

Abstract
Background
CD19 is a B-cell specific molecule that serves as a major co-stimulatory molecule for amplifying B cell receptor (BCR) responses. Bi-allelic CD19 gene mutations cause common variable immunodeficiency (CVID) in humans. BCR and TLR9 induced B-cell responses are impaired in most CVID patients.
Objective
We sought to analyze whether CD19 is required for TLR9 function in human B cells.
Methods
The expression of surface activation markers was assessed after anti-IgM or CpG stimulation using flow cytometry on B cells from patients with one or two defective CD19 alleles, which decrease or abrogate CD19 expression, respectively. The phosphorylation or interaction of signaling molecules was analyzed using phosphoflow cytometry, immunoblot or co-immunoprecipitation in CD19-deficient or control B cells and in a B cell line in which CD19 has been knocked-down using lentiviral transduced shRNA.
Results
B cells from individuals with one or two defective CD19 alleles showed defective upregulation in vitro of CD86, TACI and CD23 activation markers after TLR9 stimulation. TLR9 ligands normally induce via MYD88/PYK2/LYN complexes the phosphorylation of CD19, which allows the recruitment of PI3K and the phosphorylation of BTK and AKT in human B cells with a different kinetic than that of BCRs. In addition, inhibition of PI3K, AKT or BTK as well as BTK-deficiency also result in TLR9 activation defects in B cells similar to those in CD19 deficiency. Conclusion: CD19 is required for TLR9-induced B-cell activation. Hence, CD19/PI3K/AKT/BTK is an essential axis integrating BCRs and TLR9 signaling in human B cells.
Keywords: B cells, TLR9, CD19, PI3K, BTK, AKT, CVID
INTRODUCTION
CD19 is a B-cell specific transmembrane protein expressed from the pro-B cell stage until plasma cell differentiation in both humans and mice and functions as a B cell receptor (BCR) co-receptor. The nine tyrosines in its cytoplasmic domain represent a scaffold protein that amplifies proximal BCR signaling. When phosphorylated, the tyrosines function as Src homology 2 recognition motifs that recruit regulatory molecules (1, 2). Hence, phosphorylated CD19 binds and amplifies the function of the SRC-family kinase (SFK) LYN as well as other tyrosine kinases and recruits PI3K, thereby promoting BTK and AKT phosphorylation in B cells (3–6).
CD19 plays an essential role in regulating B-cell activation thresholds and thereby influences B-cell selection and differentiation. Altering CD19 surface expression in knockout or transgenic mice significantly changes B-cell development and function (7, 8). CD19 overexpression results in B cells that are hyper-responsive to BCR triggering, leading to a lupus-like autoimmune disease with the production of anti-nuclear antibodies (ANAs) in the serum of transgenic mice (7). The complete abrogation of CD19 expression causes defective late B-cell differentiation and decreased antibody responses in mice as well as the development of common variable immunodeficiency (CVID) in humans (7–9). Paradoxically, mutations in CD19 not only cause CVID in humans but also induced the development of autoimmune manifestations resembling systemic lupus erythematosus (SLE) (10, 11). Autoimmunity often develop in CVID patients in which BCR and TLR9 induced B-cell responses are impaired (12–14). In addition, defective BCRs and TLRs function in B cells have been associated with altered late B-cell differentiation, decreased antibody production and abnormal tolerance induction (13–16).
Human B cells mainly express the endosomal TLR7 and TLR9 that are involved in sensing RNA and DNA, respectively (17). Upon ligation with their specific ligand TLR7 and TLR9 signal through MyD88/IRAK1/4 complexes and activate the NF-κB and MAPK pathways where BCR-and TLR-signaling pathways intersect in B cells (18). TLR9 activation in human B cells induces B-cell proliferation, Ig secretion and differentiation into plasmablasts (19). By analyzing patients with primary immunodeficiencies, it has been suggested that TLR signaling pathways may play an important role for B-cell tolerance induction in humans (13, 16, 20). In addition, recent reports indicate that nucleic acid sensing by endosomal TLRs may provide negative regulation to autoreactive B cells (21). For instance, TLR9 deficiency exacerbates clinical symptoms in mouse SLE models, suggesting that defective TLR9 function in CVID may favor autoimmunity (22). However, the etiology of the abnormal TLR responses in B cells from CVID patients remains vastly unknown. Since CD19 deficiency is known to affect BCR responses altered in CVID and that CD19 mutation favors the development of autoimmunity, we assessed TLR9 function in B cells from subjects carrying one or two mutated CD19 alleles. We found that CD19 plays an essential role in the regulation of TLR9 responses in human B cells by recruiting PI3K and mediating AKT and BTK phosphorylation after ligation of nucleic acids.
METHODS
Patients and healthy donor controls
All CD19-deficient patients and CD19-heterozygous carriers have been described (9, 23). The specific mutations in BTK have been previously reported in the following family members: XLA 022 (24) and XLA 596, XLA 796 (25). All samples were collected in accordance with the institutional review board-reviewed protocols from respective hospitals.
Cell preparation and purification
Peripheral blood mononuclear cells (PBMCs) were purified from the blood of patients and control donors by density gradient centrifugation. B cells were purified using CD20-microbeads (Miltenyi Biotec, Bergisch-Gladbach, Germany), the EasySep™ Human B cell Enrichement Kit or the EasySep™ Human Naive B cell Enrichement Kit (StemCell Technologies). Plasmacytoid dendritic cells were purified using the Plasmacytoid Dendritic Cell Isolation Kit (Miltenyi Biotec).
B-cell activation, proliferation and inhibition experiments
Total CD20+ B cells or naïve B cells were plated at 150,000 cells/well in a 96-well-plate in RPMI 10% FBS and 2.5 μg/mL polyclonal F(ab′)2 anti-human IgM (Jackson Immunoresearch), 0.5 μg/mL CpG ODN2006 (TLR9 agonist, Invivogen), 2.0 μg/mL Gardiquimod or 1.0 μg/mL Loxoribine (TLR7 agonists, Invivogen). Expression of surface activation markers was analyzed after 48 hours using flow cytometry. For inhibition experiments B cell cultures were preincubated with the specific inhibitors for 45 minutes. Intracellular phosphospecific flow cytometric analysis was performed as described before (26). Cells were acquired with a FACSCalibur or LSR II (BD Biosciences) and analyzed with FlowJo software. Information on the antibodies and inhibitors used is provided in the supplemental Methods section.
