

Abstract
Fatty acid synthase (FASN), the enzyme that catalyzes de novo synthesis of fatty acids, is expressed in many cancer types. Its potential as a therapeutic target is well recognized, but inhibitors of FASN have not yet been approved for cancer therapy. Orlistat (ORL), an FDA-approved lipase inhibitor, is also an effective inhibitor of FASN. However, ORL is extremely hydrophobic and has low systemic uptake after oral administration. Thus, new strategies are required to formulate ORL for cancer treatment as a FASN inhibitor. Here, we report the development of a nanoparticle (NP) formulation of ORL using amphiphilic bioconjugates that are derived from hyaluronic acid (HA), termed Nano-ORL. The NPs were loaded with up to 20 wt % weight of ORL at greater than 95% efficiency. The direct inhibition of the human recombinant thioesterase domain of FASN by ORL extracted from Nano-ORL was similar to that of stock ORL. Nano-ORL demonstrated a similar ability to inhibit cellular FASN activity when compared to free ORL, as demonstrated by analysis of 14C-acetate incorporation into lipids. Nano-ORL treatment also disrupted mitochondrial function similarly to ORL by reducing adenosine triphosphate turnover in MDA-MB-231 and LNCaP cells. Nano-ORL demonstrated increased potency compared to ORL toward prostate and breast cancer cells. Nano-ORL decreased viability of human prostate and breast cancer cell lines to 55 and 57%, respectively, while free ORL decreased viability to 71 and 79% in the same cell lines. Moreover, Nano-ORL retained cytotoxic activity after a 24 h preincubation in aqueous conditions. Preincubation of ORL dramatically reduced the efficacy of ORL as indicated by high cell viability (>85%) in both breast and prostate cell lines. These data demonstrate that NP formulation of ORL using HA-derived polymers retains similar levels of FASN, lipid synthesis, and ATP turnover inhibition while significantly improving the cytotoxic activity against cancer cell lines.
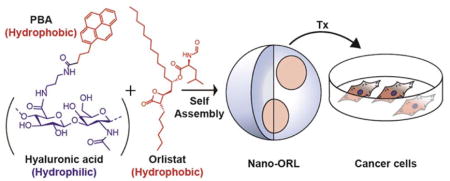
Keywords: Orlistat, tetrahydrolipstatin, fatty acid synthase, hyaluronic acid, self-assembly, nanoparticle
INTRODUCTION
Nearly all cancers show increased endogenous fatty acid synthesis, regardless of the level of circulating dietary fatty acids.1 The key enzyme responsible for this process is homodimeric and multifunctional, fatty acid synthase (FASN), which cyclically produces fatty acids, using malonyl-CoA, acetyl-CoA, and NADPH.2 The primary product of FASN is the 16-carbon fatty acid palmitate. In healthy liver and other lipogenic tissues, palmitate is produced primarily to store excess energy in the form of triglycerides, and is regulated by dietary intake.1,3 However, the activity of FASN and the fate of palmitate are quite different in cancerous tissue, where palmitate is primarily synthesized into phospholipids, the predominant class of lipids found in cell membranes, and regulation is typically independent of circulating fatty acids.1,3 Evidence has accumulated that FASN confers a survival advantage to cancers and their precursor lesions, indicating its status as a potential metabolic oncogene.1,4,5 Increased FASN expression has been shown in every major cancer type, and it has been recognized as a potential target for therapy.3,5 Several molecules that target FASN have been investigated, including cerulenin, C75, epigallocatechin-3-gallate, and triclosan,6–9 but these have off-target effects and unwanted side effects.3,8 Thus, new molecules and formulations are needed that cause fewer side effects.
Orlistat (ORL) is an FDA approved weight loss drug, which, when taken orally, inhibits gastric and pancreatic lipases.10 In addition to lipase activity, ORL has been shown to be a potent inhibitor of FASN, where ORL binds to the thioesterase domain of the enzyme, leading to the hydrolysis of the β-lactone group within ORL.11–13 In cellular assays, ORL effectively inhibits FASN, reduces cancer cell and endothelial cell proliferation, and reduces angiogenesis.13–15 Mechanisms associated with this include cell cycle blockade, ER stress, and apoptosis.16–18 In vivo tumor models have shown that ORL reduces tumor growth, metastasis, and angiogenesis, but at very high ORL concentrations.19–22 ORL is extremely hydrophobic (Figure 1); its predicted logP is 8.1.23,24 Less than 2.5% of ORL is absorbed after oral administration, of which approximately 42% of ORL is degraded into two metabolites.10,25 Due to the high hydrophobicity, low absorption, and poor metabolic stability, a means of improving delivery of ORL to tumors is needed.
Figure 1.

HA was conjugated to the hydrophobic ligand aminopropyl-1-pyrenebutanamide (PBA) to drive self-assembly in aqueous solution. During self-assembly, ORL can be entrapped in the hydrophobic domains of the NPs, effectively solubilizing it.
In order to facilitate delivery of active ORL to tumor cells we have developed a novel ORL formulation, termed Nano-ORL, in which ORL is loaded into the hydrophobic regions of self-assembled polymeric nanoparticles (NPs) derived from hyaluronic acid (HA) (Figure 1). HA is a hydrophilic, extracellular glycosaminoglycan composed of repeating units of β(1,4) D-glucuronic acid and β(1,3) N-acetyl-D-glucosamine.26,27 Polymeric NPs utilizing HA as the hydrophilic backbone have previously been shown to improve delivery of drugs and imaging agents to tumors.28–30 We have previously synthesized HA-derived NPs using the hydrophobic conjugate aminopropyl-1-pyrenebutanamide (PBA), which efficiently loaded indocyanine green and improved its delivery to tumors.28 In this study we now utilize these polymer conjugates to instead load ORL to form Nano-ORL as depicted in Figure 1. Nano-ORL was analyzed for physicochemical properties and ORL loading capability, FASN-TE domain inhibition, 14C-acetate incorporation in cancer cell lines, cytotoxicity against human prostate and breast cell lines, and metabolic analysis.
