

Abstract
The ability to detect picomolar concentrations of glucagon and amylin using fluorescently labeled mirror-image aptamers, so-called Spiegelmers, is demonstrated. Spiegelmers rival the specificity of antibodies and overcome the problem of biostability of natural aptamers in a biological matrix. Using Spiegelmers as affinity probes, noncompetitive capillary electrophoresis affinity assays of glucagon and murine amylin were developed and optimized. The detection limit for glucagon was 6 pM and for amylin was 40 pM. Glucagon-like peptide-1 and -2 did not interfere with the glucagon assay, while the amylin assay showed cross-reactivity to calcitonin gene related peptide. The developed assays were combined with a competitive immunoassay for insulin to measure glucagon, amylin, and insulin secretion from batches of islets after incubation with different glucose concentrations. The development of these assays is an important step towards incorporation into an online measurement system for monitoring dynamic secretion from single islets.
Introduction
Affinity probe capillary electrophoresis (APCE) assays coupled with laser-induced fluorescence (LIF) detection has been shown to be an effective and simple alternative to traditional heterogeneous affinity assays.1 There are two formats of APCE assays. The competitive assay format is widely applied with antibodies (Ab) as affinity probes since the separation of free labeled-antigen (Ag*) from the antibody-Ag* complex peak (Ab-Ag*) is relatively straightforward. However, these assays suffer from low sensitivity due to their dependence on the dissociation constant (Kd) of the binding reaction. In noncompetitive affinity assays, an excess of labeled affinity probe (P*) is added to the sample containing a target (T). The presence of a probe-target (P*-T) complex peak is directly proportional to the concentration of T in the sample. Important advantages of this mode include the ability to differentiate possible cross-reactive species in the sample2 and the sensitivity of the assay is less dependent on the Kd since an excess of P* is used. Noncompetitive affinity assays using fluorescently-labeled Ab as P* are rare due to the difficulty in labeling the large biomolecule homogeneously resulting in a difficult separation of bound P*-T from free P*.3
The advent of aptamers has spurred their incorporation as affinity probes into noncompetitive capillary electrophoresis (CE) affinity assays due to the facile labeling with fluorophores and predictable electrophoretic properties. For example, aptamers have been used in APCE assays to detect targets including small molecules and proteins (eg. IgE, thrombin, and ricin).3–9 Furthermore, the lengths of aptamers are tunable and can be used to modify the mobility of the protein-aptamer complex to achieve quantitation of multiple species in a single run on capillary and microfluidic devices.10, 11 A functional limitation of aptamers is biodegradation due to the omnipresence of degrading enzymes in all biological fluids.5, 12
To reduce the possibility of biodegradation, mirror-image aptamers, so-called Spiegelmers®, have been developed.12–14 These mirror-image aptamers are formed from L-oligonucleotides making them resistant to nucleases since naturally occurring enzymes are stereoselective for the naturally occurring D-enantiomeric form.15 Due to the native biostability, L-aptamers have been recognized to be viable pharmaceutical candidates, and several Spiegelmers against pharmacologically attractive targets have been developed and tested in clinical trials. In contrast to their use as potential pharmacological agents, L-aptamers have not been widely used in bioanalytical assays so far. Early work took advantage of their biostability against nucleases for use as stationary phases in liquid chromatography and chiral selectors in CE.14, 16, 17 Another example used Spiegelmers as affinity probes in electrochemical and colorimetric detection of ghrelin.18, 19 However, these bioassays involved immobilization of either ghrelin or its Spiegelmer on a solid surface and included multiple washing steps, making the assays time consuming and laborious.
In this paper, we demonstrate the use of two recently developed Spiegelmers as affinity probes in noncompetitive APCE assays for detection of glucagon and amylin secretion from islets of Langerhans in vitro. The developed assays can detect glucagon and amylin with detection limits of 6 and 40 pM, respectively. No cross-reactivity was detected for the glucagon Spiegelmer to glucagon like peptide-1 or -2 (GLP-1 or GLP-2), whereas the amylin Spiegelmer showed reactivity to human and murine amylin and calcitonin gene-related peptide (CGRP), but not to human adrenomedullin. The assays for murine amylin and glucagon were combined with a previously developed competitive immunoassay for insulin to allow measurement of insulin, glucagon, and amylin secretion from groups of murine islets upon stimulation with different glucose concentrations. In the future, the combined assays can be transferred to a microfluidic platform to monitor dynamic secretion of these endocrine hormones from islets.
Experimental
Chemicals and reagents
Ethylenediaminetetraacetic acid (EDTA), sodium hydroxide (NaOH), hydrochloric acid (HCl), calcium chloride (CaCl2), potassium chloride (KCl), Tween-20, and bovine serum albumin (BSA) were purchased from EMD Chemicals (San Diego, CA). Dextrose, sodium phosphate monobasic (NaH2PO4), and sodium tetraborate (Na2B4O7) were from Fisher Scientific (Pittsburgh, PA). Mouse amylin and glucagon were purchased from Bachem Americas, Inc. (Torrance, CA). Human amylin was from AnaSpec, Inc. (Fermont, CA). 6-FAM labeled mixed L-DNA/L-RNA anti-glucagon Spiegelmer (NOX-G16004: 5′-GCGGGOH AAATG GOHGAGOHGOH GCTAG GTOHGGAOH AOHGGAA TCTGA GCGC-3′)20 and 6-FAM labeled L-RNA-based anti-amylin Spiegelmer (NOX-A42003: 5′-GGACU GAUGG CGCGG UCCUA UUACG CCGAU AGGGU GAGGG GA-3′)13, 21 were synthesized at NOXXON Pharma. A monoclonal Ab to human insulin C-terminal was obtained from Meridian Life Science, Inc. (Saco, ME). Cy5 monofunctional N-hydroxysuccinimide ester was from GE Healthcare Bio-Sciences (Piscataway, NJ). Labeling of bovine insulin with Cy5 (Cy5-insulin) was performed as previously described. Collagenase P was obtained from Roche Diagnostics (Indianapolis, IN). RPMI 1640 was from Mediatech (Manassas, VA). Gentamicin was from Lonza (Walkersville, MD). Cosmic Calf Serum was from HyClone Laboratories (South Logan, UT). All other chemicals were purchased from Sigma-Aldrich unless otherwise stated. All solutions were prepared using ultrapure deionized water (NANOpure® Diamond TM deionization system, Barnstead International, Dubuque, IA).