Co-IP and IB
B cells were stimulated as described above and subsequently lysed in lysis buffer (50mM Tris, 1% NP-40, 2mM EDTA) including phosphatase inhibitor cocktail 2 (Sigma) and protease inhibitor (Roche). For co-immunopreciptation cells were lysed in lysis buffer (50mM Tris, 150mM NaCl, 1mM EDTA, 0.5% Triton) including protease and phosphatase inhibitors, pre-cleared with protein A/G beads, incubated with either control- or specific antibody and precipitated with protein A/G beads (Thermo Scientific). Total cell lysates or immunoprecipitates were separated by SDS page, transferred to PDVF membranes, probed with specific antibodies and a secondary antibody coupled to HRP (Cell Signaling), and detected by chemiluminescence (Amersham ECL Prime Western Blotting detection Reagent) using a GBox documentation system (Syngene). Information on the antibodies used is provided in the supplemental Methods section.
CD19 gene knockdown and Ramos B cell transduction
Ramos B cells were transduced with lentiviral constructs containing an shRNA targeting human CD19 cDNA sequence as reported before (27, 28). GFP+CD19− Ramos B cells were sorted by flow cytometry with a FACS Aria cell sorter (BD Biosciences) and retained a stable phenotype in culture.
Statistical analysis
Differences were analyzed for statistical significance with unpaired Student’s t-tests or in case of paired samples with paired Student’s t-tests, using Prism software (GraphPad). A p-value of less than 0.05 was considered significant.
RESULTS
CD19 mutations interfere with TLR9-induced B-cell activation
To investigate a role for CD19 in TLR9-induced B-cell responses, we assessed if decreased or abrogated CD19 expression affected TLR9 function in purified B cells from individuals carrying mutations in one or two CD19 alleles from two unrelated families. B cells carrying a single functional CD19 allele displayed about half the amount of CD19 expression on their cell surface compared to B cells from siblings with 2 functional alleles, whereas CD19 failed to be expressed on B cells carrying bi-allelic CD19 mutations as previously reported (9) (Fig 1, A). In agreement with CD19 being a major positive BCR co-receptor, decreased CD19 expression affected BCR-induced B-cell activation as illustrated by the defective induction of CD86 expression after BCR-triggering, which was almost absent on CD19-deficient B cells (Fig 1,B and C). The induction of CD86 after TLR9 stimulation by its agonist CpG showed a similar CD19 gene dosage dependence (Fig 1, B and C). Defective induction of TACI and CD23 was also observed on CD19-deficient B cells after CpG stimulation (Fig 1, B and C). These defects were not the result of a global failure of CD19-deficient B cells to be activated by TLR9 since they normally upregulated CD25 and CD69 expression after CpG stimulation (Fig 1, B and C). The normal expression of TLR9 in CD19-deficient EBV-immortalized B-cell lines (EBV-BCL) suggests that abnormal TLR9 responses in CD19-mutated B cells were not likely due to an altered expression of this receptor (Fig E1, A). Impaired BCR responses in CD19-deficient B cells were also not the result of decreased cell surface BCR/IgM expression, which was similar to their counterpart expressing CD19 (Fig E1, B). In addition, plasmacytoid dendritic cells (pDCs), which do not express CD19, displayed normal CD80 and CD86 induction in CD19-deficient patients after TLR9 triggering, revealing that TLR9 activation defects were restricted to B cells carrying mutated CD19 allele(s) (Fig E1, C). Decreased CD19 expression also affected TLR7-induced B-cell activation as illustrated by the defective induction of CD86 and TACI expression after Loxoribine stimulation (Fig E2). We therefore conclude that CD19 appears to play an important role in mediating BCR- but also TLR7- and TLR9-induced responses in human B cells.
FIG 1.
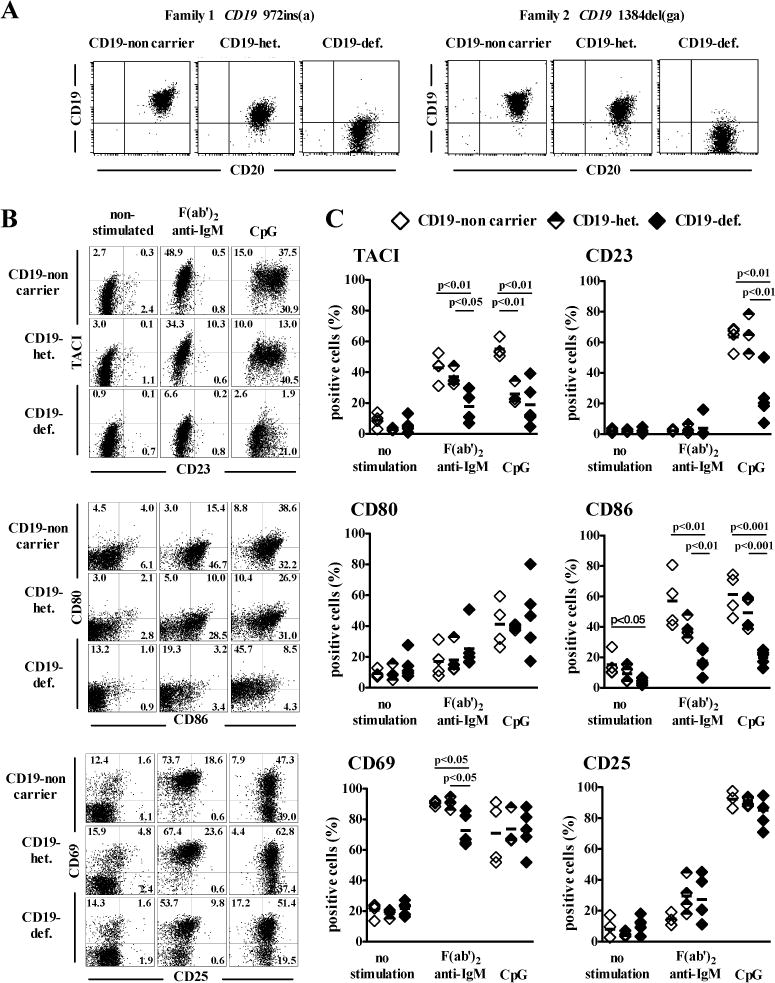
Defective BCR- and TLR9-induced B-cell activation in CD19-deficient patients. A, Decreased CD19 cell surface expression in subjects carrying CD19 mutated allele(s). The cell surface expression of CD19 and CD20 was analyzed on CD20+ purified B cells from healthy family members (CD19-non carrier), CD19-heterozygous carriers (CD19-het.) and CD19-deficient patients (CD19-def.) of two unrelated families using flow cytometry. B, Altered TLR9 responses in B cells carrying CD19 mutated allele(s). Surface expression of TACI, CD23, CD80, CD86, CD69 and CD25 on CD20+CD27− naive B cells of a healthy family member (CD19-non carrier), a CD19-heterozygous carrier (CD19-het.) and a CD19-deficient patient (CD19-def.) after no stimulation or in vitro stimulation with F(ab′)2 anti-IgM or the TLR9 ligand CpG for two days. The frequency for the various B-cell surface markers after activation for two days as analyzed above is represented in C. Each symbol represents an individual, horizontal bars display the mean.
