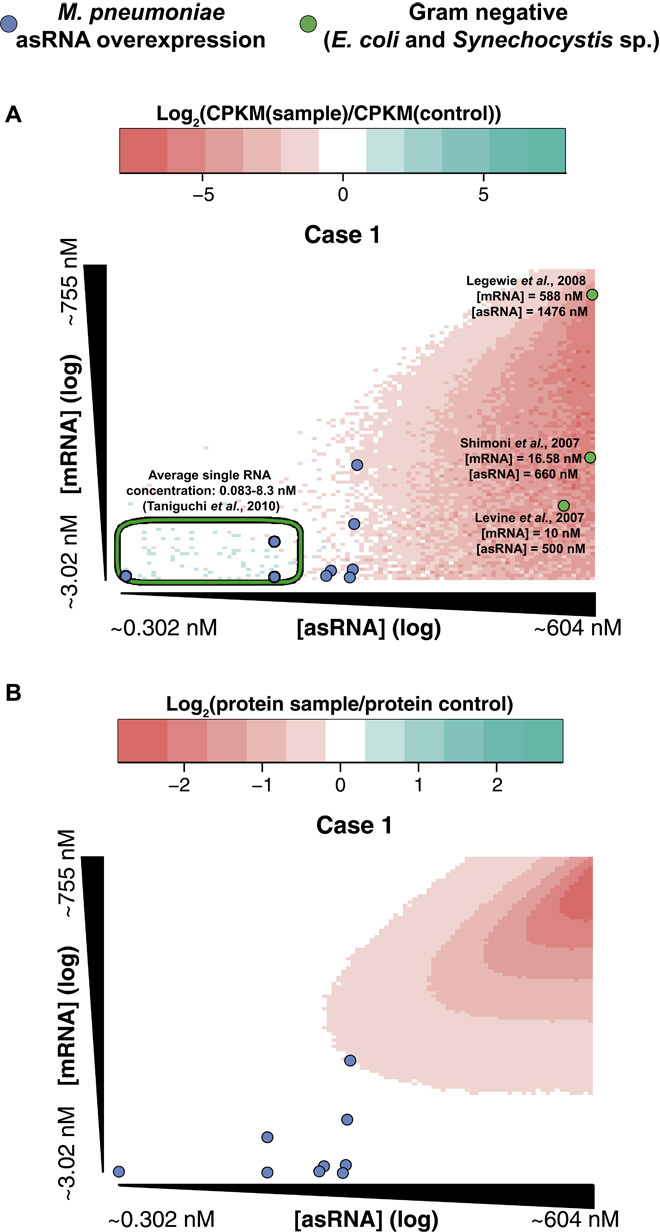
Parameters for the simulations are detailed in the Supplementary Materials. Each point of the heat maps represents the average change in the protein concentration for 100 simulations of 1000 min each, for specific parameters of asRNA and mRNA transcription rates. The remaining parameters remain constant for all the simulations. The axes represent the mRNA and asRNA concentration in the control experiments for the corresponding transcription rates scanned. (A) Changes in the mRNA concentration after 1000 min of simulation. Blue circles represent experimental data from the overexpression of asRNAs in M. pneumoniae, whereas green circles represent data from studies in Gram-negative bacteria (29–31). The green ellipse delimits the region of the concentrations of most transcripts in E. coli (28). (B) Changes in the protein concentration after 1000 min of simulation. Blue circles represent experimental data from the overexpression of asRNAs in M. pneumoniae.
To verify these results, we overexpressed nine asRNAs in the bacterium M. pneumoniae (up to sixfold; Fig. 2 and table S5). These asRNAs were selected such that they overlap different regions of their corresponding mRNA partners (5′ end, 3′ end, or center), to test different possible action mechanisms. Additionally, asRNAs with different expression levels were chosen. Shotgun proteomics of the clones revealed no significant changes in the protein levels of the overlapping genes (Fig. 3A and table S6). Also, RNA-seq (RNA sequencing) revealed no significant changes in the mRNA levels (Fig. 3B and table S7). Thus, our simulations and our experimental data do not support the hypothesis that asRNAs have a general regulatory role in bacteria replacing the function of transcription factors. Only in those exceptions in which both asRNA and mRNA are expressed over a certain threshold can a regulatory behavior be expected.
Fig. 3. Effect of the overexpression of asRNAs in their overlapping genes, measured by RNA-seq and shotgun proteomics.