Sample preparation
Spiegelmer stock and working solutions were prepared in 10 mM Tris-HCl, 1 mM EDTA, 1 mM MgCl2, pH 8.0. Before use, Spiegelmer working solutions were heated to 94 °C for 3 min and then cooled down at the rate of 0.1 °C/s to room temperature in a thermal cycler (Bibby Scientific Ltd., Burlington, NJ) to obtain the favorable thermodynamic conformation for binding. Stock solutions of mouse amylin, GLP-1, GLP-2, and mouse/rat CGRP were prepared in deionized water at 1 mg/mL. Stock solution of human amylin was prepared in 10% DMSO and 90% deionized water. Glucagon powder was reconstituted in 50 mM acetic acid that had been degassed with N2 to remove oxygen in order to minimize oxidation of glucagon. Working solutions of all peptides were diluted to 1 μM in the sample buffer which was composed of 20 mM tricine, 1 mM EDTA, 10 mM MgCl2, pH 7.4, with 0.01% (w/v) BSA and 0.01% (w/v) Tween-20.
For the optimization of incubation conditions, the sample contained a mixture of Spiegelmer and glucagon or amylin in the sample buffer. Optimization was performed by varying the incubation time and temperature of the mixture in the sample buffer. For the optimization of the glucagon assay, the sample mixture was incubated from 0 – 120 min in the CE sample tray where the temperature was controlled between 15 and 37 °C. For the glucagon calibration samples, the mixture contained 250 nM G16004 and 0 – 500 nM glucagon in the sample buffer.
To optimize the amylin assay, the incubation time was investigated from 0 – 40 hours and the incubation temperature was tested from 4 to 37 °C. Calibration samples for the simultaneous measurement of amylin and insulin contained a mixture of 250 nM A42003, 0 – 100 nM amylin, 100 nM Cy5-insulin, 40 nM anti-insulin antibody, and 0 – 500 nM insulin in sample buffer. The mixture of A42003, amylin, and insulin were incubated at the optimized amylin assay conditions first, and then Cy5-insulin and anti-insulin antibody were spiked to the desired concentrations before the analysis.
Instrumentation and separations
CE experiments were performed on a PA800 CE instrument (Beckman Coulter, Fullerton, CA) with a dual wavelength LIF detector. A 3 mW 488 nm argon-ion laser (Beckman) was used as the excitation source for glucagon and amylin assays. For experiments involving simultaneous detection of insulin, a 100 mW 635 nm laser (AixiZ, Houston, TX) with a neutral density filter (Omega, Brattleboro, VT) was also used. The power of the 635 nm laser was 3 mW prior to entry to the instrument. A dual color detection scheme was used for simultaneous assays of amylin and insulin as previously described.22, 23 All separations were performed in a 50 μm i.d. × 360 μm o.d., 30 cm long fused-silica capillary (Polymicro Technologies, Phoenix, AZ). Data was acquired at 4 Hz.
At the beginning of each day, the capillary was rinsed with 1 M HCl, 1 M NaOH, 0.1 M NaOH, and deionized water, each for 10 min at 20 psi. The capillary was then conditioned with separation buffer for 10 min at 20 psi. Between runs of the amylin assay, the capillary was rinsed with 0.1 M NaOH, deionized water, and separation buffer each for 1 min at 20 psi. For the glucagon assay, additional rinses with 1 M HCl and 1 M NaOH, each for 1 min at 20 psi, were added between runs to regenerate the capillary surface for stable electroosmotic flow. Samples were injected by applying a pressure of 0.5 psi for 5 s. Separation was performed at 20 kV with a 1 min voltage ramp time and a cartridge temperature of 15 °C. For the glucagon assay, the optimized separation buffer was 20 mM borate and 1 mM MgCl2 at pH 9.0. For the amylin assay, the optimized separation buffer was 20 mM borate at pH 9.0.
Isolation and incubation of islets
All experiments were performed under guidelines approved by the Florida State University Animal Care and Use Committee, protocol #1235. Pancreatic islets were obtained from 20 – 40 g CD-1 male mice (Charles River Laboratories Internal, Inc., Wilmington, MA) as previously described.24 Isolated islets were cultured at 37 °C, 5% CO2 in RPMI 1640 media containing 11 mM glucose, 10% calf serum, 100 units mL−1 penicillin, 100 μg mL−1 streptomycin, and 10 μg mL−1 gentamicin. The islets were used within 5 days after isolation.
Prior to experiments for measuring secretion, about 20 islets were picked from the culture media and rinsed in pre-warmed balanced salt solution (BSS) containing 3 or 20 mM glucose. In addition to glucose, the BSS contained (in mM): 125 NaCl, 5.9 KCl, 1.2 MgCl2, 2.4 CaCl2, 20 tricine at pH 7.4, with 0.01% (w/v) BSA. Islets rinsed in BSS containing 3 mM glucose were then transferred to 10 μL BSS containing 20 mM glucose. After an hour of incubation, 4 μL of the BSS was collected and diluted to 10 μL with the sample buffer that contained G16004 and incubated at the optimized conditions. For measurement of amylin and insulin, an additional 4 μL of the BSS was collected and diluted to 8 μL with A42003 in the sample buffer and incubated at the optimized condition. After incubation and immediately before the APCE separation, 1 μL of Cy5-insulin and 1 μL anti-insulin antibody in the sample buffer were spiked into the sample. The final concentration of each reagent in the sample was the same as that was used in obtaining the calibration curves. Three sets of islet experiments were performed for each glucose concentration.