EXPERIMENTAL SECTION
ORL was obtained from 120 mg Xenical capsules (Roche; Basel, CH) or in powder form (Alfa Aesar; Ward Hill, MA). Sodium hyaluronate was purchased from Lifecore Biomedical (Chaska, MN). 1-Pyrenebutyric acid, 1,3-diaminopropane, N-hydroxy succinimide, and 1-ethyl-3-(3-(dimethylamino)-propyl)carbodiimide (EDC) were obtained from Sigma-Aldrich (St. Louis, MO). Unless otherwise noted, all water was obtained from a Barnstead NANOpure Diamond (Thermo Scientific; Waltham, MA) system producing 18.2 MΩ water. Methanol, N,N-dimethylformamide (DMF), 96-well tissue culture plates (Falcon), and dialysis tubing (MWCO = 3500 Da, Spectrum Laboratories) were purchased from Fisher Scientific (Pittsburgh, PA). Ethanol was purchased from the Warner-Graham Company (Cockeysville, MD). Penicillin/streptomycin and other medium reagents (Gibco) were purchased from the Cell and Viral Vector Core Laboratory of the Comprehensive Cancer Center of Wake Forest School of Medicine. Cell lines were obtained from American Type Culture Collection (Manassas, VA) and were grown in RPMI-1640 (PC-3 and LNCaP) or EMEM (MDA-MB-231) with 10% fetal bovine serum, 1% penicillin/streptomycin.
Synthesis of Amphiphilic HA
Synthesis of PBA and conjugation to HA was performed as described previously.28 Briefly, 1-pyrenebutyric acid (1.0 g, 3.5 mmol) was dissolved in methanol and refluxed at 65 °C for 6 h. 1-Pyrenebutyric methyl ester was then isolated and refluxed in 1,3-diaminopropane at 130 °C for 6 h. The product, PBA, was then precipitated with cold water and washed with cold water using vacuum filtration, followed by drying in a vacuum desiccator. Conjugation of PBA to HA was then performed by separately dissolving PBA (10 mg, 29 μmol) in 25 mL of DMF and HA (90 mg) in 25 mL of H2O. 0.8 mmol of EDC and NHS were then added to the HA solution, and the PBA was added dropwise under constant stirring. The reaction was allowed to proceed for 24 h. This was followed by dialysis into 50:50 H2O:EtOH for 24 h, dialysis into pure H2O for 48 h, and then lyophilization.
Preparation of Nano-ORL
ORL was isolated from Xenical capsules by mixing capsule contents into 10 mL of EtOH, followed by vortexing and centrifugation at 1500 rpm for 5 min. Supernatant containing ORL was removed from the precipitated filler material, followed by a second separation and centrifugation. Stock 12 mg/mL (24.2 mM) ORL solution was then aliquoted and stored at −20 °C. Alternatively, stock solution was also prepared from powdered ORL (Alfa Aesar) by dissolution into ethanol or methanol to a concentration of 12 mg/mL followed by storage at −20 °C. ORL was loaded into NPs by dissolution of 18.0 mg of HA into 10 mL of H2O, followed by addition of 10 mL of ethanol. ORL (2.0 mg, 4.0 μmol, 20 wt % loading) was then added, and the solution was dialyzed against pure water for 24 h. Material was then filtered through PD-10 columns (GE Lifesciences; Pittsburgh, PA) to remove free ORL, followed by lyophilization and storage at −20 °C. This material is termed Nano-ORL. Empty NPs formed via self-assembly upon dissolution in aqueous solution, as described previously.28
ORL loading efficiency was determined by extraction from Nano-ORL followed by HPLC quantification. Extraction of ORL was performed following the protocol for extraction of ORL from human plasma as described in the literature.31 A standard curve of ORL extraction from NPs was obtained by spiking solutions of empty NPs with known quantities of ORL from stock solution and performing the following extraction protocol. Empty NPs were dissolved into pure H2O to a concentration of 1.0, 0.5, or 0.25 mg/mL. Stock ORL was then added to the empty NP solutions to a concentration of 0.20, 0.10, or 0.05. mg/mL. 1.0 mL aliquots of these solutions were then mixed with 1.0 mL of acetonitrile, to which 5 mL of n-hexane was added. This solution was then stirred for 30 min, at which time the hexane layer was removed and evaporated under vacuum. Extracted ORL was dissolved in 100% HPLC grade MeOH and run on a Beckman Ultrasphere ODS 4.6 × 250 mm column with 5 μm particle size using a mobile phase of 82.5:17.5 methanol:acetonitrile-0.01% trifluoroacetic acid with a Waters 510 pump, 717+ autosampler, and 2998 PDA UV/vis detector.32,33 Separation was isocratic, and ORL was measured at 200–205 nm. Each ORL concentration was performed in quadruplicate. ORL content in Nano-ORL was then measured by performing these extraction methods and comparing to the standard curve. Loading efficiency of Nano-ORL was determined by resuspension of Nano-ORL into pure H2O at a concentration of 0.50 mg/mL and extracted and analyzed in quadruplicate as described above. Extracted values were then analyzed using the standard curve.
Physicochemical Characterization
5 wt %, 10 wt %, and 20 wt % Nano-ORL formulations were synthesized in order to test the effect of ORL loading on NP size and zeta-potential. 0 wt % (empty NPs), 5 wt %, 10 wt %, and 20 wt % ORL formulations were dissolved in pure H2O to a concentration of 1.0 mg/mL. Hydrodynamic diameter was then determined using a ZetaPlus system with onboard dynamic light scattering (DLS) analyzer (Brookhaven Instruments Corporation; Holtsville, NY). Zeta potential was determined using a Zetasizer Nano ZS90 (Malvern Instruments; Worcestershire, U.K.).
Inhibition of FASN and Lipid Synthesis
The C-terminal thioesterase domain of FASN (FASN-TE, residues 2200–2510) was expressed and purified as previously reported.11 The inhibition assay using 4-methylumbelliferyl heptanoate (MUH) as the substrate was slightly modified from what was previously reported.34,35 Each 100 μL reaction contained 250 nM FASN-TE, 100 mM Tris pH 7.4, 50 mM NaCl, 1 mM EDTA, 120 μM MUH, 1% DMSO, and Orlistat 0.1–10 μM. Orlistat was preincubated with FASN-TE for 30 min at 37 °C prior to starting the reaction with MUH. The samples within a 96-well plate were read at 30 s intervals for 30 min on a Tecan Safire2 instrument (excitation, 350 nm; emission, 450 nm).