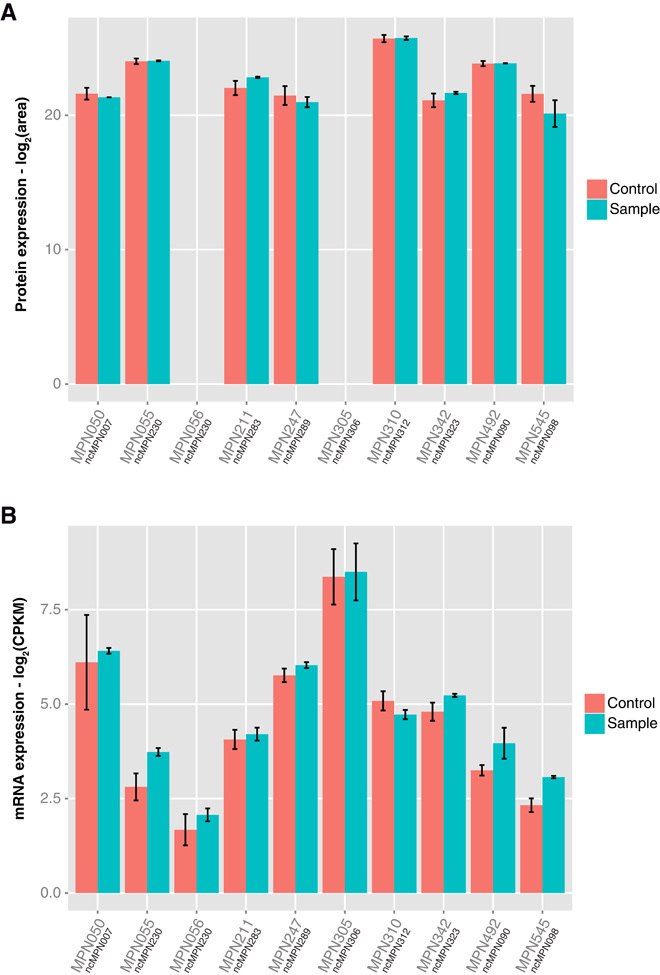
(A) Protein levels of the genes overlapping each asRNA under control conditions and in the strains transformed with the antisense constructs. Error bars represent the SD of the samples. Two of the proteins, MPN056 and MPN305, were not detected in any of the strains of M. pneumoniae. (B) mRNA levels of the genes overlapping each asRNA under control (wild-type) conditions and in the strains overexpressing the antisense transcripts. Error bars represent the SD of the samples.
Our findings support the idea that most of the asRNAs are a consequence of transcriptional noise, rather than of tightly regulated events. The distribution of asRNAs in bacteria with distinct AT content and the lack of capability of replacing transcription factors support this idea. Probably, the bias toward AT mutations in bacteria (21) generates spurious promoter sequences that are able to trigger transcription. However, spurious expression of asRNAs is not incompatible, with some being functional, as described elsewhere (1, 2, 6–8, 12). Indeed, asRNAs claimed to be functional are expressed at much higher rates than the average (28–31). Despite the observed general trend, we should not ignore that, in some bacteria, there are proteins [such as RNA chaperone Hfq (32)] that help to stabilize asRNAs or the duplexes they form with mRNAs. In such cases, even low expressed asRNAs may exert a regulatory function. Nevertheless, this protein is not conserved throughout the bacteria in our study, and although it is conserved in some species, it is not essential. Therefore, we cannot expect such a mechanism to be general but rather an adaptation for specific cases. This suggests that asRNAs may accumulate in bacterial genomes because of transcriptional noise and a lack of negative selection, probably due to the low energy needed for their transcription and the absence of deleterious effects. Some of these asRNAs may afterward gain a function. Additionally, pervasive noncoding transcription may as well have unspecific functional roles, such as buffering the RNA polymerase levels inside the bacterial cell. Our results are likely to be valid throughout the bacterial kingdom, and according to a recent study (33), they may also apply to eukaryotes.
MATERIALS AND METHODS
Bacterial strains and growth conditions
M. hyopneumoniae
Culture samples from M. hyopneumoniae were obtained from batch fermentation in exponential growth. Culture (50 ml) was centrifuged for 3 min at 9000g in a cooled centrifuge (2° to 8°C). Supernatant was removed and the cell pellet was stabilized using RNAlater (Ambion). Stabilized cell pellets were stored at 2° to 8°C until RNA extraction.
B. aphidicola
Cedar aphids were collected from a population maintained in the facilities of the Institut Cavanilles de Biodiversitat i Biologia Evolutiva (ICBiBE) at the University of Valencia (Paterna, Valencia, Spain) (34).
M. pneumoniae
M. pneumoniae was grown in 50 ml of modified Hayflick medium supplemented with glucose at 37°C as previously described (35). To select mycoplasma cells expressing the sRNAs, the medium was supplemented with tetracycline (2 μg ml−1).
M. mycoides
M. mycoides JCVI syn1.0 (36) was grown in 50 ml of SP4 medium containing 17% fetal bovine serum at 37°C and harvested during the mid-log phase as previously described (37).
RNA extraction
M. hyopneumoniae
RNA was extracted from a bacterial pellet stabilized with RNAlater (Ambion) using the Quick-RNA MiniPrep (Zymo Research) following the manufacturer’s protocol.
B. aphidicola
The bacteriomes of 200 adult wingless parthenogenetic insects were dissected under a Wild Heerbrugg Plan 1× microscope and preserved on RNAlater (Ambion) at −80°C until its use. The bacteriome sample was defrosted and washed with phosphate-buffered saline (PBS) [137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 2 mM KH2PO4 (pH 7.2)], and total RNA was purified using the TRI Reagent Solution Kit (Ambion).
M. pneumoniae
After growing M. pneumoniae strains for 6 hours at 37°C, cells were washed twice with PBS and lysed with 700 μl of Qiazol buffer. Then, samples were lysed with 700 μl of Qiazol buffer. RNA extractions were performed by using the miRNeasy Mini Kit (Qiagen) following the instructions of the manufacturer.
M. mycoides
Cells were centrifuged from culture medium and washed twice in Hepes-buffered saline containing 20% sucrose. Cell pellets were stabilized with RNAprotect (Qiagen) until extraction with UltraClean RNA isolation kits (MO BIO).