Data analysis
Data were analyzed using the 32 Karat™ software supplied with the CE instrument. Peak height percentages of the G16004-glucagon or peak area percentages of A42003-amylin complex were background subtracted by a blank electropherogram containing only 250 nM Spiegelmer. For glucagon assays, the peak height percentage was used for quantification since the peaks of P*-T and P* was not baseline resolved. Calibration curves of glucagon or amylin were obtained by plotting the background subtracted peak height or area percentages against the concentrations of glucagon or amylin with a linear regression fit, respectively. Limit of detection (LOD) was determined as the concentration of peptide that would produce a signal that was equal to three times the standard deviation of the peak height or area percentages from a blank sample, which contained only Spiegelmer. The dissociation constants (Kd) of G16004 to glucagon was extracted from the full calibration curve by fitting a one site binding model using nonlinear regression analysis (Origin 8.0, Northampton, MA).25 Insulin data were analyzed as previously described.23 Each sample was injected three times and results are shown as the mean ± 1 standard deviation. For the islet secretion data, the concentrations of each peptide were quantified with its respective calibration curve, converted to mass, and normalized to number of incubated islets. Statistical significance was tested with a one-tail Student’s t-test and results were considered different with a p-value < 0.05.
Results and discussion
Noncompetitive assays are preferred over competitive assays since the former offers the advantages of simplicity, lower detection limits, and larger dynamic range. Despite these advantages, development of noncompetitive assays is limited by two challenges associated with the most commonly used affinity reagent, antibodies: (1) difficulty in homogeneous labeling and (2) unpredictable electrophoresis properties. To circumvent these limitations, aptamers have been evaluated as affinity reagents for the detection of numerous proteins. Spiegelmers, as a new generation of aptamers, were designed to overcome the problem of biostability of conventional aptamers in biological fluids. In contrast to aptamers, they consist of mirror image building blocks, i.e. L-DNA or L-RNA nucleotides, and are therefore resistant to nucleases.
In this report, fluorescently-labeled versions of Spiegelmers against glucagon (NOX-G16004) and amylin (NOX-A42003) were used. NOX-G16004, is a fluorescently labeled, modified version of a previously identified Spiegelmer, NOX-G15. NOX-G15 is a PEGylated 39 nucleotide mixed DNA/RNA L-aptamer. The exchange of L-deoxyribonucleotides by the corresponding L-ribonucleotides in six positions led to a higher affinity.20 The dissociation constant for NOX-G15 to L-glucagon was found to be 3 nM. G16004 has a similar sequence (with an additional deoxyribonucleotide to ribonucleotide exchange), is FAM-labeled, and unPEGylated. NOX-A42003, the Spiegelmer used for the amylin assays, is a FAM-labeled 42 nucleotide L-RNA aptamer modified from STAR-F12-Δ43–48, an aptamer originally developed to bind to calcitonin gene-related peptide (CGRP).13, 21 In addition to the 6-FAM labeling, two nucleotide bases were exchanged (A→U) to the STAR-F12-Δ43–48 aptamer in order to improve production yield.
Both glucagon and amylin are two hormones secreted from pancreatic islets that help regulate glucose homeostasis and their autocrine and paracrine interactions have triggered the demand to measure their secretion simultaneously to understand the mechanisms of glucose homeostasis. Glucagon and/or amylin assays have been developed in the competitive immunoassay format using Ab as affinity ligands22, 23 and transferred to microfluidic devices to monitor their dynamic secretion from islets of Langerhans.26 However, the LOD of the developed competitive immunoassays were at unsatisfactorily high levels, which as stated before, were intrinsically determined by the Kd of the Ab-Ag* binding reaction. In this report, we developed noncompetitive affinity assays for glucagon and amylin using their respective Spiegelmers as affinity probes, and applied this method to measure hormone secretions from islets of Langerhans.
Optimization of glucagon affinity assay
Optimization of separation and incubation conditions
A mixture of 250 nM G16004 and 100 nM glucagon was used to optimize the separation and incubation conditions of the glucagon assay. In noncompetitive APCE assays where the P* concentration is above the Kd of binding between P* and T and excessively above the concentration of T, the signal intensity of P*-T after separation is a direct measure of the concentration of T in the sample.27 Therefore, resolution of the P*-T from the free P* is important to achieve accurate quantification and low detection limits. Different separation buffers including 20 mM phosphate (pH 7.4), 50 mM tricine (pH 8.5), and 20 mM borate (pH 9.0), were tested to achieve the best separation resolution. Compared to the other two buffers, 20 mM borate separation buffer provided the fastest separation, highest signal intensity, and most narrow peaks (data not shown). The short separation time and higher signal intensity was attributed to the high pH of the borate buffer compared to the other two buffers. Therefore, 20 mM borate, pH 9.0 was chosen as the optimal separation buffer.
Figure 1A i & ii are representative electropherograms of (i) 250 nM G16004 alone and (ii) with 100 nM glucagon. The peak labeled with a star is free G16004; addition of glucagon resulted in the decrease of the free G16004 peak and an increase of the complex peak of G16004 formed with glucagon. However, the resolution between the free G16004 and complex peak was not satisfactory for quantification of glucagon at low concentrations. To improve the resolution between the complex and free G16004 peak, different effective separation lengths (10 cm vs. 20 cm) were compared. From Figure 1A ii & iii, it can be seen that a 20 cm separation length provided better resolution of the complex from the free G16004. At the same time, a longer separation time resulted in fronting of the free G16004 peak, which could be due to nonspecific binding between G16004 with the capillary, or different conformations of G16004. We did not see evidence for dissociation of the complex since the peak profiles of G16004 in the absence and presence of glucagon were similar. G16004 is a guanine rich sequence, which may form an intramolecular G-quadruplex structure as has been observed with other G-rich aptamers.28 Magnesium is an important ion for stabilizing the conformation of oligonucleotides29, 30 and has been used as an additive in separation buffers to interact with the aptamer and improve separation.11 Here, the effect of MgCl2 was investigated by adding 0 – 10 mM MgCl2 into the separation buffer. Higher concentrations of MgCl2 provided better resolution, but also slowed down electroosmotic flow and distorted the peak shapes (data not shown). Addition of 1 mM MgCl2 to the separation buffer resulted in a better resolution and a higher signal intensity (Figure 1A iv) compared with that when the effective separation length was doubled and without MgCl2 in the separation buffer (Figure 1A iii). The higher signal intensity could be due to the interaction between Mg2+ with Spiegelmer. However, addition of Mg2+ to the separation buffer lead to irreproducible migration times (RSD: ~16% for 6 consecutive runs), which may have resulted from the interaction between the Mg2+ with the silica surface of the capillary.31 To regenerate the capillary surface, additional conditioning of the capillary with 1 M HCl and 1M NaOH were added between runs and the migration time was then reproducible with an RSD of ~0.4%. Therefore, the optimized separation conditions for the glucagon assay were 20 mM borate, 1 mM MgCl2, pH 9.0 in an effective separation length of 10 cm at a separation voltage of 20 kV.