TLR9 stimulation induces CD19 phosphorylation
BCR-triggering induces a rapid phosphorylation of specific tyrosine residues of CD19 followed by recruitment of signaling transduction molecules (29). To analyze whether TLR9 triggering also activates the CD19 pathway, we first tested the phosphorylation of CD19 by immunoblot (IB) analysis using lysates from primary human B cells after BCR-triggering or TLR9 stimulation with CpG. The activation of purified human B cells isolated from the peripheral blood of healthy controls by anti-BCR or CpG induced CD19 phosphorylation at residue Y531, a tyrosine that is deleted in the truncated mutant product of both CD19-deficient families (Fig 2, A and B). However, the kinetics of phosphorylation induced by BCR or TLR9 triggering were different in that the former resulted in early phosphorylation events occurring between 2–10 minutes whereas CpG induced CD19 phosphorylation peaking at much later 20–40 minute time points (Fig 2, A and B). Activation of B cells by the TLR7 ligand Gardiquimod also induced phosphorylation of CD19 with similar kinetics as CpG stimulation (Fig E3).
FIG 2.
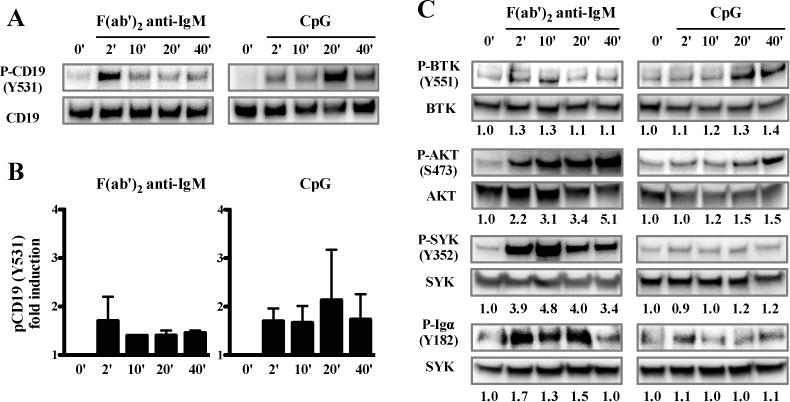
TLR9 stimulation induces phosphorylation of CD19, BTK and AKT in human B cells. A, Phosphorylation of CD19 was assessed by immunoblot in lysates from purified healthy control peripheral blood B cells stimulated or not for various time points with F(ab′)2 anti-IgM or TLR9 ligand CpG. B, Fold induction of CD19 phosphorylation after F(ab′)2 anti-IgM or TLR9 stimulation (mean±SEM; n=3). Phosphorylation of BTK, AKT, SYK and Igα was assessed in C. The numbers below each immunoblot indicate the fold induction of phosphorylation compared to the unstimulated sample. Data are representative of two independent experiments with similar results.
To decipher CD19-dependent TLR7 and TLR9 pathways in human B cells, we analyzed the phosphorylation of signaling molecules mediating CD19 function. BCR- or TLR9-triggering induced BTK phosphorylation at residue Y551 and AKT phosphorylation at residue S473, with phosphorylation peaking later for TLR9 stimulation compared to BCR triggering (Fig 2, C). A similar trend was observed when stimulating the B cells with the TLR7-ligand Gardiquimod (Fig E3). However, not all kinases involved in BCR signaling seemed to be phosphorylated in B cells after TLR7 or TLR9 stimulation. Indeed, BCR triggering strongly induced SYK phosphorylation at residue Y352, whereas CpG or Gardiquimod stimulation only had a marginal effect on the phosphorylation of SYK (Fig 2, C and Fig E3). In addition, CpG stimulation did not induce phosphorylation of the immunoglobulin-associated protein alpha (Igα, CD79A), which is required for initiation of the signal transduction cascade activated by BCRs (Fig 2, C). Hence, in agreement with CD19 mediating some TLR7 and TLR9 functions in B cells, stimulation of these TLRs induces CD19 phosphorylation as well as the phosphorylation of a group of signaling molecules known to mediate CD19 function after BCR triggering.
TLR9-induced AKT and BTK phosphorylation is dependent on CD19 and PI3K function
CD19 is one of the main regulators of PI3K activity, which converts phosphatidylinositol-(3,4)-biphosphate (PIP2) to phosphatidylinositol-(3,4,5)-triphosphate (PIP3) that bind molecules containing pleckstrin homology domains, such as AKT and BTK (30). We therefore tested if TLR9 induced AKT and BTK phosphorylation depended on CD19 and/or PI3K function by analyzing the phosphorylation by flow cytometry of AKT and BTK in CD20+CD21+CD27− naïve B cells from CD19-deficient patients and healthy controls after BCR-triggering or CpG stimulation. Since TLR7 triggering induced weaker but qualitatively similar phosphorylation signals than TLR9 (Fig 2 and Fig E3), we decided to focus on TLR9. Phosphorylation of AKT at S473 was still induced after BCR triggering in CD19-deficient B cells compared with control B cells but was found reduced after CpG stimulation (Fig 3, A and B). In addition, CD19-deficient B cells showed reduced phosphorylation of BTK at Y551 after both, BCR-triggering or CpG stimulation (Fig 3, A and B). To confirm the role of CD19 in mediating TLR9 induced BTK and AKT phosphorylation, we generated a Ramos B-cell line in which CD19 expression has been knocked down using a specific shRNA (Fig E4) and assessed AKT and BTK phosphorylation after BCR or TLR9 triggering using IB analysis (Fig 3, C and D). In agreement with our observations in freshly isolated B cells, AKT phosphorylation at S473 was strongly reduced in the absence of CD19 after CpG stimulation whereas CD19 knock-down B cells only showed slightly decreased AKT phosphorylation after BCR-triggering (Fig 3, C and D). BTK phosphorylation at Y551 was also diminished and virtually abrogated in the absence of CD19 expression after BCR or CpG stimulation, respectively (Fig 3, C and D). To further characterize the involvement of CD19 on the TLR9 signaling pathway, we analyzed the activation of MAPK and NF-κB in the CD19 knock down B cells after CpG stimulation. In contrast to AKT and BTK, the phosphorylation of p38 MAPK and JNK as well as the phosphorylation/degradation of Iκ-Bα were not altered in the absence of CD19 after CpG stimulation (Fig 3, E and F).