For analysis of the inhibition of cellular fatty acid synthesis by Nano-ORL, PC-3 cells were seeded in two 24-well plates, 7 × 104 cells per well, and treated 2 days later with inhibitors or vehicle controls, in quadruplicate for 18 h. [2-C14]Acetate (0.037 MBq, 1 μCi; PerkinElmer, Boston, MA) was added to wells in one plate for 2 h. The cells were trypsinized, washed, and lysed in hypotonic buffer (20 mM Tris-HCl, pH 7.5, 1 mM EDTA, 1 mM DTT). Lipids were extracted in chloroform:methanol (2:1). The organic fraction was washed twice with PBS and transferred to vials for scintillation counting (Beckman Coulter LS 6500). Protein concentration was determined by the Lowry method (Bio-Rad, Hercules, CA) in cells treated in the duplicate plate.
Cell Viability
PC-3, LNCaP, and MDA-MB-231 cell lines were seeded separately into 96-well plates at concentrations of 2000 cells/well and allowed to adhere overnight. Empty NPs and Nano-ORL were resuspended in culture media, while stock ORL was diluted into normal medium from ethanol. Cells were then treated with either normal medium, empty NPs (0.062 mg/mL), ORL (0.0124 mg/mL, 25 μM), or Nano-ORL (0.062 mg/mL, theoretical [ORL] = 25 μM, actual [ORL] = 24 μM due to loading efficiency) and allowed to incubate at 37 °C for 48 h. After 48 h incubation, cells were analyzed by CCK-8 assay (Dojindo, Japan) and results were normalized to the standard medium group.
Similar methods were used to study the effect of preincubation on ORL and Nano-ORL. ORL and Nano-ORL were first dissolved in serum-free medium at concentrations of 0.124 mg/mL (250 μM) and 0.62 mg/mL, respectively. These solutions were then incubated for 24 h at 37 °C. ORL and Nano-ORL solutions were then diluted to 0.0124 mg/mL (25 μM) and 0.062 mg/mL, respectively, in serum-containing medium. Empty NPs were treated in a manner identical to Nano-ORL. Nonincubated ORL and Nano-ORL solutions were also formed at identical concentrations to preincubated ORL and Nano-ORL. All nonincubated solutions, including standard medium, were diluted to the same FBS and penicillin/streptomycin concentrations as preincubated solutions. Solutions were then applied to PC-3, LNCaP, or MDA-MB-231 cancer cell lines in 96-well plates and allowed to incubate for 24 h. At 24 h, solutions were reapplied (both preincubated and nonincubated) in order to maintain concentrations of free ORL and Nano-ORL. CCK-8 assay was performed at 48 h, and all groups were normalized to the standard medium group.
Cellular Recovery after Exposure to ORL Formulations
Cells were seeded at 1 × 103 cells per well (PC-3, LNCaP) or 3 × 103 cells per well (MDA-MB-231) in 96-well plates. Cells were allowed to adhere for 24 h prior to treatment with test formulations. After 24 h, treatment medium was removed and replaced with fresh medium without ORL or Nano-ORL. Cells were allowed to recover from treatment for 24 h before 20 μL of CellTiter 96 AQueous One reagent was added to each well. Cells incubated in the dark for 1 h at 37 °C/5% CO2. Absorbance was measured at 490 nm in a plate reader (VersaMax Tunable Microplate Reader, Molecular Devices, LLC; Sunnyvale, CA, United States) to determine the formazan concentration. Results are reported as % viability compared to control cells.
Mitochondrial Function Assays and Bioenergetics Measurement
All cellular oxygen consumption rates (OCR) were measured using the XF24 Extracellular Flux Analyzer (Seahorse Bioscience; Massachusetts, United States) per manufacturer’s protocols. Prior to plating with LNCaP cells, XF24-well plates were coated with sterile 22.4 μg/mL Cell-Tak (Corning; Corning, New York, United States) for approximately 12 h at room temperature. LNCaP and MDA-MB-231 cells were plated at 4 × 104 cells/well in RPMI-1640 with 10% FBS/1% PS. Cells were treated with identical media containing either vehicle or test compounds for 16 h. Before OCR measurements were taken, cells were washed with Seahorse assay medium supplemented with fresh sodium pyruvate and glucose per published Seahorse protocol and incubated at 37 °C without CO2 for 1 h. After equilibration, three 3 min measurements were recorded to measure the basal level of oxygen consumption. Oligomycin (1 μM), Carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP) (0.5 μM), and Rotenone/Antimycin A (1 μM each) were injected into each well sequentially with three 3 min measurements after each injection to measure the amount of oxygen consumption for adenosine triphosphate (ATP) production (coupling efficiency), the level of proton leak (non-ATP-linked oxygen consumption), maximal respiration capacity, and the level of nonmitochondrial respiration. The following metrics were used in accordance with published Seahorse protocols and previous publications36 to analyze results:
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
Immediately postassay, cells from each treatment group were collected and counted and their viability was determined by trypan blue exclusion. Basal OCR levels were normalized to average number of live cells per well.
Statistical Analysis
All statistical analyses were performed with GraphPad Prism 6.0 software (San Diego, CA). Extracted stock ORL measured by HPLC was plotted and analyzed using linear regression to form a standard curve. Extracted Nano-ORL was quantified using this standard curve. Average hydrodynamic diameters and zeta-potential of Nano-ORL with different loading wt % ORL were plotted and analyzed using linear regression. FASN inhibition, 14C-acetate incorporation, cytotoxicity, cellular recovery, and mitochondrial respiration assays were analyzed separately using two-way ANOVA with multiple comparisons using Tukey’s post-test.
RESULTS
Drug Loading and Physical Characterization
Figure S1 shows the standard curve and extracted Nano-ORL values. An R2 = 0.98 was achieved over the range from 0.05 mg/mL to 0.20 mg/mL. Analysis of the extracted Nano-ORL demonstrated a loaded wt % of 19.3 wt % ± 0.4 wt %, corresponding to a 96.7% ± 2.4% loading efficiency. The effect of ORL loading on NP size was determined by loading 0, 5, 10, and 20 wt % ORL. Figure 2A shows the linear effect that ORL has on Nano-ORL effective diameter (R2 = 0.99) indicating that ORL loading has a direct influence on the size of Nano-ORL, with hydrodynamic diameters ranging from approximately 290 (±7) to 580 (±13) nm, from 0 wt % ORL to 20 wt % ORL, respectively. Dispersity, as measured by DLS, ranged from 0.09 (10 wt % ORL) to 0.2 (0 wt % ORL), and did not depend on ORL content. Zeta-potential of Nano-ORL formulations ranged from −27 to −35 mV and did not correlate with ORL content (Figure 2B).