Fig. 1.

Optimization of glucagon assay. (A) Electropherograms for the optimization of the separation conditions of glucagon assay: i. 250 nM G16004; ii – iv. 250 nM G16004 with 100 nM glucagon at different separation conditions. The effective separation length was 10 cm for i, ii, and iv, while it was 20 cm for iii. The separation buffer was 20 mM borate, pH 9.0 for i, ii, and iii, and 20 mM borate with 1 mM Mg2+, pH 9.0 for iv. The free G16004 is starred in each electropherogram and the G16004-glucagon complex is present in runs ii – iv. The G16004 and glucagon mixture was incubated for at least an hour at 15 °C prior to injection. (B) Effect of the incubation temperature and time on the peak height percentages of the G16004-glucagon complex is shown. 250 nM G16004 was incubated with 100 nM glucagon at 15 (squares), 25 (circles), and 37(triangles) °C for incubation times ranging from 0 – 120 min. The other conditions were the same as in Figure 1A iv. Each data point represents the mean of three runs at the same conditions with error bars corresponding to ± 1 SD.
The incubation conditions of the glucagon assay were then optimized for the highest height percentage of the complex peak. The original selection buffer for G15 contained MgCl2, CaCl2, surfactant, serum albumin, other factors, and was at physiological pH.20 The incubation buffer tested in this report was similar and produced an adequate signal of the complex, so further optimization of the incubation buffer composition was not performed. A mixture of 250 nM G16004 and 100 nM glucagon was incubated in the CE sample tray, and three different incubation temperatures were tested (15, 25, 37°C). Three replicate samples incubated at each temperature were then injected and separated at the optimized separation conditions. Figure 1B shows the average peak height percentages of the complex peak at different incubation temperatures as a function of reaction time. As can be seen, compared to 25 and 37°C, an incubation temperature of 15°C resulted in the highest peak percentage with the smallest RSD and the percentage of bound reached a plateau in 30 min. The runs at 25 and 37°C had higher RSDs than those at 15°C (10–50% compared to 1–3%, respectively), likely due to unstable G16004-glucagon complexes at the higher temperatures. Therefore, incubation of the mixture at 15°C for one hour was chosen as the optimal binding condition.
Calibration curve of glucagon assay
Using the optimized incubation and separation conditions, a series of solutions containing 250 nM G16004 and 0 – 500 nM glucagon were incubated, injected, and separated. Figure 2A shows representative electropherograms at different concentrations of glucagon indicated in the figure. The average peak height percentage obtained as a function of glucagon concentration is summarized in Figure 2B. Each point is the average of three replicates and the RSD of the peak height percentages of the complex peak ranged from 1.0 – 5.8%. From this data, the Kd of G16004 to glucagon was extracted and estimated to be 144 ± 38 nM. From 0 to 100 nM glucagon (Figure 2C), the complex peak height percentage increased linearly (Y = 0.5249 X) with a regression coefficient (r2) of 0.9968. From this curve, the LOD was found to be 6 pM, which rivals the detection limits of commercial heterogeneous immunoassays of glucagon.32 Figure 2D is a zoomed-in view of the 0.1 nM glucagon run, highlighted in Figure 2A. As can be seen, 0.1 nM glucagon was easily detected using the developed method. Picomolar detection limits indicated the LOD was less dependent on the Kd, which is expected in noncompetitive assays as the concentration of P* can be experimentally increased to promote formation of the P*-T complex.2
Fig. 2.
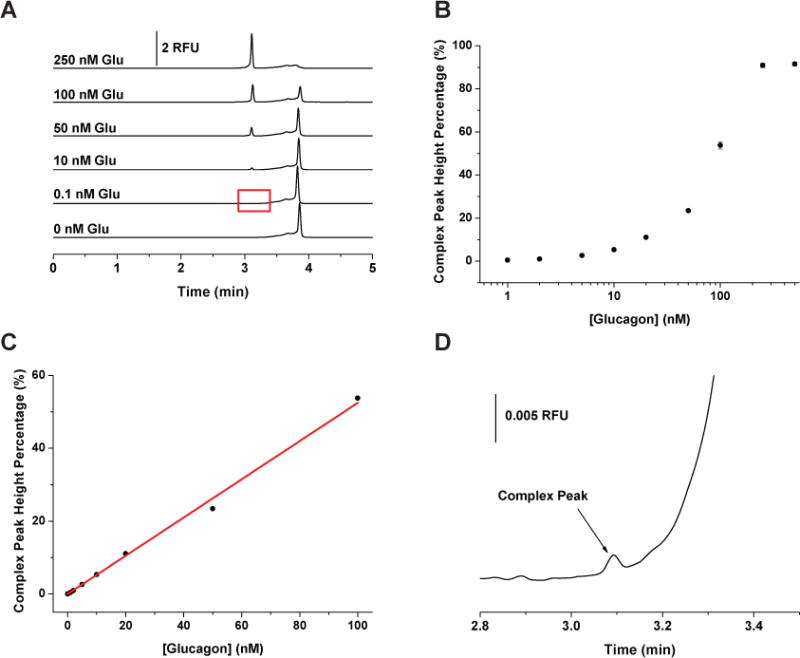
Calibration of glucagon assay. (A) Representative electropherograms when G16004 was held constant at 250 nM while the concentration of glucagon was increased from 0 to 250 nM from the bottom to the top as indicated in the figure. (B) Complex peak height percentages as a function of glucagon concentrations. (C) Calibration curve of complex peak height percentages vs. glucagon concentration from 0 to 100 nM. The data followed a linear response, Y = 0.5249 X, r2 = 0.9968. (D) Zoom in view of the G16004-glucagon complex peak for 0.1 nM glucagon. This run is highlighted by the red box in (A). All samples were incubated at 15 °C for at least 1 hour before injection. Each data point represents the mean of three runs with error bars corresponding to ± 1 SD.