FIG 3.
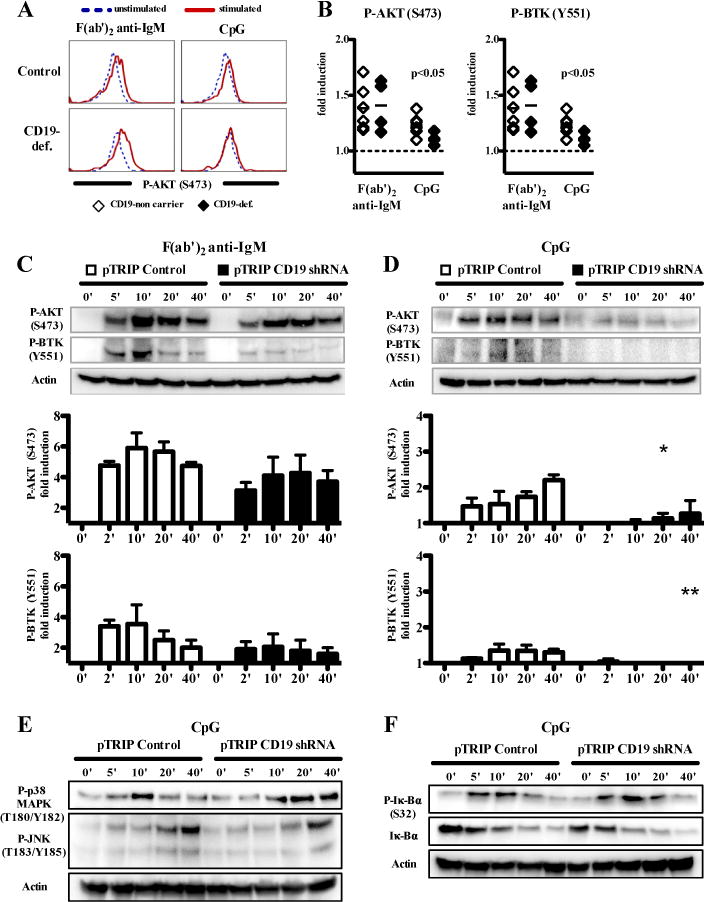
TLR9 induced AKT and BTK signaling in human B cells is defective in the absence of CD19. A and B, The phosphorylation of AKT and BTK after F(ab′)2 anti-IgM or CpG stimulation for 15 minutes was analyzed by flow cytometry in CD20+CD21+CD27- naïve B cells from CD19-deficient patients and healthy controls. Overlays of a representative experiment are shown in A. The fold induction of AKT and BTK phosphorylation in naïve B cells of CD19-def. patients (n=4) and healthy controls (n=6) after F(ab′)2 anti-IgM or CpG stimulation compared to the unstimulated sample is shown in B. C and D, Phosphorylation of AKT and BTK was analyzed in lysates of a Ramos B-cell line transduced with lentiviral constructs containing no shRNA (pTRIP control) or a shRNA targeting CD19 (pTRIP CD19 shRNA) and stimulated for various time points with F(ab′)2 anti-IgM or CpG. Each bar shows the fold induction of phosphorylation at a given time point (mean±SEM; n=3; *p<0.05, *p<0.01, compared with pTRIP control). E and F, Phosphorylation of p38 and JNK or Iκ-Bα was assessed as described in A–D. Data are representative of two independent experiments with similar results.
We then assessed if PI3K, which mediates most CD19 function, interacts with CD19 after TLR9 stimulation by immunoprecipitating CD19 from non-stimulated or CpG-stimulated human tonsil B cells and probing for p85 PI3K. We found that CpG stimulation induced interaction of p85 PI3K with CD19 (Fig 4, A). Interfering with PI3K function the LY294002 inhibitor strongly reduced or even blocked the phosphorylation of AKT and its downstream transcription factor FoxO1 as well as BTK in purified peripheral blood B cells of healthy controls after CpG stimulation (Fig 4, B). Similarly to CD19 knock-down in B cells, inhibition of PI3K function had only little or no effects on the phosphorylation of p38 MAPK and JNK or the degradation of Iκ-Bα, respectively (Fig 4, C). In summary, CD19 and PI3K are required for the induction of AKT and BTK phosphorylation after TLR9 stimulation, whereas these molecules seem more dispensable for TLR9-induced MAPK and NF-κB activation.
FIG 4.
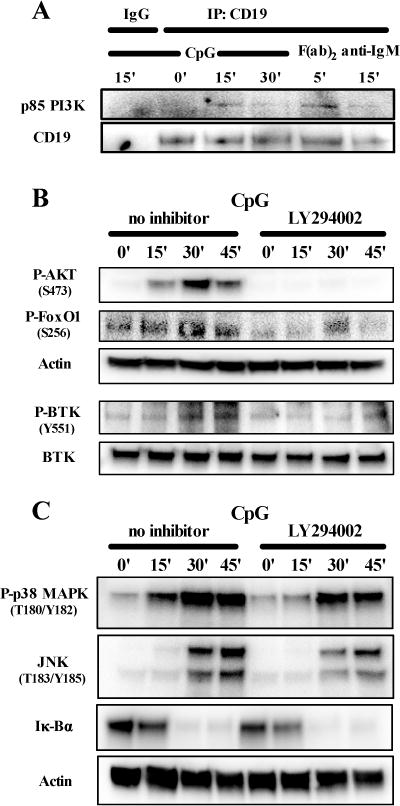
PI3K mediates CD19-dependent AKT and BTK phosphorylation after TLR9 stimulation in human B cells. A, Lysates of a healthy control CD20+ tonsil B cells stimulated with CpG or F(ab)2 anti-IgM for various time points were immunprecipitated with anti-CD19 or an isotype control antibody (IgG) followed by immunblot analysis of p85 PI3K. B, C, and D, The phosphorylation of AKT, FoxO1 and BTK, p38 and JNK or the degradation of Iκ-Bα was analyzed in lysates of purified peripheral blood B cells from a healthy control stimulated for various time points with CpG with or without addition of a PI3K inhibitor (LY294002). Data are representative of two independent experiments with similar results.