Figure 2.

(A) Effective diameters, as determined by dynamic light scattering, increased with loading content of ORL. Empty NPs were in the range of 290 nm, increasing linearly up to 580 nm with approximately 20 wt % ORL. (B) The zeta-potential of Nano-ORL formulations ranged from −27 to −35 mV, and was independent of ORL content.
Inhibition of FASN and Fatty Acid Synthesis
To determine that Nano-ORL remained able to inhibit FASN, ORL was extracted from Nano-ORL and tested for inhibition of FASN-TE to ensure that Nano-ORL retained the same molecular action as free ORL (Figure 3A). At 10 μM, extracted stock ORL inhibited 93.1% ± 5.4% of FASN-TE activity and extracted Nano-ORL inhibited 93.4% ± 6.4% of FASN-TE activity. At 1 μM and 0.1 μM, extracted stock ORL and Nano-ORL showed lower degrees of inhibition. At 1 μM, extracted stock ORL inhibited 31.0% ± 1.1% and extracted Nano-ORL inhibited 36.4% ± 15.0%. At 0.1 μM extracted stock ORL inhibited 0.0% ± 2.6% and extracted Nano-ORL inhibited 7.3% ± 34.5%. No significant difference was found between stock ORL or ORL extracted from the NP formulation (p ≥ 0.05). These data indicate that ORL packaged in the NP formulation retains the same ability to inhibit FASN-TE as pure ORL. Inhibition of FASN-TE activity by Nano-ORL also translated to inhibition of cellular lipid synthesis by analysis of 14C-acetate incorporation (Figure 3B). Nano-ORL reduced lipid synthesis in PC-3 cells by 51% (p = 0.0003), whereas free ORL reduced lipid synthesis by 62% (p ≤ 0.0001), with no significant difference between free ORL or Nano-ORL (p = 0.59). There was no significant difference between empty NPs and control medium (p = 0.99), demonstrating that the observed effect of Nano-ORL results solely from ORL incorporated into the NPs and its subsequent intracellular release.
Figure 3.

(A) FASN-TE was directly inhibited using ORL extracted from Nano-ORL or stock ORL that underwent the extraction protocol. The efficacy of extracted Nano-ORL was not significantly different from that of extracted stock ORL, indicating that Nano-ORL retains the same molecular mechanism as free ORL. (B) 14C-Acetate incorporation in lipid was measured when PC-3 cells were exposed to medium, 0.062 mg/mL empty NPs, Nano-ORL (equivalent to 25 μM ORL), or free ORL (25 μM ORL) for 18 h. Cells treated with empty NPs did not show a decrease in lipid synthesis, while both Nano-ORL and ORL inhibited lipid synthesis to a similar degree.
Effect on Cell Viability
The cytotoxicity of Nano-ORL was next tested against human cancer cell lines. As seen in Figure 4A–C both ORL and Nano-ORL significantly reduced cell viability. In PC-3 and LNCaP prostate cancer cell lines, Nano-ORL reduced cell viability to 54% and 31%, respectively, while ORL reduced viability to 71% and 51% respectively. Similarly, Nano-ORL reduced MDA-MB-231 viability to 57% while ORL reduced viability to 79%. These differences between Nano-ORL and ORL were statistically significant (p ≤ 0.0001) and indicate that NP formulations of ORL increase toxicity by an additional 24%, 39%, and 28% for PC3, LNCaP, and MDA-MB-231 cells, respectively. In LNCaP cells, empty NPs decreased viability by 15% relative to untreated cells (p ≤ 0.0001).
Figure 4.

Treatment with Nano-ORL (25 μM ORL) results in significantly lower relative viability of cancer cell lines PC-3 (A), MDA-MB-231 (B), and LNCaP (C) compared with free ORL (25 μM) after 48 h of treatment. PC-3 (D), MDA-MB-231 (E), and LNCaP (F) cells were then treated with ORL (25 μM) and Nano-ORL (25 μM ORL) which had been preincubated for 24 h prior cell exposure. ORL was significantly less toxic after preincubation, while Nano-ORL did not lose efficacy, and both preincubated and nonincubated Nano-ORL exhibited higher toxicities than free ORL. ****: p ≤ 0.0001; NS = not significant.
To test the duration of Nano-ORL activity compared to free ORL, ORL, Nano-ORL, and empty NPs were preincubated for 24 h at 37 °C prior to application to cells (designated with “PI” superscript). Medium was reapplied at 24 h (after an additional 24 h preincubation) to ensure a consistent level of preincubated drugs and materials, and plates were tested after a total of 48 h exposure. The data in Figure 4D–F indicate that ORL and Nano-ORL are cytotoxic without preincubation, although Nano-ORL was significantly more toxic than ORL (p ≤ 0.0001 for MDA-MB-231, p ≤ 0.01 for LNCaP, and p ≤ 0.01 for PC-3). Interestingly, when NanoORL and ORL were preincubated for 24 h in medium, Nano-ORL provided a formulation where cytotoxicity was maintained under these harsher conditions, while free ORL had little potency under these same conditions (86%, 105%, and 95% viability in PC-3, LNCaP, and MDA-MB-231 cells, respectively; p ≤ 0.0001 compared to cells treated with ORL and no preincubation). These results indicate that the Nano-ORL formulation results in significantly higher activity against cancer cell lines and thus may improve therapeutic efficacy.
Inhibition of 14C-acetate incorporation into lipids was then further examined using preincubated ORL and Nano-ORL to determine if preincubation in aqueous solution had an effect on FASN inhibition. Conditions were identical to the previous 14C-acetate incorporation study in Figure 3 with the exception that two groups were added, free ORLPI and Nano-ORLPI, each receiving 24 h preincubation. Figure S2 shows that all groups containing free ORL or Nano-ORL inhibit lipid synthesis to a similar degree within each cell line, regardless of whether the formulation had been preincubated or not. Between groups containing a formulation of ORL, only Nano-ORL (preincubated) had slightly more 14C incorporation than free ORL (not incubated) in MDA-MB-231 cells (p = 0.025).