Library preparation and RNA sequencing
M. hyopneumoniae
rRNA was removed using the Ribo-Zero Kit (Epicentre). rRNA-depleted RNA was fragmented with an average length of 100 to 200 base pairs (bp) and converted to double-stranded complementary DNA (cDNA). Library preparation was done using a protocol based on the “dUTP (deoxyuridine triphosphate) method,” to generate strand-specific mRNA-seq libraries including barcoding (38, 39). The Illumina stranded TruSeq RNA-seq library preparation kit was used. Sequencing of the library was done using the Illumina HiSeq: single-end reads, one lane, 50 cycles, two samples per lane. The sequencing data produced were processed, removing low-quality sequence reads. Furthermore, the sequence data in FastQ format were additionally filtered and trimmed on the basis of Phred quality scores.
B. aphidicola
The samples were mRNA-enriched using the MicrobExpress Kit (Ambion) and the Ribo-Zero Magnetic Kit (Epicentre) to remove rRNA of bacterial and eukaryote origin, respectively, following the manufacturer’s protocol. Library preparation was done with the SOLiD Total RNA-Seq Kit (Life Technologies), and sequencing was performed with an ECC Module on a 5500 XL Genetic Analyzer (Life Technologies) at the sequencing facility of the University of Valencia.
M. pneumoniae
Libraries for RNA-seq were prepared following directional RNA-seq library preparation and sequencing as previously described (23).
M. mycoides
cDNA libraries were constructed with ScriptSeq Complete Gold Kits (Epicentre) and were sequenced on an Illumina HiSeq instrument.
Data analysis
Reads from all RNA-seq experiments detailed above were mapped to their corresponding reference genomes using MAQ (40). All reads were treated as single-end reads. For paired-end sequencing reads, only fragment 1 was considered, and read 2 was considered a technical replicate. After mapping, only reads mapping to a unique position in the reference genome were used. Pileups were obtained using custom-designed software and visualized on the Integrative Genomics Viewer [IGV (41)] for manual annotation of sRNAs. An example of this manual annotation using IGV can be found in fig. S3.
To filter out noise and define the regions corresponding to sRNAs, we first determined the expression levels of all ORFs in the different bacteria analyzed. Expression levels were calculated using a custom-made script to determine the CPKM (counts per kilobase per million counts) values, a measure that is similar to RPKM (reads per kilobase per million mapped reads) for single-end reads. For each genome, we used the expression values of genes with known function. The lower 0.05 quantile of this distribution was chosen as a threshold to determine expression of new nonannotated features. trans-Encoded sRNAs and asRNAs above this threshold were manually identified and annotated.
Regarding the published data from other bacterial species, whenever the sRNA annotation was available, we mapped the noncoding transcripts to the genome to determine how many of them overlapped a gene in antisense and how many corresponded to trans-encoded sRNAs. To do so, we used the reference annotations from the National Center for Biotechnology Information to define ORFs. Partial or total overlap was considered, and for bacteria with more than one replicon, only features in the largest replicon were considered. In some cases, the numbers of asRNAs and trans-encoded sRNAs differ from the numbers reported in the different publications. This is due to the usage of different annotation versions, the inclusion or exclusion of untranslated regions, and the consideration of all the replicons in the different publications.
Calculation of the energy cost of noncoding RNA transcription
To determine the energy cost of transcribing noncoding RNAs, we estimated the relative cost compared to the transcription of mRNAs, rRNAs, and tRNAs. The cost of transcription was assumed to be proportional to the average length of the RNAs multiplied by their transcription rates. In M. pneumoniae, there are 738 ORFs, 3 rRNAs, and 37 tRNAs. The average length of each of these groups is 981.38, 1516, and 77.91 bp, respectively. Transcription rates for each group were estimated from an equilibrium situation, as follows
where [m] is the mRNA concentration, αm is the transcription rate, and km is the decay rate. In equilibrium
RNA concentrations in exponential growth were estimated using the copy numbers previously reported (42) and extrapolating to all RNAs in the cell according to experimental RNA-seq data. RNA decay rates were experimentally determined using novobiocin, a DNA gyrase inhibitor, which releases the RNA polymerase from the chromosome (Junier et al., under review). After the treatment with this inhibitor, RNA from the cells was extracted at different time points and RNA concentrations were determined (Junier et al., under review). RNA decay in the cell population was thus modeled following an exponential decay, as follows
After the treatment
Solving this, we fitted our experimental data to the following exponential decay
and obtained the degradation rate values, km. Averages for mRNAs and asRNAs not overlapping other transcripts were used to ensure that no other factors participate in the degradation. However, we compared the transcription and decay rates determined experimentally between genes overlapped by asRNAs and genes not overlapped by any other transcript, and strikingly, we found no statistically significant differences in transcription rates (P = 0.29, Mann-Whitney U test) or decay rates (P = 0.053, Mann-Whitney U test).
Transcription rates were estimated to be, on average, 0.016, 0.966, and 0.061 molecules/min for mRNAs, rRNAs, and tRNAs, respectively. An estimate of the energy that the cell spends in transcribing these molecules can be obtained by multiplying their number by their length and their transcription rate. Multiplying these values, we obtained an estimate of 16,157.37 [arbitrary units (a.u.)]. Following the same logic for sRNAs, in M. pneumoniae, there are 251 sRNAs, with an average length of 270.597 bp and a transcription rate of 0.007 molecules/min. Multiplying these values, we obtained an estimate of 475.43 (a.u.), equivalent to 2.94% of the energy spent in transcribing mRNAs, tRNAs, and rRNAs together.
Previous studies report that the energy spent in transcribing total RNA (referring to mRNAs, tRNAs, and rRNAs), in terms of number of ATPs required, is ~5000 units of ATP per second per cell (24). This implies that, according to our calculations, the number of ATPs required for sRNA transcription would be ~147 units per cell per second. This number, compared with the total ATP produced by the cell [~60,000 units per second in mid-exponential growth (24)], results in only 0.24% of the cell’s generated energy.