TLR9-induced CD19 phosphorylation and interaction with p85 PI3K is dependent on PYK2 and SRC family kinases
CD19 becomes phosphorylated by the Src family kinase (SFK) LYN after BCR-triggering, allowing the recruitment of PI3K (31). It has been suggested that the tyrosine kinase PYK2 interacts with MyD88 and might induce LYN phosphorylation after TLR9-stimulation (32). In addition, BTK has been shown to interact with MyD88 in myeloid cells and malignant B cells and mediate proximal TLR signaling (33, 34). We therefore tested if LYN, PYK2 or BTK were mediating CD19 phosphorylation after TLR9 stimulation in primary human B cells. Both PP2 (SFK inhibitor) and AG-17 (PYK2-inhibitor) blocked CpG-induced CD19 phosphorylation at residue Y531, whereas PCI32765 (BTK inhibitor) had no significant effect on the phosphorylation of CD19 after CpG stimulation (Fig 5, A).
FIG 5.
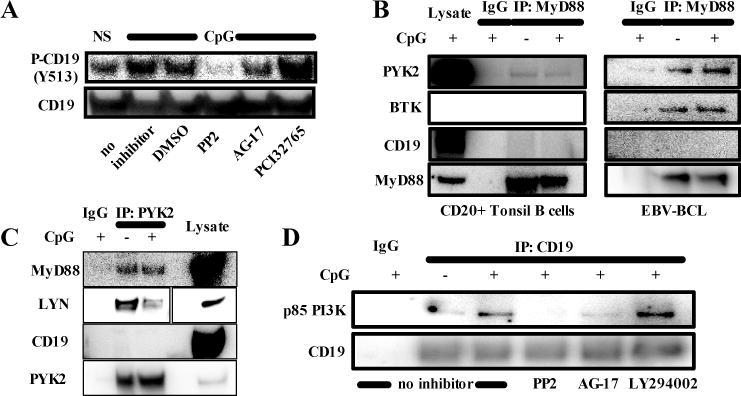
A MyD88/PYK2/LYN complex mediates TLR9 induced p85 PI3K recruitment to CD19. A, The phosphorylation of CD19 was analyzed in lysates of purified peripheral blood B cells from a healthy control stimulated with CpG with or without addition of a src family kinase inhibitor (PP2), a PYK2 inhibitor (AG-17) or a BTK inhibitor (PCI32765). Lysates of healthy control CD20+ tonsil B cells (B) or EBV-transformed B-cell lines (C) stimulated or not with CpG for 30 minutes were immunprecipitated with anti-MyD88 (B), anti-PYK2 (B) or an isotype control antibody (IgG) followed by immunoblot analysis. D, EBV-transformed B-cells from a healthy control were stimulated or not for 30 min with CpG without inhibitor or in the presence of a src family kinase inhibitor (PP2) or a PYK2 inhibitor (AG-17). Lysates were immunoprecipitated with anti-CD19 or an isotype control antibody (IgG) followed by immunoblotting against p85 PI3K.
To determine if these signaling molecules might interact with each other, we immunoprecipitated MyD88 from non stimulated or CpG-stimulated human primary tonsil B cells or EBV-BCL derived from healthy donors and probed for CD19, BTK and PYK2. We found that MyD88 interacted with BTK and PYK2 but not with CD19 (Fig 5, B). This interaction was already present in unstimulated B cells and was not modified by CpG stimulation (Fig 5, B). Of note, we could not find an interaction between TLR9 and CD19 (data not shown). We also precipitated PYK2 from EBV-BCL and probed against LYN. Interestingly, LYN seemed to be released from PYK2/MYD88 complexes upon TLR9 activation (Fig 5, C). Moreover, CpG stimulation induced the recruitment of PI3K p85 to CD19 and this association was dependent on PYK2 and SFK function, demonstrating that PYK2 and SFKs are involved in the phosphorylation of tyrosines from CD19, which are critical for its binding to PI3K p85 (Fig 5, D). Thus, PYK2 and SFK including LYN are responsible for the phosphorylation of CD19 and the recruitment of PI3K following TLR9 stimulation.
Inhibition of PI3K, AKT and BTK mimic B-cell activation defects induced by CD19-deficiency
We assessed if the inhibition of PI3K, BTK or AKT, which are molecules involved in the CD19 signaling pathway induced TLR9-activation defects resembling those resulting from the absence of functional CD19. PI3K, BTK or AKT blockade using chemical inhibitors mimicked CD19-deficiency in that it decreased BCR- and TLR9-induced CD86, TACI and CD23 induction in human B cells (Fig 6, A and B). However, similar to CD19-deficient B cells, CD69 induction on B cells after TLR9 stimulation remained intact after PI3K, BTK or AKT inhibition, whereas inhibition of PI3K and BTK did block CD69 induction after BCR triggering (Fig 6, A and B). Similar observations were obtained when stimulating B cells with the TLR7-ligand Gardiquimod and PI3K, BTK or AKT blockade (Fig E5). In addition, these findings could be reproduced with a second set of chemically unrelated inhibitors (LY294002 PI3K-inhibitor, LMF-A13 BTK-inhibitor and MK-2206 AKT-inhibitor; data not shown). Since the specificity of kinase inhibitors may always be argued, we analyzed B-cell responses in X-linked agammaglobulinemia (XLA) patients who display non-functional BTK molecules (25). We found that activation of CD20+CD27− naïve B cells from XLA patients after BCR triggering was completely abrogated as illustrated by the lack of CD86 and CD69 induction, further demonstrating BTK requirement for BCR functions (Fig 7, A). In contrast, BTK-deficient CD20+CD27− naïve B cells stimulated by CpG were similar to CD19-deficient B cells in that they normally upregulated CD69 but failed to induce CD86 expression (Fig 7, A and B). The majority of circulating CD20+CD27− naïve B cells in XLA patients displays a phenotype similar to new emigrant/transitional B cells with high expression of IgM and CD38 whereas mature naïve B cells predominate in healthy controls (35). To rule out that defective TLR9- activation of BTK-deficient B cells might be explained by B-cell developmental differences and a predominance of new emigrant/transitional B cells in these patients we compared B-cell activation after CpG stimulation between sorted new emigrant/transitional and mature naïve B cells of healthy controls. We could not find significant differences in the extent of B-cell activation between both B-cell subsets (Fig 7, C). Moreover, the XLA patient displayed in Figure 7A was unique in that he only displayed a milder reduction in peripheral CD19+CD10−CD21+IgM+CD27− mature naïve B cells compared to other XLA patients. These BTK-deficient mature naive B cells failed to upregulate CD86 after TLR9 stimulation but normally upregulated CD69 (Fig 7, A). Hence, abnormal PI3K, AKT or BTK function reproduced the defects observed in CD19-deficient B cells after TLR9 stimulation, further revealing that these 3 molecules mediate CD19-dependent TLR9 function in human B cells.