PC-3, MDA-MB-231, and LNCaP cell lines were also tested for recovery after ORL treatment. Cells were treated with 12.5, 25, or 50 μM free ORL or Nano-ORL for 24 h, followed by an additional 24 h in standard medium without ORL or Nano-ORL. The data in Figure 5 illustrate concentration and treatment dependent effect. PC-3 cells with 50 μM free ORL (49.3% reduction, p ≤ 0.01) or Nano-ORL (45% reduction, p ≤ 0.01) for 24 h caused reduced viability after 24 h recovery, while only treatment with 50 μM Nano-ORL significantly reduced viability in MDA-MB-231 cells (29.9% reduction, p ≤ 0.001). Cell viability of LNCaP cells was significantly reduced at all concentrations using both ORL or Nano-ORL (p ≤ 0.05).
Figure 5.

PC3 (A), MDA-MB-231 (B), and LNCaP (C) cell lines were subjected to treatment of 12.5 μM, 25 μM, and 50 μM ORL or Nano-ORL for 24 h followed by 24 h of recovery in standard medium. After recovery, cell viability was assessed by MTS assay. Vehicle group was EtOH for ORL and empty NPs for Nano-ORL. (*, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001).
Mitochondrial Stress Analysis
FASN activity and the subsequent effects of FASN inhibition have been linked to mitochondrial metabolism. Therefore, mitochondrial stress assays were performed to examine the impact of Nano-ORL treatment on cellular energy production in MDA-MB-231 cells (Figure 6A–D) and LNCaP cells (Figure 6E–H). Cells were treated with 50 μM ORL, 50 μM Nano-ORL, or vehicle for 16 h followed by Seahorse XF24 analysis. Figures 6A and 6E show the OCR of MDA-MB-231 and LNCaP cells over the course of the experiment. ORL treatment of MDA-MB-231 cells did not affect basal respiration to a statistically significant level (Figure 6B), whereas Nano-ORL treatment decreased basal respiration of MDA-MB-231 cells from 2.69 pmol of O2/min/1000 live cells to 0.92 pmol of O2/min/1000 live cells (Figure 6B, p ≤ 0.001). Neither ORL nor Nano-ORL treatment significantly decreased basal respiration of LNCaP cells when compared to vehicle (Figure 6F). Coupling efficiency of both cell lines was impaired by ORL treatment (Figure 6C,G). ORL treatment of MDA-MB-231 cells reduced coupling efficiency from 75.40% to 34.03% of respective basal OCR (Figure 6C, p ≤ 0.05). Similarly, coupling efficiency of Nano-ORL treated MDA-MB-231 cells was reduced from 62.11% to 31.70% of respective basal OCR (Figure 6C, p ≤ 0.05). This result was reflected in LNCaP cells as ORL treatment reduced coupling efficiency from 71.57% to 35.55% of respective basal OCR (Figure 6G, p ≤ 0.001) and Nano-ORL treatment reduced coupling efficiency from 60.63% to 33.29% of respective basal OCR (Figure 6G, p ≤ 0.005). No difference in spare respiratory capacity was seen in either cell line with either treatment method (Figures 6D and 6H).
Figure 6.

4 × 104 MDA-MB-231 cells (A–D) or LNCaP cells (E–H) were plated in an XF24 well plate and allowed to adhere before treatment with vehicle (4:1 DMSO:EtOH), empty NPs, (NP), 50 μM free ORL, or 50 μM Nano-ORL for 16 h. Oxygen consumption rates were measured (A, E), and values representing basal respiration (B, F), coupling efficiency (C, G), and spare respiratory capacity (D, H) were quantified. Nano-ORL reduced basal mitochondrial respiration (B) and coupling efficiency (C) in MDA-MB-231 cell, and reduced coupling efficiency in LNCaP cells (G). ORL reduced coupling efficiency in both MDA-MB-231 and LNCaP cells. Nano-ORL was superior to ORL at reducing basal respiration in MDA-MB-231 cells, and overall showed a similar metabolic inhibition as ORL by decreasing basal metabolism and coupling efficiency by 50% or more. (*, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001).
DISCUSSION
Previous studies have demonstrated that ORL inhibits FASN, triggers apoptosis, and inhibits tumor growth, metastasis, and angiogenesis.13,21,22 A growing body of evidence suggests that reliance on FASN in cancer is nearly ubiquitous and thus presents a valuable target for therapy.1,3,5 Indeed, ORL has been used in preclinical investigations against several cancer types, including prostate, gastric, oral, skin, and leukemia.13,19–21,37 In combination, the low bioavailability through enteric delivery, high degree of hydrophobicity, and potential for off-target effects present significant barriers to clinical use of ORL and require novel developments in new ORL formulations.10,23–25
In our previous work, we demonstrated that HA modified with PBA could entrap the near-infrared fluorophore indocyanine green and preferentially deliver the dye to tumors.28 We postulated that the PBA modified HA polymers could load ORL through self-assembly of the amphiphilic polymeric bioconjugate resulting in a new ORL formulation (Figure 1). ORL, as demonstrated here, is effectively loaded into the amphiphilic HA NPs. The high loading efficiency observed with Nano-ORL (96.7% ± 2.4%) shows a strong association between ORL and the NPs, likely through interaction with hydrophobic domains created by PBA.28 Since NP biodistribution is dependent on diameter, the ability to control NP size based on ORL content could prove useful in optimizing biodistribution, circulation time, and delivery of Nano-ORL to tumors.
The direct inhibition of FASN and decreased lipid synthesis (Figure 3) demonstrate that Nano-ORL has the same molecular activity as free ORL. Nano-ORL had similar FASN-TE inhibition compared to free ORL (Figure 3A). This strongly indicates that the molecular action of Nano-ORL retains the same FASN inhibitory activity as free ORL. Inhibition of cellular lipid synthesis, as indicated by 14C-acetate incorporation, was also similar between free ORL and Nano-ORL, further demonstrating that Nano-ORL retains the molecular action of free ORL and that Nano-ORL effectively inhibits new lipid synthesis in cancer cells.