A similar calculation was performed in Escherichia coli. The genome of E. coli codes for 4067 genes, with an average length of 907.09 bp, and 1005 asRNAs (43). Because of the lack of a complete annotation of these asRNAs, we used the average length of the sRNAs in M. pneumoniae. The approximate transcription rate used was ~0.0602 to 0.602 molecules/min (44). Assuming that both genes and asRNAs are transcribed at 0.602 molecules/min, the energy E. coli spends in antisense transcription is 6.7% of that spent in sense transcription. If we consider that transcription of asRNAs occurs at a lower rate of 0.0602 molecules/min, this percentage decreases to 0.67% of energy spent in antisense transcription.
Mathematical modeling of the effect of the asRNAs
Three putative effects of the asRNAs were considered: in case 1, the binding of the asRNA to the corresponding mRNA induces degradation of the duplex. In case 2, the binding of the asRNA to the mRNA induces degradation of the mRNA, but not of the asRNA. In case 3, the mRNA and the asRNA bind reversibly to form a stable duplex, preventing translation of the mRNA. In the three cases, binding to the ribosome protects the mRNA from the effect of the asRNA. The three cases were modeled as follows
Case 1
Case 2
Case 3
In the equations above, [m] stands for the mRNA concentration, [s] for the asRNA concentration, and [p] for the protein concentration. [rib] stands for the ribosome concentration, [dup] for the duplex concentration, and [mrib] for the mRNA-ribosome complex concentration. The values of all the parameters of the model are summarized in table S8, and most of them were determined specifically for M. pneumoniae. RNA decay rates were determined experimentally (see the previous section; Junier et al., under review). Using the experimental decay rates and assuming an equilibrium situation (see the previous section), we determined experimental transcription rates for all RNAs in M. pneumoniae. RNA concentrations used in the calculations had been previously reported (42). All simulations were run for a time of 1000 min using Matlab. An SBML (Systems Biology Markup Language) version of each of the models was generated using COPASI (COmplex PAthway SImulator) (45) and has been submitted to the BioModels database (46).
DNA manipulations and transformation of M. pneumoniae
Different sRNAs encoded by the M. pneumoniae genome were amplified by polymerase chain reaction (PCR) using primers described in table S9. All 5′ primers included sequence of the constitutive promoter (P438) that drove the overexpression of sRNAs. PCR fragments were inserted into the pMTnTetM438 minitransposon (47) by Gibson Assembly. Transformation of the M. pneumoniae M129 strain was performed as previously described (35), and clones were selected by supplementing the medium with tetracycline (2 μg ml−1) at 37°C in 5 % CO2.
Proteomics data acquisition and analysis
M. pneumoniae strain M129 was grown for 6 hours at 37°C. The medium was then removed, and cells were washed twice with PBS. Total protein extract was obtained by breaking the cells with 200 μl of lysis buffer [4% SDS, 0.1 M dithiothreitol (DTT), and 0.1 M Hepes]. Total protein extracts of two biological replicates were analyzed by mass spectrometry (MS).
Each fraction (with amounts ranging from 20 to 486 μg) was trypsin-digested. Briefly, samples were dissolved in 6 M urea, reduced with DTT (10 mM at 37°C for 60 min), and alkylated with iodoacetamide (20 mM at 25°C for 30 min). Samples were diluted 10-fold with 0.2 M NH4HCO3 before being digested at 37°C overnight with trypsin (with a protein/enzyme ratio of 10:1). Peptides generated in the digestion were desalted, evaporated to dryness, and dissolved in 300 μl of 0.1% formic acid. An aliquot of 2.5 μl of each fraction (amounts ranging from 0.17 to 4 μg) was run on an LTQ-Orbitrap Velos (Thermo Fisher) fitted with a nanospray source (Thermo Fisher) after a nanoLC separation in an EasyLC system (Proxeon). Peptides were separated in a reversed-phase column, 75 μm × 150 mm (Nikkyo Technos Co. Ltd.), with a gradient of 5 to 35% acetonitrile in 0.1% formic acid for 60 min at a flow of 0.3 ml/min. The Orbitrap Velos was operated in positive ion mode with nanospray voltage set at 2.2 kV and source temperature at 325°C. The instrument was externally calibrated using Ultramark 1621 for the Fourier transform mass analyzer, and the background polysiloxane ion signal at m/z (mass/charge ratio) 445.120025 was used as lock mass. The instrument was operated in data-dependent acquisition mode, and full-MS scans were acquired in all experiments over a mass range of m/z 350 to 2000 with detection in the Orbitrap mass analyzer set at a resolution setting of 60,000. Fragment ion spectra produced via collision-induced dissociation were acquired in the ion trap mass analyzer. In each cycle of data-dependent analysis, the top 20 most intense ions with multiple charged ions above a threshold ion count of 5000 were selected for fragmentation at a normalized collision energy of 35% following each survey scan. All data were acquired with Xcalibur 2.1 software. Total extract (20 μg) was also digested and desalted, and 1 μg of the resulting peptides was analyzed on an Orbitrap Velos Pro under the same conditions as the fractions but with a longer gradient (120 min).