FIG 6.
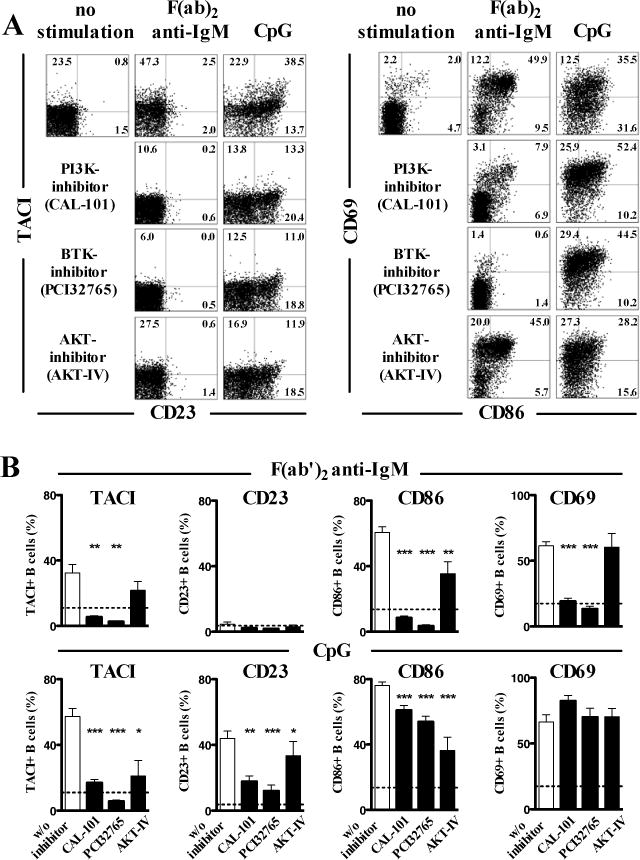
Inhibition of PI3K, AKT or BTK mimics TLR9 induced B-cell activation defects observed in CD19-deficient B cells. Surface expression of TACI, CD23, CD86 and CD69 on purified CD19+CD27− naive B cells of healthy individuals after in vitro stimulation with F(ab′)2 anti-IgM or TLR9 ligand CpG for two days with or without addition of a PI3K-inhibitor (CAL-101), a BTK-inhibitor (PCI32765) or an AKT inhibitor (AKT-IV inhibitor) was analyzed by flow cytometry. Dot blots of a representative experiments are shown in A and the data of five independent experiments is summarized in B. Each bar represents the mean ± SEM frequency, horizontal dashed lines represent the mean of the unstimulated samples. (* p<0.05; ** p<0.01; *** p<0.001).
FIG 7.
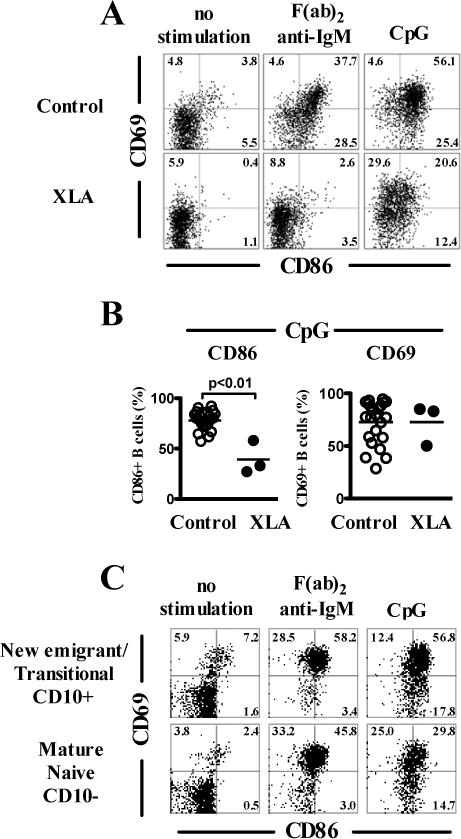
Defective TLR9 induced B-cell activation in BTK-deficient XLA patients. A, Surface expression of B-cell activation markers on CD19+CD27− naïve B cells of a XLA-patient harboring a BTK mutation after no stimulation or in vitro stimulation with F(ab′)2 anti-IgM or TLR9 ligand CpG for two days. B, Quantification of CD86 and CD69 expression on CD19+CD27− naïve B cells of healthy controls (n=23) or XLA-patients (n=3) after in vitro stimulation with the TLR9 ligand CpG for two days. Each bar represents the mean ± SEM frequency.
C, Surface expression of the B-cell activation markers CD69 and CD86 on purified CD19+CD10+CD27− new emigrant/transitional or CD19+CD10−CD27− mature naïve B cells from a healthy control after in vitro stimulation as described above.
DISCUSSION
The recognition of nucleic acid containing antigens by human B cells through TLR9 induces robust activation, proliferation and Ig production (19). Impaired or altered TLR9 responses have been associated with the development of antibody deficiencies (e.g. CVID) and autoantibody production (e.g. in SLE) (13, 14, 36). We reported herein that CD19 plays an essential role in mediating TLR9 functions in human B cells. Indeed, CD19-deficient human B cells displayed impaired activation responses after TLR9 stimulation whereas pDCs from the same patients that do not express CD19 showed normal TLR9-induced activation, excluding a general and unspecific defect of TLR9-signaling in these patients.