Analysis of the cytotoxic effect of Nano-ORL against prostate (PC-3 and LNCaP) and breast (MDA-MB-231) cancer cell lines demonstrated the improved efficacy of the Nano-ORL formulation over free ORL (Figure 4). 48-h cytotoxicity analysis resulted in higher levels of toxicity from Nano-ORL as compared to free ORL (Figure 4A). The increase in cytotoxicity of Nano-ORL may result from increased solubility of ORL due to its entrapment within NPs, which could result in a higher availability. It is also possible that Nano-ORL experiences greater internalization due to binding with HA, which can be endocytosed and subsequently deliver therapeutics intracellularly.38 In order to show that Nano-ORL retains its activity longer than free ORL, both ORL and Nano-ORL were preincubated for 24 h prior to exposure to cells (Figure 4B). This period allowed for a number of processes to potentially occur, including self-association and aggregation, precipitation, and surface adsorption of ORL, which could impact cytotoxicity. Preincubation profoundly demonstrated the maintenance of the cytotoxic effect of Nano-ORL with no change in relative viability observed; in comparison, the relative viability of free ORL-treated cells increased by 26–39% with preincubation depending on the cell line. Further demonstration of equivalent or higher toxicity of Nano-ORL was observed by recovery analysis of cells receiving serial dilutions of ORL or Nano-ORL. Nano-ORL-treated cells showed equal or lower levels of viability after 24 h recovery time from treatment (Figure 5). This data also indicates a negative correlation between cell viability and the concentration of ORL and Nano-ORL. These results demonstrate the potential advantages of NP encapsulation of ORL include maintaining ORL in a colloidally stable form available for cell uptake.
Interestingly, inhibition of lipid synthesis was not observed to be different over the 18 h incubation when cells were treated with either preincubated or non-preincubated ORL or Nano-ORL formulations (Figure S2). While this appears to be in contrast to the increased relative viability of cells when treated with preincubated free ORL, the 18 h time point was chosen specifically to avoid complicating cytotoxic effects, thereby uncoupling the effects of cytotoxicity and lipid synthesis inhibition. Detailed metabolic analysis also shows that Nano-ORL decreases basal respiration and coupling efficiency equal to, or better than, free ORL (Figure 6). This indicates that both the total O2 consumed and ATP turnover, respectively, are reduced in cells treated with Nano-ORL or free ORL. Thus, both ORL and Nano-ORL exhibit a combination of reduced lipid synthesis and mitochondrial metabolism, which results in a cytotoxic effect against cancer cell lines. Nano-ORL, however, shows superior stability and solubility in aqueous solution, which results in higher cytotoxicity over extended periods of time. Overall, these results show that Nano-ORL has the same molecular action as free ORL, but provides improved bioavailability due to the soluble, amphiphilic HA-NP formulation.
The Nano-ORL NPs developed here ranged from 400 nm (average HD) with 5 wt % ORL loading to 580 nm (average HD) with 20 wt % ORL loaded. While these NPs may be outside the optimal biodistribution size range, we believe they are nevertheless viable candidates to deliver ORL to tumors. Bae and Park39 and Petros and DeSimone40 report that 500 nm is the approximate maximum diameter for particles to be capable of utilizing the EPR effect, and 200–500 nm to avoid clearance by the spleen. Therefore, Nano-ORL formulations with lower ORL loading would fit into the optimized size regime for the EPR effect. Future studies aimed at determining the biodistribution of this and next generation Nano-ORL formulations, which identify strategies to maintain ORL loading while tuning NP size, are required. Specific strategies will include reducing Nano-ORL size either mechanically, e.g., using filtration, and/or chemically, such as using lower molecular weight polymers and conjugating alternative hydrophobic ligands to improve ORL packing within NPs.
In summary, we have developed a new formulation of ORL by physicochemical entrapment in a self-assembled HA-NP. The NP formulation of ORL retained equal activity against FASN-TE and reduction of 14C-acetate incorporation. Furthermore, Nano-ORL demonstrated improved cytotoxic activity against human prostate and breast cancer cell lines over free ORL and reduced the metabolic activity and recovery of treated cells. These results show the potential of NP formulations of ORL and warrant continued investigation of Nano-ORL as a therapeutic. A comprehensive evaluation of Nano-ORL treatment of tumors in vivo is currently underway, which will include the next generation of Nano-ORL formulations. The study will evaluate Nano-ORL dosing amount and frequency, relationship between ORL loading (high vs low) and toxicity, Nano-ORL composed of varying HA molecular weight, different hydrophobic moieties structurally optimized to be more efficiently loaded with ORL, dye-labeled HA to longitudinally track NP components, and key toxicological outcomes.
Supplementary Material
Acknowledgments
This work was supported in part by the National Institutes of Health, Grants R00 CA153916 (A.M.M.), R01 EB019449 (A.M.M.), R01 CA161503 (S.J.K.), T32 CA079488 and T32 HL091797 (A.L.D.), P30 CA036727 (Fred & Pamela Buffett Cancer Center Support Grant), and a joint pilot grant from the Wake Forest-Virginia Tech School of Biomedical Engineering and Sciences and the Comprehensive Cancer Center of Wake Forest University (P30 CA012197, A.M.M. and S.J.K.). T.K.H. is thankful for the support of Mike and Lucy Robbins Fellowship. The Waters Q-TOF mass spectrometer was purchased with funds from NIH Shared Instrumentation Grant 1S10RR17846 and the Thermo Electron TSQ Quantum XLS GC/MS/MS from NIH Shared Instrumentation Grant 1S10RR027940.
ABBREVIATIONS USED
- FASN
fatty acid synthase
- ORL
Orlistat
- HA
hyaluronic acid
- NP
nanoparticle
- FASN-TE
recombinant FASN thioesterase domain
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.molpharmaceut.5b00447.