Protein identification was performed with Proteome Discoverer software v.1.3 (Thermo Fisher) using MASCOT v2.4.01 (Matrix Science) as a search engine (48). Tandem mass spectrometry spectra were searched against a HomoConTrans19 database comprising all putative M. pneumoniae proteins longer than 19 (after in silico translation of the M. pneumoniae genome in the six putative frames) and a list of the common contaminants (599 entries). We set a precursor ion mass tolerance of 15 parts per million at the MS1 level and a fragment ion mass tolerance of 0.5 daltons. We allowed up to three miscleavages for trypsin. Oxidation of methionine and protein acetylation at the N terminus were defined as variable modifications, whereas carbamidomethylation on cysteines was set as a fixed modification. False discovery rates in peptide identification were evaluated using a decoy database set to a maximum of 5%.
RNA-seq and shotgun proteomics data analysis
RNA-seq
Reads were mapped as explained above to obtain the log2(CPKM) values. Data from the nine experiments were quantile-normalized. Each experiment (one biological replicate and two technical replicates) was compared to the rest of the experiments, which were thus used as internal controls. Comparison was performed twofold, by calculating the fold changes in gene expression and performing a t test between the samples and the internal controls. A multiple-test correction was applied to correct the P values of the t test. We considered only those changes with absolute fold changes larger than 0.8 and corrected P values smaller than 0.05 as biologically significant.
Shotgun proteomics
To obtain reliable protein expression values, only unique peptides (those uniquely mapping to a single protein) were considered. The three largest areas from peptides of the same protein (top three peptides) were averaged to obtain a single value for each protein. Areas were rescaled so that each experiment would have the same expression baseline. Comparison was performed twofold, by calculating the fold changes of the areas (in log2) and by performing a t test between the samples (one biological replicate and two technical replicates for each experiment) and the internal controls (the rest of the samples of the experiment). We applied a multiple-test correction to the P values of the t test. Again, we considered only those changes with absolute fold changes larger than 0.8 and corrected P values smaller than 0.05 as significant.
Supplementary Material
Acknowledgments
We thank the Genomics, Proteomics, and Protein Technologies Core Facilities at the Centre for Genomic Regulation. Funding: This work was supported by the European Union Seventh Framework Programme (FP7/2007–2013), through the European Research Council (232913); Fundación Botín, the Spanish Ministry of Economy and Competitiveness (BIO2007-61762); National Plan of R + D + i; ISCIII—Subdirección General de Evaluación y Fomento de la Investigación (PI10/01702); European Regional Development Fund (to the Institució Catalana de Recerca i Estudis Avançats research professor L.S.); and Spanish Ministry of Economy and Competitiveness, “Centro de Excelencia Severo Ochoa 2013–2017” (SEV-2012-0208). A.L. received grant BFU2012-39816-C02-01 from the Spanish Ministry of Economy and Competitivity cofinanced by FEDER (Fondo Europeo de Desarrollo Regional) funds. Author contributions: M.L.-S., L.S., J.I.G., W.-H.C., and P.B. conceived the study and contributed to the experimental design. V.L.-R. performed the comparative analysis, the gene expression analyses, and the modeling, and wrote the manuscript. J.C. manually annotated the sRNAs de novo in three bacterial species in this study. M.L.-S. performed the overexpression experiments in M. pneumoniae. R.G. and A.L. provided the data from B. aphidicola. T.K. provided the data from M. hyopneumoniae. J.I.G. provided the data from M. mycoides. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper, the Supplementary Materials, and/or the public databases. Models were deposited to the BioModels database (8) (identifiers MODEL1511170000, MODEL1511170001, and MODEL1511170002). The mass spectrometry proteomics data were deposited to the ProteomeXchange Consortium via the PRIDE partner repository (identifier PXD003231). RNA-seq data are available in the ArrayExpress database (www.ebi.ac.uk/arrayexpress) under accession number E-MTAB-4081. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/3/e1501363/DC1
Table S1. sRNA annotation of B. aphidicola.
Table S2. sRNA annotation of M. hyopneumoniae.
Table S3. sRNA annotation of M. mycoides.
Table S4. Bacterial strains used in this study.
Table S5. asRNAs overexpressed in M. pneumoniae.
Table S6. Shotgun proteomics results of the whole proteome of the nine clones of M. pneumoniae overexpressing asRNAs.
Table S7. RNA-seq results of the whole transcriptome of the nine clones of M. pneumoniae overexpressing asRNAs.
Table S8. Parameters and initial conditions used in the simulations of the asRNA effects.
Table S9. Primers used in this study to clone the asRNAs.
Fig. S1. Different regulatory mechanisms of sRNAs.
Fig. S2. Theoretical and real numbers of TANAAT boxes in bacteria.
Fig. S3. Manual annotation of sRNAs in M. hyopneumoniae.
Fig. S4. Dependency on the AT content of different types of sRNAs.
Fig. S5. Transcript levels of asRNAs and mRNAs in different bacteria.
Fig. S6. Relationship between asRNAs and transcription factors in bacteria.
Fig. S7. Simulation of the effect of the asRNAs, assuming that the pairing asRNA-mRNA causes mRNA degradation (case 2) or translation inhibition (case 3).