TLR9 engagement in B cells is followed by phosphorylation of IRAK1 and 4, recruitment of these kinases to MyD88, and interaction with TRAF6, which then triggers downstream signaling resulting in the activation of NF-κB and MAPKs (18) (Fig 8). However, phosphorylation of Iκ-Bα and p38 MAPK after CpG stimulation was only slightly affected in B cells in which CD19 expression has been silenced by shRNA, suggesting that CD19 does not appear to play an essential role in mediating TLR9-induced activation of NF-κB and MAPK pathways in human B cells. In contrast, the phosphorylation of BTK and AKT after CpG stimulation was decreased in the absence of CD19, suggesting that CD19 mainly controls BTK- and AKT-dependent TLR9-signaling pathways in human B cells. The specific requirement of CD19 in TLR9-induced BTK and AKT phosphorylation was demonstrated in B cells from CD19-deficient patients that displayed impaired BTK and AKT phosphorylation after TLR9 stimulation. In addition, B-cell stimulation by CpG induced recruitment of p85 PI3K to CD19 and inhibition of PI3K function severely diminished CpG induced BTK and AKT phosphorylation. The inhibition of PI3K, BTK or AKT function using small molecular inhibitors resulted in TLR9-induced B-cell activation defects similar to those observed in CD19-deficient B cells, further demonstrating the important role of CD19/PI3K/BTK/AKT axis in mediating TLR9 functions in B cells. These conclusions were further supported by the activation defects induced by the lack of functional BTK in B cells from XLA patients following TLR9 stimulation and which reproduced those of CD19 deficiency or induced by BTK inhibitor. Moreover, the analysis of TLR7 responses in CD19-deficient B cells or when blocking PI3K, BTK or AKT function revealed activation defects similar to those after TLR9 stimulation. Hence, CD19 is essential for complete B-cell activation after TLR7 or TLR9 stimulation in human B cells and depends on a PI3K-mediated BTK and AKT signaling pathway (Fig 8).
FIG 8.
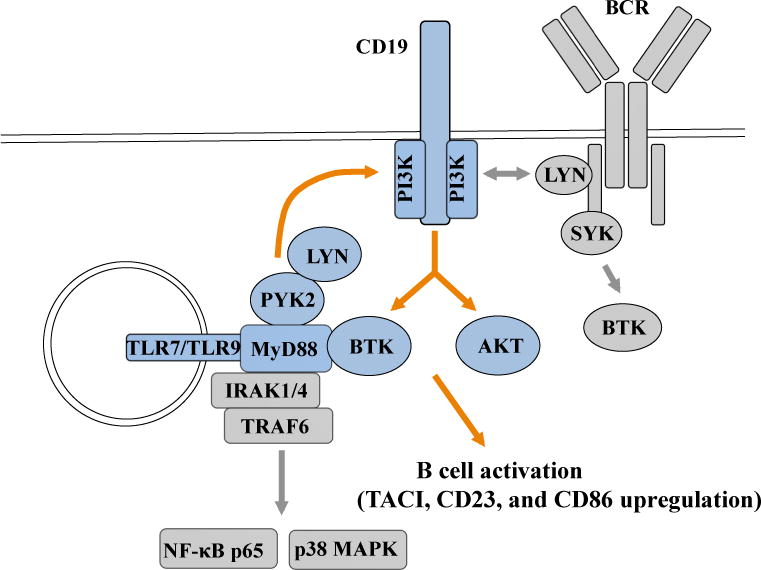
Scheme of CD19-dependent TLR7 and TLR9 signaling pathways in B cells. TLR7 or TLR9 stimulation induces the PYK2/LYN dependent phosphorylation of CD19. CD19 phosphorylation allows the recruitment of PI3K and mediates AKT and BTK phosphorylation required for proper B cell activation and proliferation. Early BCR signaling pathways involving CD19 are also represented.
In contrast to TLR9 activation, the absence of CD19 had only small effects on BTK and AKT phosphorylation after BCR-triggering. This is in agreement with previous studies in CD19-deficient mouse models or cell lines, suggesting that CD19 is not required for AKT phosphorylation after BCR-triggering and is involved in prolongation or amplification of BTK-mediated signaling rather than serving as a mandatory molecule for BTK activation after BCR-triggering (6). Despite some controversial data regarding the involvement of CD19 in mediating BTK and AKT activation after BCR-triggering (37, 38), it is generally believed that PI3K function is essential for activation of AKT-dependent signaling pathways after BCR-triggering (39). Interestingly, other adaptor molecules such as BCAP and Nck have been shown to be involved in controlling the PI3K-AKT pathway after BCR-triggered B-cell activation and might serve as adaptor molecule in addition to CD19 (31, 40). Although phosphorylation of AKT and BTK after TLR9-stimulation was significantly decreased or almost abolished in the absence of CD19, we cannot rule out that - similar to the BCR-pathway – other adaptor molecules than CD19 might also be involved in mediating the TLR9-PI3K-AKT pathway in human B cells. However, it is tempting to speculate that the BCR and TLR9 signaling might intersect at the level of CD19 and branch into a PI3K mediated signaling pathway involving BTK and AKT (Fig 8).
PI3K blockade in B cells impaired activation following TLR9-stimulation, suggesting that CD19 and PI3K are key molecules for TLR9 signaling and amplify its responses in human B cells. In contrast, PI3K activation downstream of TLR ligation mediated by the signaling adaptor BCAP in myeloid cells inhibits macrophage activation and promotes negative regulation of TLR responses (41, 42). Hence, different cell type specific receptors and adaptor molecules may be responsible for mediating the functions of the same TLRs in different cell types. In line with this hypothesis, CD14 acts as a positive co-receptor for TLR7 and TLR9 in macrophages (43). In addition, integrin CD11b, which is highly expressed in monocytes and macrophages, is a negative regulator of TLR signaling in these cells (44). Interestingly, CD19 has been reported to play a role in mediating the signaling of the TLR family receptor RP105 (CD180) in murine B cells (45). Moreover, the paired Ig like receptor B (PIR-B) negatively regulates TLR9 signaling in murine B-1 cells by dephosphorylating BTK, further identifying BTK as a key player mediating both TLR9 and BCR signaling in B cells (46).