Methods (PDF)
References
- 1.Menendez JA, Lupu R. Fatty Acid Synthase and the Lipogenic Phenotype in Cancer Pathogenesis. Nat Rev Cancer. 2007;7(10):763–777. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]
- 2.Maier T, Jenni S, Ban N. Architecture of Mammalian Fatty Acid Synthase at 4.5 Å Resolution. Science. 2006;311(5765):1258–1262. doi: 10.1126/science.1123248. [DOI] [PubMed] [Google Scholar]
- 3.Kuhajda FP. Fatty Acid Synthase and Cancer: New Application of an Old Pathway. Cancer Res. 2006;66(12):5977–5980. doi: 10.1158/0008-5472.CAN-05-4673. [DOI] [PubMed] [Google Scholar]
- 4.Flavin R, Peluso S, Nguyen PL, Loda M. Fatty Acid Synthase as a Potential Therapeutic Target in Cancer. Future Oncol. 2010;6(4):551–562. doi: 10.2217/fon.10.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pandey PR, Liu W, Xing F, Fukuda K, Watabe K. Anti-Cancer Drugs Targeting Fatty Acid Synthase (FAS) Recent Pat Anti-Cancer Drug Discovery. 2012;7(2):185–197. doi: 10.2174/157489212799972891. [DOI] [PubMed] [Google Scholar]
- 6.Alli PM, Pinn ML, Jaffee EM, McFadden JM, Kuhajda FP. Fatty Acid Synthase Inhibitors Are Chemopreventive for Mammary Cancer in Neu-N Transgenic Mice. Oncogene. 2005;24(1):39–46. doi: 10.1038/sj.onc.1208174. [DOI] [PubMed] [Google Scholar]
- 7.Li J, Gorospe M, Chrest F, Kumaravel T. Pharmacological Inhibition of Fatty Acid Synthase Activity Produces Both Cytostatic and Cytotoxic Effects Modulated by p53. Cancer Res. 2001;61:1493–1499. [PubMed] [Google Scholar]
- 8.Lupu R, Menendez JA. Pharmacological Inhibitors of Fatty Acid Synthase (FASN)–Catalyzed Endogenous Fatty Acid Biogenesis: A New Family of Anti-Cancer Agents? Curr Pharm Biotechnol. 2006;7(6):483–493. doi: 10.2174/138920106779116928. [DOI] [PubMed] [Google Scholar]
- 9.Pizer E, Thupari J, Han W, Pinn M. Malonyl-Coenzyme-A Is a Potential Mediator of Cytotoxicity Induced by Fatty-Acid Synthase Inhibition in Human Breast Cancer Cells and Xenografts. Cancer Res. 2000;60:213–218. [PubMed] [Google Scholar]
- 10.Filippatos T, Derdemezis C, Gazi I. Orlistat-Associated Adverse Effects and Drug Interactions- A Critical Review. Drug Saf. 2008;31(1):53–65. doi: 10.2165/00002018-200831010-00005. [DOI] [PubMed] [Google Scholar]
- 11.Pemble CW, Johnson LC, Kridel SJ, Lowther WT. Crystal Structure of the Thioesterase Domain of Human Fatty Acid Synthase Inhibited by Orlistat. Nat Struct Mol Biol. 2007;14(8):704–709. doi: 10.1038/nsmb1265. [DOI] [PubMed] [Google Scholar]
- 12.Fako VE, Zhang J-T, Liu J-Y. Mechanism of Orlistat Hydrolysis by the Thioesterase of Human Fatty Acid Synthase. ACS Catal. 2014;4:3444–3453. doi: 10.1021/cs500956m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kridel SJ, Axelrod F, Rozenkrantz N, Smith JW. Orlistat Is a Novel Inhibitor of Fatty Acid Synthase with Antitumor Activity. Cancer Res. 2004;64(6):2070–2075. doi: 10.1158/0008-5472.can-03-3645. [DOI] [PubMed] [Google Scholar]
- 14.Browne CD, Hindmarsh EJ, Smith JW. Inhibition of Endothelial Cell Proliferation and Angiogenesis by Orlistat, a Fatty Acid Synthase Inhibitor. FASEB J. 2006;20(12):2027–2035. doi: 10.1096/fj.05-5404com. [DOI] [PubMed] [Google Scholar]
- 15.Grube S, Dünisch P, Freitag D, Klausnitzer M, Sakr Y, Walter J, Kalff R, Ewald C. Overexpression of Fatty Acid Synthase in Human Gliomas Correlates with the WHO Tumor Grade and Inhibition with Orlistat Reduces Cell Viability and Triggers Apoptosis. J Neuro-Oncol. 2014;118(2):277–287. doi: 10.1007/s11060-014-1452-z. [DOI] [PubMed] [Google Scholar]
- 16.Menendez Ja, Vellon L, Lupu R. Antitumoral Actions of the Anti-Obesity Drug Orlistat (XenicalTM) in Breast Cancer Cells: Blockade of Cell Cycle Progression, Promotion of Apoptotic Cell Death and PEA3-Mediated Transcriptional Repression of Her2/neu (erbB-2) Oncogene. Ann Oncol. 2005;16(8):1253–1267. doi: 10.1093/annonc/mdi239. [DOI] [PubMed] [Google Scholar]
- 17.Chuang HY, Chang YF, Hwang JJ. Antitumor Effect of Orlistat, a Fatty Acid Synthase Inhibitor, Is via Activation of Caspase-3 on Human Colorectal Carcinoma-Bearing Animal. Biomed Pharmacother. 2011;65(4):286–292. doi: 10.1016/j.biopha.2011.02.016. [DOI] [PubMed] [Google Scholar]
- 18.Little JL, Wheeler FB, Fels DR, Koumenis C, Kridel SJ. Inhibition of Fatty Acid Synthase Induces Endoplasmic Reticulum Stress in Tumor Cells. Cancer Res. 2007;67(3):1262–1269. doi: 10.1158/0008-5472.CAN-06-1794. [DOI] [PubMed] [Google Scholar]
- 19.Dowling S, Cox J, Cenedella RJ. Inhibition of Fatty Acid Synthase by Orlistat Accelerates Gastric Tumor Cell Apoptosis in Culture and Increases Survival Rates in Gastric Tumor Bearing Mice in Vivo. Lipids. 2009;44(6):489–498. doi: 10.1007/s11745-009-3298-2. [DOI] [PubMed] [Google Scholar]
- 20.Agostini M, Almeida LY, Bastos DC, Ortega RM, Moreira FS, Seguin F, Zecchin KG, Raposo HF, Oliveira HCF, Amoedo ND, Salo T, Coletta RD, Graner E. The Fatty Acid Synthase Inhibitor Orlistat Reduces the Growth and Metastasis of Orthotopic Tongue Oral Squamous Cell Carcinomas. Mol Cancer Ther. 2014;13(3):585–595. doi: 10.1158/1535-7163.MCT-12-1136. [DOI] [PubMed] [Google Scholar]
- 21.Seguin F, Carvalho MA, Bastos DC, Agostini M, Zecchin KG, Alvarez-Flores MP, Chudzinski-Tavassi aM, Coletta RD, Graner E. The Fatty Acid Synthase Inhibitor Orlistat Reduces Experimental Metastases and Angiogenesis in B16-F10 Melanomas. Br J Cancer. 2012;107(6):977–987. doi: 10.1038/bjc.2012.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carvalho MA, Zecchin KG, Seguin F, Bastos DC, Agostini M, Rangel ALCa, Veiga SS, Raposo HF, Oliveira HCF, Loda M, Coletta RD, Graner E. Fatty Acid Synthase Inhibition with Orlistat Promotes Apoptosis and Reduces Cell Growth and Lymph Node Metastasis in a Mouse Melanoma Model. Int J Cancer. 2008;123(11):2557–2565. doi: 10.1002/ijc.23835. [DOI] [PubMed] [Google Scholar]
- 23.Orlistat: DB01083. http://www.drugbank.ca/drugs/DB01083.