REFERENCES AND NOTES
- 1.Altuvia S., Identification of bacterial small non-coding RNAs: Experimental approaches. Curr. Opin. Microbiol. 10, 257–261 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Thomason M. K., Storz G., Bacterial antisense RNAs: How many are there and what are they doing? Annu. Rev. Genet. 44, 167–188 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lybecker M., Bilusic I., Raghavan R., Pervasive transcription: Detecting functional RNAs in bacteria. Transcription 5, e944039 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wade J. T., Grainger D. C., Pervasive transcription: Illuminating the dark matter of bacterial transcriptomes. Nat. Rev. Microbiol. 12, 647–653 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Raghavan R., Sloan D. B., Ochman H., Antisense transcription is pervasive but rarely conserved in enteric bacteria. mBio 3, e00156-12 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gottesman S., Storz G., Bacterial small RNA regulators: Versatile roles and rapidly evolving variations. Cold Spring Harb. Perspect. Biol. 3, a003798 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Storz G., Vogel J., Wassarman K. M., Regulation by small RNAs in bacteria: Expanding frontiers. Mol. Cell 43, 880–891 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gottesman S., Micros for microbes: Non-coding regulatory RNAs in bacteria. Trends Genet. 21, 399–404 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Gripenland J., Netterling S., Loh E., Tiensuu T., Toledo-Arana A., Johansson J., RNAs: Regulators of bacterial virulence. Nat. Rev. Microbiol. 8, 857–866 (2010). [DOI] [PubMed] [Google Scholar]
- 10.Papenfort K., Vogel J., Regulatory RNA in bacterial pathogens. Cell Host Microbe 8, 116–127 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Chabelskaya S., Gaillot O., Felden B., A Staphylococcus aureus small RNA is required for bacterial virulence and regulates the expression of an immune-evasion molecule. PLOS Pathog. 6, e1000927 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Repoila F., Darfeuille F., Small regulatory non-coding RNAs in bacteria: Physiology and mechanistic aspects. Biol. Cell 101, 117–131 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Georg J., Hess W. R., cis-Antisense RNA, another level of gene regulation in bacteria. Microbiol. Mol. Biol. Rev. 75, 286–300 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tijsterman M., Plasterk R. H. A., Dicers at RISC; the mechanism of RNAi. Cell 117, 1–3 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Horvath P., Barrangou R., CRISPR/Cas, the immune system of bacteria and archaea. Science 327, 167–170 (2010). [DOI] [PubMed] [Google Scholar]
- 16.Zebec Z., Manica A., Zhang J., White M. F., Schleper C., CRISPR-mediated targeted mRNA degradation in the archaeon Sulfolobus solfataricus. Nucleic Acids Res. 42, 5280–5288 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hüttenhofer A., Schattner P., Polacek N., Non-coding RNAs: Hope or hype? Trends Genet. 21, 289–297 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Costa F. F., Non-coding RNAs: Lost in translation? Gene 386, 1–10 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Mendoza-Vargas A., Olvera L., Olvera M., Grande R., Vega-Alvarado L., Taboada B., Jimenez-Jacinto V., Salgado H., Juárez K., Contreras-Moreira B., Huerta A. M., Collado-Vides J., Morett E., Genome-wide identification of transcription start sites, promoters and transcription factor binding sites in E. coli. PLOS One 4, e7526 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Voskuil M. I., Chambliss G. H., The –16 region of Bacillus subtilis and other gram-positive bacterial promoters. Nucleic Acids Res. 26, 3584–3590 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hershberg R., Petrov D. A., Evidence that mutation is universally biased towards AT in bacteria. PLOS Genet. 6, e1001115 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wodke J. A. H., Alibés A., Cozzuto L., Hermoso A., Yus E., Lluch-Senar M., Serrano L., Roma G., MyMpn: A database for the systems biology model organism Mycoplasma pneumoniae. Nucleic Acids Res. 43, D618–D623 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lluch-Senar M., Delgado J., Chen W.-H., Lloréns-Rico V., O’Reilly F. J., Wodke J. A. H., Unal E. B., Yus E., Martínez S., Nichols R. J., Ferrar T., Vivancos A., Schmeisky A., Stülke J., van Noort V., Gavin A.-C., Bork P., Serrano L., Defining a minimal cell: Essentiality of small ORFs and ncRNAs in a genome-reduced bacterium. Mol. Syst. Biol. 11, 780 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wodke J. A. H., Puchałka J., Lluch-Senar M., Marcos J., Yus E., Godinho M., Gutiérrez-Gallego R., dos Santos V. A. P. M., Serrano L., Klipp E., Maier T., Dissecting the energy metabolism in Mycoplasma pneumoniae through genome-scale metabolic modeling. Mol. Syst. Biol. 9, 653 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mehta P., Goyal S., Wingreen N. S., A quantitative comparison of sRNA-based and protein-based gene regulation. Mol. Syst. Biol. 4, 221 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ortet P., De Luca G., Whitworth D. E., Barakat M., P2TF: A comprehensive resource for analysis of prokaryotic transcription factors. BMC Genomics 13, 628 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pérez-Rueda E., Collado-Vides J., Segovia L., Phylogenetic distribution of DNA-binding transcription factors in bacteria and archaea. Comput. Biol. Chem. 28, 341–350 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Taniguchi Y., Choi P. J., Li G.-W., Chen H., Babu M., Hearn J., Emili A., Xie X. S., Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science 329, 533–538 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Legewie S., Dienst D., Wilde A., Herzel H., Axmann I. M., Small RNAs establish delays and temporal thresholds in gene expression. Biophys. J. 95, 3232–3238 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shimoni Y., Friedlander G., Hetzroni G., Niv G., Altuvia S., Biham O., Margalit H., Regulation of gene expression by small non-coding RNAs: A quantitative view. Mol. Syst. Biol. 3, 138 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levine E., Hwa T., Small RNAs establish gene expression thresholds. Curr. Opin. Microbiol. 11, 574–579 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aiba H., Mechanism of RNA silencing by Hfq-binding small RNAs. Curr. Opin. Microbiol. 10, 134–139 (2007). [DOI] [PubMed] [Google Scholar]