Optimization of amylin affinity assay
Optimization of separation and incubation conditions
For the amylin assay, a mixture of A42003 and murine amylin was tested with the same buffers used for the glucagon assay. The optimal separation buffer was chosen as 20 mM borate buffer, pH 9.0 because it provided a clean background for the complex peak (Figure 3 i & iv). Addition of 50 nM amylin to 250 nM A42003 resulted in a decrease of the free A42003 peak and the addition of a new peak. The resolution between the A42003-amylin peak and free A42003 was better compared to the glucagon assay at the same separation conditions (Figure 1A ii). This may be in part due to the amylin being more positively charged compared to glucagon at the same conditions (pI of amylin and glucagon, 9.2 and 6.1, respectively). Since this resolution was satisfactory for quantification, 20 mM borate, pH 9.0 was selected as the separation buffer without further optimization.
Fig. 3.
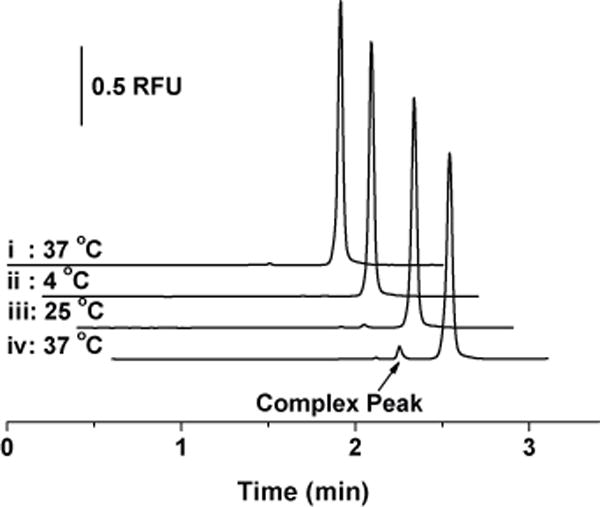
Effect of temperature on A42003-amylin complex peak formation. i: 250 nM A42003 after incubation for 16 hours at 37 °C. ii–iv: 250 nM A42003 with 50 nM murine amylin after incubation for 16 hours at 4 °C (ii), 25 °C (iii), and 37 °C (iv).
The same sample buffer was used as the incubation buffer for the amylin assay and the incubation temperature was optimized from 4 to 37 °C as shown in Figure 3 ii – iv after incubation for 16 hours. As demonstrated, an incubation temperature of 37 °C provided the highest complex peak area compared to the other two temperatures. Incubation times from 0 – 40 hours were tested and the peak area percentages increased linearly over the course of this time (data not shown). To compromise between a short incubation time and high sensitivity, 16 hours was chosen as the incubation time for the amylin assay. We found no experimental evidence for why the amylin assay required a longer incubation time, necessitating further experiments to understand the differences in the reaction kinetics that were observed. Therefore, the optimal amylin assay conditions were selected as 20 mM borate, pH 9.0 as the separation buffer and incubation of 250 nM A42003 with amylin at 37°C for 16 hours.
Calibration curve of amylin assay
Representative electropherograms of the amylin assay at the optimized conditions are shown in Figure 4A. The complex peak area percentage measured from 0 – 100 nM amylin is shown in Figure 4B. From 0 to 10 nM amylin, the peak area percentages increased linearly (Y = 0.1191 X) with a regression coefficient of 0.9990 (Figure 4C). Similar to glucagon, the peak area percentages were reproducible with RSDs < 5.3%. The detection limit was determined to be 40 pM from the calibration curve and Figure 4D is a zoomed-in view of an electropherogram obtained for the detection of 0.5 nM amylin. Because equilibrium binding between A42003 and amylin was not achieved, Kd values were not determined for this interaction.
Fig. 4.
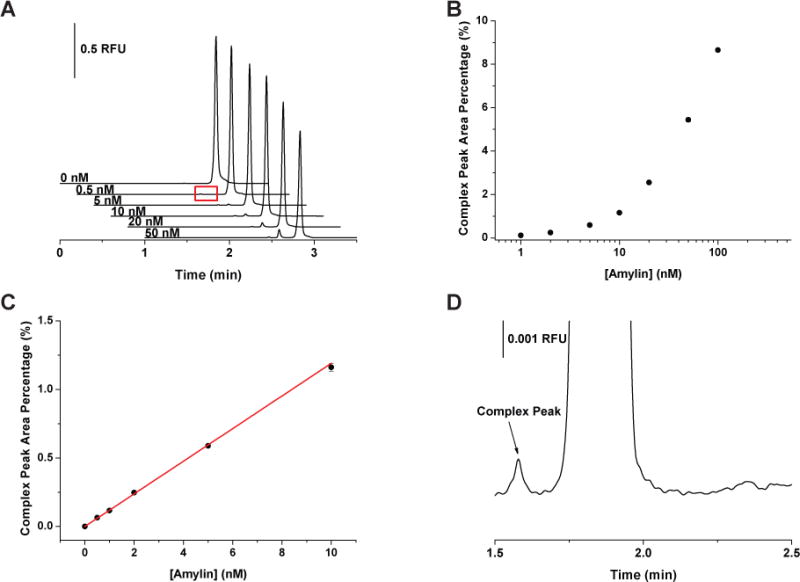
Effect of varying amylin concentrations on the peak area percentages of the A42003-amylin complex peak. (A) Representative electropherograms of amylin assay. The concentration of A42003 was fixed at 250 nM while the concentration of murine amylin increased from 0 to 50 nM as indicated in the figure. (B) The variation of the complex peak area percentage obtained as a function of amylin concentration. (C) Calibration curve of complex peak area percentages vs. amylin concentrations from 0 to 10 nM. The data followed a linear response, Y = 0.1191 X, r2 = 0.9990. (D) A zoomed-in view of the complex peak produced by incubation with 0.5 nM amylin, highlighted (A). All samples were incubated at 37 °C for 16 hours before injection. Each point represents the average of 3 runs with error bars corresponding to ± 1 SD.