- 24.Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P, Chang Z, Woolsey J. DrugBank: A Comprehensive Resource for in Silico Drug Discovery and Exploration. Nucleic Acids Res. 2006;34(Database issue):D668–D672. doi: 10.1093/nar/gkj067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhi J, Melia AT, Funk C, Viger-Chougnet A, Hopfgartner G, Lausecker B, Wang K, Fulton JS, Gabriel L, Mulligan TE. Metabolic Profiles of Minimally Absorbed Orlistat in Obese/overweight Volunteers. J Clin Pharmacol. 1996;36(11):1006–1011. doi: 10.1177/009127009603601104. [DOI] [PubMed] [Google Scholar]
- 26.Hargittai I, Hargittai M. Molecular Structure of Hyaluronan: An Introduction. Struct Chem. 2008;19(5):697–717. [Google Scholar]
- 27.Laurent T, Fraser J. Hyaluronan. FASEB J. 1992;6:2397–2404. [PubMed] [Google Scholar]
- 28.Hill TK, Abdulahad A, Kelkar SS, Marini FC, Long TE, Provenzale JM, Mohs AM. Indocyanine Green-Loaded Nanoparticles for Image-Guided Tumor Surgery. Bioconjugate Chem. 2015;26(2):294–303. doi: 10.1021/bc5005679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cho HJ, Yoon HY, Koo H, Ko SH, Shim JS, Lee JH, Kim K, Kwon IC, Kim DD. Self-Assembled Nanoparticles Based on Hyaluronic Acid-Ceramide (HA-CE) and Pluronic® for Tumor-Targeted Delivery of Docetaxel. Biomaterials. 2011;32(29):7181–7190. doi: 10.1016/j.biomaterials.2011.06.028. [DOI] [PubMed] [Google Scholar]
- 30.Choi KY, Yoon HY, Kim JH, Bae SM, Park RW, Kang YM, Kim IS, Kwon IC, Choi K, Jeong SY, Kim K, Park JH. Smart Nanocarrier Based on PEGylated Hyaluronic Acid for Cancer Therapy. ACS Nano. 2011;5(11):8591–8599. doi: 10.1021/nn202070n. [DOI] [PubMed] [Google Scholar]
- 31.Bennett PK, Li YT, Edom R, Henion J. Quantitative Determination of Orlistat (tetrahydrolipostatin, Ro 18–0647) in Human Plasma by High-Performance Liquid Chromatography Coupled with Ion Spray Tandem Mass Spectrometry. J Mass Spectrom. 1997;32(7):739–749. doi: 10.1002/(SICI)1096-9888(199707)32:7<739::AID-JMS526>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 32.Mohammadi a, Haririan I, Rezanour N, Ghiasi L, Walker RB. A Stability-Indicating High Performance Liquid Chromatographic Assay for the Determination of Orlistat in Capsules. J Chromatogr A. 2006;1116(1–2):153–157. doi: 10.1016/j.chroma.2006.03.038. [DOI] [PubMed] [Google Scholar]
- 33.Souri E, Jalalizadeh H, Kebriaee-Zadeh A, Zadehvakili B. HPLC Analysis of Orlistat and Its Application to Drug Quality Control Studies. Chem Pharm Bull. 2007;55(2):251–254. doi: 10.1248/cpb.55.251. [DOI] [PubMed] [Google Scholar]
- 34.Odens H, Lowther T, Kridel S, Watts L, Filipponi L, Schmitt J. Inhibition of the Thioesterase Activity of Human Fatty Acid Synthase by 1,4- and 9,10-Diones. Chem Pharm Bull. 2014;62(9):933–936. doi: 10.1248/cpb.c13-00809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Purohit VC, Richardson RD, Smith JW, Romo D. Practical, Catalytic, Asymmetric Synthesis of Beta-Lactones via a Sequential Ketene Dimerization/hydrogenation Process: Inhibitors of the Thioesterase Domain of Fatty Acid Synthase. J Org Chem. 2006;71(12):4549–4558. doi: 10.1021/jo060392d. [DOI] [PubMed] [Google Scholar]
- 36.Hill BG, Dranka BP, Zou L, Chatham JC, Darley-Usmar VM. Importance of the Bioenergetic Reserve Capacity in Response to Cardiomyocyte Stress Induced by 4-Hydroxynonenal. Biochem J. 2009;424(1):99–107. doi: 10.1042/BJ20090934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pallasch CP, Schwamb J, Königs S, Schulz a, Debey S, Kofler D, Schultze JL, Hallek M, Ultsch a, Wendtner C-M. Targeting Lipid Metabolism by the Lipoprotein Lipase Inhibitor Orlistat Results in Apoptosis of B-Cell Chronic Lymphocytic Leukemia Cells. Leukemia. 2008;22(3):585–592. doi: 10.1038/sj.leu.2405058. [DOI] [PubMed] [Google Scholar]
- 38.Choi KY, Min KH, Yoon HY, Kim K, Park JH, Kwon IC, Choi K, Jeong SY. PEGylation of Hyaluronic Acid Nanoparticles Improves Tumor Targetability in Vivo. Biomaterials. 2011;32(7):1880–1889. doi: 10.1016/j.biomaterials.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 39.Bae YH, Park K. Targeted Drug Delivery to Tumors: Myths, Reality and Possibility. J Controlled Release. 2011;153(3):198–205. doi: 10.1016/j.jconrel.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petros RA, DeSimone JM. Strategies in the Design of Nanoparticles for Therapeutic Applications. Nat Rev Drug Discovery. 2010;9(8):615–627. doi: 10.1038/nrd2591. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.