- 33.Palazzo A. F., Lee E. S., Non-coding RNA: What is functional and what is junk? Front. Genet. 6, 2 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lamelas A., Gosalbes M. J., Manzano-Marín A., Peretó J., Moya A., Latorre A., Serratia symbiotica from the aphid Cinara cedri: A missing link from facultative to obligate insect endosymbiont. PLOS Genet. 7, e1002357 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yus E., Güell M., Vivancos A. P., Chen W.-H., Lluch-Senar M., Delgado J., Gavin A.-C., Bork P., Serrano L., Transcription start site associated RNAs in bacteria. Mol. Syst. Biol. 8, 585 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gibson D. G., Glass J. I., Lartigue C., Noskov V. N., Chuang R.-Y., Algire M. A., Benders G. A., Montague M. G., Ma L., Moodie M. M., Merryman C., Vashee S., Krishnakumar R., Assad-Garcia N., Andrews-Pfannkoch C., Denisova E. A., Young L., Qi Z.-Q., Segall-Shapiro T. H., Calvey C. H., Parmar P. P., Hutchison C. A. III, Smith H. O., Venter J. C., Creation of a bacterial cell controlled by a chemically synthesized genome. Science 329, 52–56 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Lartigue C., Glass J. I., Alperovich N., Pieper R., Parmar P. P., Hutchison C. A. III, Smith H. O., Venter J. C., Genome transplantation in bacteria: Changing one species to another. Science 317, 632–638 (2007). [DOI] [PubMed] [Google Scholar]
- 38.Parkhomchuk D., Borodina T., Amstislavskiy V., Banaru M., Hallen L., Krobitsch S., Lehrach H., Soldatov A., Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 37, e123 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Levin J. Z., Yassour M., Adiconis X., Nusbaum C., Thompson D. A., Friedman N., Gnirke A., Regev A., Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat. Methods 7, 709–715 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li H., Ruan J., Durbin R., Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 18, 1851–1858 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Robinson J. T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E. S., Getz G., Mesirov J. P., Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maier T., Schmidt A., Güell M., Kühner S., Gavin A.-C., Aebersold R., Serrano L., Quantification of mRNA and protein and integration with protein turnover in a bacterium. Mol. Syst. Biol. 7, 511 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dornenburg J. E., DeVita A. M., Palumbo M. J., Wade J. T., Widespread antisense transcription in Escherichia coli. mBio 1, e00024-10 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Levine E., Zhang Z., Kuhlman T., Hwa T., Quantitative characteristics of gene regulation by small RNA. PLOS Biol. 5, e229 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoops S., Sahle S., Gauges R., Lee C., Pahle J., Simus N., Singhal M., Xu L., Mendes P., Kummer U., COPASI—A COmplex PAthway SImulator. Bioinformatics 22, 3067–3074 (2006). [DOI] [PubMed] [Google Scholar]
- 46.Chelliah V., Juty N., Ajmera I., Ali R., Dumousseau M., Glont M., Hucka M., Jalowicki G., Keating S., Knight-Schrijver V., Lloret-Villas A., Natarajan K. N., Pettit J.-B., Rodriguez N., Schubert M., Wimalaratne S. M., Zhao Y., Hermjakob H., Le Novère N., Laibe C., BioModels: Ten-year anniversary. Nucleic Acids Res. 43, D542–D548 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pich O. Q., Burgos R., Planell R., Querol E., Piñol J., Comparative analysis of antibiotic resistance gene markers in Mycoplasma genitalium: Application to studies of the minimal gene complement. Microbiology 152, 519–527 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Perkins D. N., Pappin D. J. C., Creasy D. M., Cottrell J. S., Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 (1999). [DOI] [PubMed] [Google Scholar]
- 49.Arnvig K. B., Comas I., Thomson N. R., Houghton J., Boshoff H. I., Croucher N. J., Rose G., Perkins T. T., Parkhill J., Dougan G., Young D. B., Sequence-based analysis uncovers an abundance of non-coding RNA in the total transcriptome of Mycobacterium tuberculosis. PLOS Pathog. 7, e1002342 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hotto A. M., Schmitz R. J., Fei Z., Ecker J. R., Stern D. B., Unexpected diversity of chloroplast noncoding RNAs as revealed by deep sequencing of the Arabidopsis transcriptome. G3 1, 559–570 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nicolas P., Mäder U., Dervyn E., Rochat T., Leduc A., Pigeonneau N., Bidnenko E., Marchadier E., Hoebeke M., Aymerich S., Becher D., Bisicchia P., Botella E., Delumeau O., Doherty G., Denham E. L., Fogg M. J., Fromion V., Goelzer A., Hansen A., Härtig E., Harwood C. R., Homuth G., Jarmer H., Jules M., Klipp E., Le Chat L., Lecointe F., Lewis P., Liebermeister W., March A., Mars R. A., Nannapaneni P., Noone D., Pohl S., Rinn B., Rügheimer F., Sappa P. K., Samson F., Schaffer M., Schwikowski B., Steil L., Stülke J., Wiegert T., Devine K. M., Wilkinson A. J., van Dijl J. M., Hecker M., Völker U., Bessières P., Noirot P., Condition-dependent transcriptome reveals high-level regulatory architecture in Bacillus subtilis. Science 335, 1103–1106 (2012). [DOI] [PubMed] [Google Scholar]