The higher LOD of the amylin assay compared to the glucagon assay was due to the increased difficulty of detecting the A42003-amylin complex. This could be due to the amylin assay having a higher Kd, a lower quantum yield of A42003-amylin complex, and/or a slower binding reaction producing less of the complex at the incubation time used.
Specificity of assays
The binding specificity of the glucagon assay was tested by the addition of GLP-1 and GLP-2 to G16004. GLP-1 and GLP-2 are generated from the cleavage of proglucagon in intestinal L cells and are homologous peptide hormones to glucagon but have different biological functions. Figure 5A shows the representative electropherograms of 250 nM G16004 with (i) 20 nM glucagon, (ii) 100 nM GLP-1, and (iii) 100 nM GLP-2. As can be seen, there is no complex peak formed or significant decrease of the free G16004 peak after adding GLP-1 or GLP-2. The lack of cross-reactivity between G16004 and GLP-1 or GLP-2 is in agreement with the previous report describing the development of the glucagon Spiegelmer NOX-G15 from which NOX-G16004 was derived.20
Fig. 5.
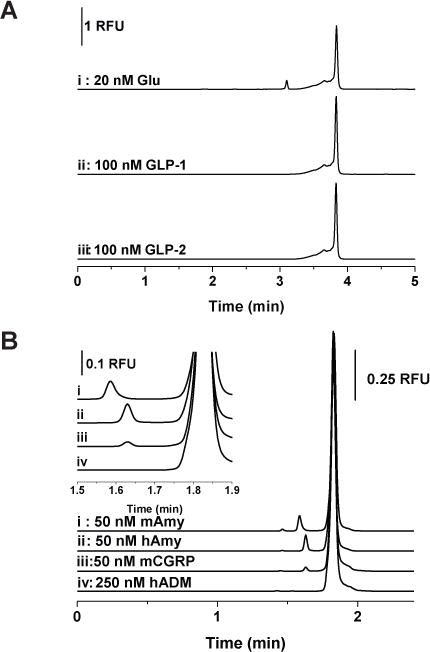
Specificity of the developed assays. (A) 250 nM G16004 incubated with (i) 20 nM glucagon, (ii) 100 nM GLP-1, and (iii) 100 nM GLP-2. (B) 250 nM A42003 incubated with (i) 50 nM murine amylin (mAmy), (ii) 50 nM human amylin (hAmy), (iii) 50 nM murine CGRP (mCGRP), and (iv) 50 nM human adrenomedullin (hADM).
The specificity of the amylin assay was first examined by testing the binding to human amylin, which has six substituted amino acids compared with murine amylin. Figure 5B shows the electropherograms of 250 nM A42003 with (i) 50 nM murine amylin and (ii) 50 nM human amylin. The complex peak formed between A42003 and human amylin has a similar peak height but migrates slower compared to the complex with murine amylin. This demonstrated A42003 has comparable reactivity with human amylin, and the slower mobility could be attributed to the difference in pI between murine and human amylin (pI of murine and human amylin, 9.2 vs. 8.3, respectively). These results indicate that the developed amylin assay can be applied to measure human amylin samples.
Other tested peptides for the specificity of amylin assay were two from the calcitonin gene peptide superfamily, CGRP and adrenomedullin.33–35 CGRP is a neuropeptide that acts as a strong vasodilator; besides peripheral and central neurons, it can also be found in rat islet cells and mouse pancreas.36 The sequence of A42003 was originally developed to bind to CGRP; it was also found to effectively block the excitatory effect of amylin in vivo.21 The cross reactivity of A42003 with murine CGRP and human adrenomedullin is shown in Figure 5B iii & iv. As expected due to the similarity with CGRP, a complex peak was observed, but with a smaller peak area and a slower migration time compared to that formed with murine amylin. The smaller peak area could have resulted from a lower affinity between the modified Spiegelmer to the original target of CGRP under the experimental conditions applied here. Although not shown in this report, the resolution between the complex peaks of A42003 formed with murine amylin and CGRP could be optimized to measure both peptides simultaneously. This is another demonstration that noncompetitive affinity assays can be applied to measure the cross-reactivity of the affinity probes with different analytes. Adrenomedullin has only structural but no sequence similarities to CGRP.37 As can be seen from Figure 5B iv, no complex peak was formed between A42003 and adrenomedullin even when the concentration of the peptide was increased to 250 nM. This indicates that the Spiegelmer A42003 specifically recognizes the amylin sequence and homologouspeptides.
Measurement of hormones secreted from islets
To better understand the mechanism of hormone-regulated glucose homeostasis, it would be ideal to measure the low levels of multiple hormones, simultaneously with insulin, secreted from islets of Langerhans. A previously developed Ab-based competitive immunoassay for insulin was added to the Spiegelmer amylin assay using a two-color detection scheme.23 The reason for choosing to integrate the amylin assay with the insulin instead of the glucagon assay was the shorter separation time compared to glucagon (2.5 vs. 4 min), which is favorable to preserve the insulin complex peak during the separation. To obtain the highest sensitivity of the insulin assay, the reagents were spiked into the sample after the 16 hour incubation of amylin with A42003. Control experiments showed that addition of the insulin immunoassay reagents (Cy5-labeled insulin, non-labeled insulin, and anti-insulin Ab) to the sample did not affect the amylin assay (data not shown).
To measure the secretion of the three hormones, 20 islets were incubated in 3 mM and subsequently in 20 mM glucose for an hour and the supernatants were collected and analyzed with the developed assays. As shown in Figure 6, secretion levels of amylin and insulin increased significantly, whereas glucagon secretion decreased significantly, when the glucose concentration was increased from 3 to 20 mM. Amylin secretion levels were 4.78 ± 0.95 pg islet−1 and 33.28 ± 3.16 pg islet−1 at 3 and 20 mM glucose, respectively. Insulin secretion amounts were 264.1 ± 20.6 pg islet−1 and 787.3 ± 18.8 pg islet−1 at the same glucose values. Glucagon secretion levels were 2.51 ± 0.59 pg islet−1 and 0.53 ± 0.63 pg islet−1 at 3 and 20 mM glucose, respectively. These results are in line with the expected biological responses to glucose stimulation and with the range of previously reported secretion levels.38–40 Transfer of the developed method to a microfluidic device will allow the capture of more details of the secretion dynamics.
Fig. 6.
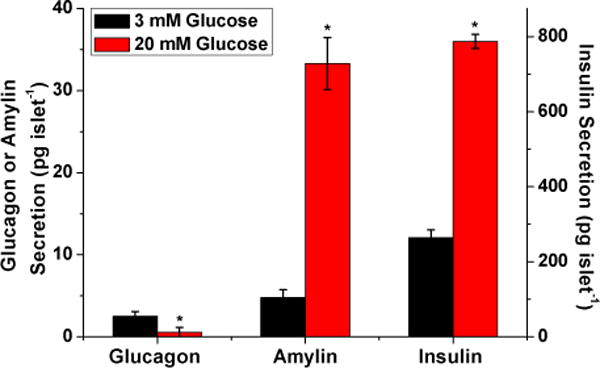
Measured amounts of amylin, glucagon, and insulin secretion from 20 islets in response to different glucose concentrations. The secretion levels measured after incubation in 3 mM glucose are shown in black and in 20 mM glucose in red. The average values of three replicate trials are plotted with error bars corresponding to ±1 standard deviation of the mean. Glucagon and amylin values are shown on the left y-axis and insulin values are shown on the right y-axis. The values of glucagon, amylin, and insulin measured at 20 mM glucose were all significantly different (p < 0.05) compared to those measured at 3 mM glucose.
Conclusions
In this report, Spiegelmers were used as affinity probes to develop noncompetitive affinity assays of glucagon and amylin, and the developed assays were successfully used to measure hormone secretion from islets. Spiegelmers provide a viable alternative to antibodies in terms of synthesis, labeling efficiency, and stability. The developed assays require incubation of a minimum number of reagents (labeled Spiegelmers) with the sample, and separation with capillary electrophoresis provides detection limits of picomolar concentrations for both glucagon and amylin. This provides new alternative assays for selective and sensitive detection of glucagon and amylin in comparison to competitive immunoassays or traditional ELISA or radioimmunoassay methods. Labeling one of the Spiegelmers with a different fluorophore and integration with a two-color detection scheme will allow simultaneous detection of both hormones. Further miniaturization of both assays onto microfluidic devices will further delineate the dynamic secretion of hormones from islets, which will be useful to understand the mechanism of glucose homeostasis regulated by multiple hormones in health and disease.
Acknowledgments
This work was supported by a grant from the National Institutes of Health (R01 DK080714). The authors wish to thank Dr. Axel Vater from NOXXON Pharma AG for helpful discussions.
Footnotes
® The term Spiegelmer is a trademark of NOXXON Pharma AG
References
- 1.Schultz NM, Kennedy RT. Anal Chem. 1993;65:3161–3165. [Google Scholar]
- 2.Shimura K, Karger BL. Anal Chem. 1994;66:9–15. doi: 10.1021/ac00073a004. [DOI] [PubMed] [Google Scholar]
- 3.German I, Buchanan DD, Kennedy RT. Anal Chem. 1998;70:4540–4545. doi: 10.1021/ac980638h. [DOI] [PubMed] [Google Scholar]
- 4.Buchanan DD, Jameson EE, Perlette J, Malik A, Kennedy RT. Electrophoresis. 2003;24:1375–1382. doi: 10.1002/elps.200390176. [DOI] [PubMed] [Google Scholar]
- 5.Haes AJ, Giordano BC, Collins GE. Anal Chem. 2006;78:3758–3764. doi: 10.1021/ac060021x. [DOI] [PubMed] [Google Scholar]
- 6.Eaton RM, Shallcross JA, Mael LE, Mears KS, Minkoff L, Scoville DJ, Whelan RJ. Anal Bioanal Chem. 2015;407:6965–6973. doi: 10.1007/s00216-015-8665-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pavski V, Le XC. Anal Chem. 2001;73:6070–6076. doi: 10.1021/ac0107305. [DOI] [PubMed] [Google Scholar]
- 8.Song M, Zhang Y, Li T, Wang Z, Yin J, Wang H. J Chromatogr A. 2009;1216:873–878. doi: 10.1016/j.chroma.2008.11.085. [DOI] [PubMed] [Google Scholar]
- 9.Zhu Z, Ravelet C, Perrier S, Guieu V, Roy B, Perigaud C, Peyrin E. Anal Chem. 2010;82:4613–4620. doi: 10.1021/ac100755q. [DOI] [PubMed] [Google Scholar]
- 10.Zhang H, Li XF, Le XC. J Am Chem Soc. 2008;130:34–35. doi: 10.1021/ja0778747. [DOI] [PubMed] [Google Scholar]