- 52.Ramachandran V. K., Shearer N., Jacob J. J., Sharma C. M., Thompson A., The architecture and ppGpp-dependent expression of the primary transcriptome of Salmonella Typhimurium during invasion gene expression. BMC Genomics 13, 25 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gómez-Lozano M., Marvig R. L., Tulstrup M. V. L., Molin S., Expression of antisense small RNAs in response to stress in Pseudomonas aeruginosa. BMC Genomics 15, 783 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gómez-Lozano M., Marvig R. L., Molin S., Long K. S., Genome-wide identification of novel small RNAs in Pseudomonas aeruginosa. Environ. Microbiol. 14, 2006–2016 (2012). [DOI] [PubMed] [Google Scholar]
- 55.Sharma C. M., Hoffmann S., Darfeuille F., Reignier J., Findeiss S., Sittka A., Chabas S., Reiche K., Hackermüller J., Reinhardt R., Stadler P. F., Vogel J., The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 464, 250–255 (2010). [DOI] [PubMed] [Google Scholar]
- 56.Moody M. J., Young R. A., Jones S. E., Elliot M. A., Comparative analysis of non-coding RNAs in the antibiotic-producing Streptomyces bacteria. BMC Genomics 14, 558 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu J. M., Livny J., Lawrence M. S., Kimball M. D., Waldor M. K., Camilli A., Experimental discovery of sRNAs in Vibrio cholerae by direct cloning, 5S/tRNA depletion and parallel sequencing. Nucleic Acids Res. 37, e46 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pfeifer-Sancar K., Mentz A., Rückert C., Kalinowski J., Comprehensive analysis of the Corynebacterium glutamicum transcriptome using an improved RNAseq technique. BMC Genomics 14, 888 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dugar G., Herbig A., Förstner K. U., Heidrich N., Reinhardt R., Nieselt K., Sharma C. M., High-resolution transcriptome maps reveal strain-specific regulatory features of multiple Campylobacter jejuni isolates. PLOS Genet. 9, e1003495 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schlüter J.-P., Reinkensmeier J., Daschkey S., Evguenieva-Hackenberg E., Janssen S., Jänicke S., Becker J. D., Giegerich R., Becker A., A genome-wide survey of sRNAs in the symbiotic nitrogen-fixing alpha-proteobacterium Sinorhizobium meliloti. BMC Genomics 11, 245 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xu W., Chen H., He C.-L., Wang Q., Deep sequencing-based identification of small regulatory RNAs in Synechocystis sp. PCC 6803. PLOS One 9, e92711 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee K., Huang X., Yang C., Lee D., Ho V., Nobuta K., Fan J.-B., Wang K., A genome-wide survey of highly expressed non-coding RNAs and biological validation of selected candidates in Agrobacterium tumefaciens. PLOS One 8, e70720 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hasselbring B. M., Jordan J. L., Krause R. W., Krause D. C., Terminal organelle development in the cell wall-less bacterium Mycoplasma pneumoniae. Proc. Natl. Acad. Sci. U.S.A. 103, 16478–16483 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Persson C., Wagner E. G., Nordström K., Control of replication of plasmid R1: Structures and sequences of the antisense RNA, CopA, required for its binding to the target RNA, CopT. EMBO J. 9, 3767–3775 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cisse I. I., Kim H., Ha T., A rule of seven in Watson-Crick base-pairing of mismatched sequences. Nat. Struct. Mol. Biol. 19, 623–627 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cortes T., Schubert O. T., Rose G., Arnvig K. B., Comas I., Aebersold R., Young D. B., Genome-wide mapping of transcriptional start sites defines an extensive leaderless transcriptome in Mycobacterium tuberculosis. Cell Rep. 5, 1121–1131 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gama-Castro S., Salgado H., Santos-Zavaleta A., Ledezma-Tejeida D., Muñiz-Rascado L., García-Sotelo J. S., Alquicira-Hernández K., Martínez-Flores I., Pannier L., Castro-Mondragón J. A., Medina-Rivera A., Solano-Lira H., Bonavides-Martínez C., Pérez-Rueda E., Alquicira-Hernández S., Porrón-Sotelo L., López-Fuentes A., Hernández-Koutoucheva A., Del Moral-Chávez V., Rinaldi F., Collado-Vides J., RegulonDB version 9.0: High-level integration of gene regulation, coexpression, motif clustering and beyond. Nucleic Acids Res. 44, D133–D143 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/3/e1501363/DC1
Table S1. sRNA annotation of B. aphidicola.
Table S2. sRNA annotation of M. hyopneumoniae.
Table S3. sRNA annotation of M. mycoides.
Table S4. Bacterial strains used in this study.
Table S5. asRNAs overexpressed in M. pneumoniae.
Table S6. Shotgun proteomics results of the whole proteome of the nine clones of M. pneumoniae overexpressing asRNAs.
Table S7. RNA-seq results of the whole transcriptome of the nine clones of M. pneumoniae overexpressing asRNAs.
Table S8. Parameters and initial conditions used in the simulations of the asRNA effects.
Table S9. Primers used in this study to clone the asRNAs.
Fig. S1. Different regulatory mechanisms of sRNAs.
Fig. S2. Theoretical and real numbers of TANAAT boxes in bacteria.
Fig. S3. Manual annotation of sRNAs in M. hyopneumoniae.
Fig. S4. Dependency on the AT content of different types of sRNAs.
Fig. S5. Transcript levels of asRNAs and mRNAs in different bacteria.
Fig. S6. Relationship between asRNAs and transcription factors in bacteria.
Fig. S7. Simulation of the effect of the asRNAs, assuming that the pairing asRNA-mRNA causes mRNA degradation (case 2) or translation inhibition (case 3).