- 11.Lin X, Chen Q, Liu W, Yi L, Li H, Wang Z, Lin JM. Biosens Bioelectron. 2015;63:105–111. doi: 10.1016/j.bios.2014.07.013. [DOI] [PubMed] [Google Scholar]
- 12.Eulberg D, Klussmann S. Chembiochem. 2003;4:979–983. doi: 10.1002/cbic.200300663. [DOI] [PubMed] [Google Scholar]
- 13.Vater A, Jarosch F, Buchner K, Klussmann S. Nucleic Acids Res. 2003;31:e130. doi: 10.1093/nar/gng130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brumbt A, Ravelet C, Grosset C, Ravel A, Villet A, Peyrin E. Anal Chem. 2005;77:1993–1998. doi: 10.1021/ac048344l. [DOI] [PubMed] [Google Scholar]
- 15.Vater A, Klussmann S. Drug Discov Today. 2015;20:147–155. doi: 10.1016/j.drudis.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 16.Ruta J, Ravelet C, Grosset C, Fize J, Ravel A, Villet A, Peyrin E. Anal Chem. 2006;78:3032–3039. doi: 10.1021/ac060033i. [DOI] [PubMed] [Google Scholar]
- 17.Ruta J, Grosset C, Ravelet C, Fize J, Villet A, Ravel A, Peyrin E. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;845:186–190. doi: 10.1016/j.jchromb.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 18.Mascini M, Papamichael K, Mevola I, Pravda M, Guilbault GG. Anal Lett. 2007;40:403–430. [Google Scholar]
- 19.Mascini M, Guilbault GG, Lebrun SJ, Compagnone D. Anal Lett. 2007;40:1386–1399. [Google Scholar]
- 20.Vater A, Sell S, Kaczmarek P, Maasch C, Buchner K, Pruszynska-Oszmalek E, Kolodziejski P, Purschke WG, Nowak KW, Strowski MZ, Klussmann S. J Biol Chem. 2013;288:21136–21147. doi: 10.1074/jbc.M112.444414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bilik KU, Ergüven E, Klussmann S, Jarosch F, Wielinga PY, Lutz TA, Riediger T. Neuroreport. 2007;18:1855–1859. doi: 10.1097/WNR.0b013e3282f1ab04. [DOI] [PubMed] [Google Scholar]
- 22.Guillo C, Roper MG. Electrophoresis. 2008;29:410–416. doi: 10.1002/elps.200700399. [DOI] [PubMed] [Google Scholar]
- 23.Guillo C, Truong TM, Roper MG. J Chromatogr A. 2011;1218:4059–4064. doi: 10.1016/j.chroma.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yi L, Wang X, Dhumpa R, Schrell AM, Mukhitov N, Roper MG. Lab Chip. 2015;15:823–832. doi: 10.1039/c4lc01360c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jameson EE, Cunliffe JM, Neubig RR, Sunahara RK, Kennedy RT. Anal Chem. 2003;75:4297–4304. doi: 10.1021/ac0342976. [DOI] [PubMed] [Google Scholar]
- 26.Lomasney AR, Yi L, Roper MG. Anal Chem. 2013;85:7919–7925. doi: 10.1021/ac401625g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moser AC, Hage DS. Electrophoresis. 2008;29:3279–3295. doi: 10.1002/elps.200700871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Viglasky V, Hianik T. Gen Physiol Biophys. 2013;32:149–172. doi: 10.4149/gpb_2013019. [DOI] [PubMed] [Google Scholar]
- 29.Pyle AM. J Biol Inorg Chem. 2002;7:679–690. doi: 10.1007/s00775-002-0387-6. [DOI] [PubMed] [Google Scholar]
- 30.Turel I, Kljun J. Curr Top Med Chem. 2011;11:2661–2687. doi: 10.2174/156802611798040787. [DOI] [PubMed] [Google Scholar]
- 31.Datta S, Conlisk AT, Li HF, Yoda M. Mech Res Commun. 2009;36:65–74. doi: 10.1016/j.mechrescom.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bak MJ, Albrechtsen NW, Pedersen J, Hartmann B, Christensen M, Vilsbøll T, Knop FK, Deacon CF, Dragsted LO, Holst JJ. Eur J Endocrinol. 2014;170:529–538. doi: 10.1530/EJE-13-0941. [DOI] [PubMed] [Google Scholar]
- 33.Wimalawansa SJ. Crit Rev Neurobiol. 1997;11:167–239. doi: 10.1615/critrevneurobiol.v11.i2-3.40. [DOI] [PubMed] [Google Scholar]
- 34.Rossowski WJ, Jiang NY, Coy DH. Eur J Pharmacol. 1997;336:51–63. doi: 10.1016/s0014-2999(97)01252-1. [DOI] [PubMed] [Google Scholar]
- 35.Cornish J, Naot D. Curr Pharm Des. 2002;8:2009–2021. doi: 10.2174/1381612023393341. [DOI] [PubMed] [Google Scholar]
- 36.Ahrén B, Sundler F. Cell Tissue Res. 1992;269:315–322. doi: 10.1007/BF00319623. [DOI] [PubMed] [Google Scholar]
- 37.Hoehlig K, Johnson KW, Pryazhnikov E, Maasch C, Clemens-Smith A, Purschke WG, Vauléon S, Buchner K, Jarosch F, Khiroug L, Vater A, Klussmann S. Br J Pharmacol. 2015;172:3086–3098. doi: 10.1111/bph.13110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karlsson E, Stridsberg M, Sandler S. Biochem Pharmacol. 1998;56:1339–1346. doi: 10.1016/s0006-2952(98)00194-4. [DOI] [PubMed] [Google Scholar]
- 39.Karlsson E, Stridsberg M, Sandler S. Regul Pept. 1996;63:39–45. doi: 10.1016/0167-0115(96)00025-0. [DOI] [PubMed] [Google Scholar]
- 40.Hellman B, Salehi A, Grapengiesser E, Gylfe E. Biochem Biophys Res Commun. 2012;417:1219–1223. doi: 10.1016/j.bbrc.2011.12.113. [DOI] [PubMed] [Google Scholar]