How does TLR9 initiate its CD19/PI3K-dependent signaling pathway? Co-immunoprecipitation experiments revealed that TLR9 and MyD88 do not interact with CD19 directly. However, we identified an association between MyD88 and BTK in human B cells and confirmed MyD88 interactions with PYK2/LYN complexes (32). Since inhibition of PYK2 and SFK but not BTK function blocked TLR9-induced CD19 phosphorylation, we propose that PYK2/LYN complexes, which bind MyD88, are responsible for TLR9-induced CD19-phosphorylation (Fig 8). It has been reported recently that DOCK8 functions as an adaptor that recruits PYK2/LYN to MyD88 and links TLR9/MyD88 to a SYK-STAT3 signaling pathway, essential for B-cell proliferation and differentiation into memory B cells (32). However, the normal up-regulation of CD86 and CD23 in DOCK8-deficient B cells after TLR9 stimulation suggests that the MyD88-PYK2/LYN/DOCK8-SYK-STAT3 pathway does not overlap with the MyD88-PYK2/LYN-CD19-PI3K/BTK/AKT pathway that controls the induction of these molecules after TLR9 triggering (32, 47). Hence, PYK2/LYN associated with MyD88 in human B cells might initiate distinct signaling pathways mediating various TLR9 functions. In agreement with this hypothesis, DOCK8 mediates TLR9-induced and STAT3-dependent B-cell proliferation, whereas CD19 controls PI3K/AKT-dependent early expression of B-cell surface molecules that include TACI, CD23 and CD86. Similarly to CD19-deficient B-cells, TLR9 induced upregulation of CD86 is decreased in individuals carrying TACI mutated alleles (13, 14). In addition, TACI can interact with MyD88 and both TLR7 and TLR9 upon engagement of these receptors (13, 48). TLR7 and TLR9 may then recognize RNA- and DNA-containing complexes, leading to TLR proteolytic activation and interaction with TACI, which in turn may allow the amplification of IRAK4/MYD88–dependent signals. Since up-regulation of TACI following TLR7 or TLR9 stimulation is impaired in CD19-deficient B cells and CVID patients with TACI- or CD19-mutations display an overlapping immunological and clinical phenotype, one might speculate that activation of TACI and interaction with TLR7/MYD88 or TLR9/MYD88 might also be involved in the amplification of the described CD19-dependent TLR7 or TLR9 signaling pathway in human B cells.
In summary, we found that CD19 mediates not only BCR but also TLR7 and TLR9 signaling pathways in human B cells. This novel CD19-dependent TLR pathway is mediated by a MyD88/PYK2/LYN and CD19/PI3K loop, which amplifies BTK and AKT phosphorylation, and controls both early B cell activation and proliferation.
Supplementary Material
Supplementary Figures
FIG E1. Defective TLR9-induced B-cell activation in CD19-deficient patients is B-cell specific and not caused by altered TLR9 expression levels. A, Immunoblot analysis of lysates from EBV-immortalized B-cell lines derived from a CD19-deficient patient and two unrelated healthy controls. B, Surface IgM expression levels on CD20+ CD10-CD21+CD27− mature naïve B cells from a CD19-def. patient and a healthy control were analyzed by flow cytometry. C, Surface expression of CD80 and CD86 on purified CD303+CD4+ plasmacytoid dendritic cells of a healthy control and a CD19-deficient patient after no stimulation or in vitro stimulation with the TLR9 ligand CpG or the TLR7 ligand Gardiquimod for two days was analyzed by flow cytometry.
FIG E2. Defective TLR7-induced B-cell activation in CD19-deficient patients. Surface expression of TACI, CD23, CD86 and CD80 on CD20+CD27- naïve B cells of a healthy family member (CD19-non carrier), a CD19-heterozygous carrier (CD19-het.) and a CD19-deficient patient (CD19-def.) after no stimulation or in vitro stimulation with the TLR7 ligand Loxorobine for two days.
FIG E3. TLR7 stimulation induces phosphorylation of CD19, BTK and AKT in human B cells. Phosphorylation of CD19, BTK, AKT and SYK was assessed by immunoblot in lysates from purified healthy control peripheral blood B cells stimulated or not for the indicated time with TLR7 ligand Gardiquimod.
FIG E4. CD19 knock-down in Ramos B cells. Ramos B cells were transduced with lentiviral constructs containing no shRNA (pTRIP control) or an shRNA targeting CD19 (pTRIP CD19 shRNA) and GFP. GFP+CD19+ (control) or GFP+CD19− (CD19 knockdown) Ramos B cells were sorted and expanded in culture. CD19 expression was determined by flow cytometry, dashed lines display the isotype control staining (MFI, mean fluorescence intensity). CD19 expression was also determined by immunoblot analysis of lysates from control or CD19 shRNA transduced Ramos B-cell lines.
FIG E5. Inhibition of PI3K, AKT or BTK mimics TLR7 induced B-cell activation defects observed in CD19-deficient B cells. Surface expression of TACI, CD23, CD86 and CD69 on purified CD19+CD27− naive B cells of healthy individuals after in vitro stimulation with the TLR7 ligand Gardiquimod for two days with or without addition of PI3K-inhibitor (CAL-101), BTK-inhibitor (PCI32765) or AKT inhibitor (AKT-IV inhibitor) was analyzed by flow cytometry. Dot blots of a representative experiments are shown in A and the data of five independent experiments is summarized in B. Each bar represents the mean ± SEM frequency, horizontal dashed lines represent the mean of the unstimulated samples. (* p<0.05; ** p<0.01; *** p<0.001).
Key messages.
CD19 controls TLR9 function in human B cells
TLR9 induces CD19/PI3K-interaction and activation of BTK and AKT pathways
The CD19/PI3K axis integrates BCR and TLR9 pathways
Acknowledgments
The author would like to thank Drs. Andrea Cerutti and Maurizio Gentile for sharing immunoprecipitiation protocols. We thank Dr. Pierre Charneau and Pauline Soulas-Sprauel for kindly providing the pTRIP-Ubi-GFP lentiviral vector for shRNA delivery.
Supported by National Institute of Health (NIH) grants AI061093, AI071087, AI082713 and AI095848 (to E. M.) and a German Research Foundation grant MO 2160/2-1 (to H.M.).
Abbreviations
- ANA
Anti-nuclear antibody
- BCR
B-cell receptor
- CVID
Common variable immune deficiency
- pDC
plasmacytoid dendritic cell
- IB
immunoblot
- SFK
Src family kinase
- SLE
Systemic lupus erythematosus
- TACI
Transmembrane Activator and CAML Interactor
- TLR
Toll-like receptor
- XLA
X-linked agammaglobulinemia
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures of potential conflict of interests: The authors declare that they have no relevant conflicts of interest.
References
- 1.Carter RH, Fearon DT. CD19: lowering the threshold for antigen receptor stimulation of B lymphocytes. Science. 1992 Apr 3;256(5053):105–7. doi: 10.1126/science.1373518. [DOI] [PubMed] [Google Scholar]
- 2.Tedder TF, Inaoki M, Sato S. The CD19-CD21 complex regulates signal transduction thresholds governing humoral immunity and autoimmunity. Immunity. 1997 Feb;6(2):107–18. doi: 10.1016/s1074-7613(00)80418-5. [DOI] [PubMed] [Google Scholar]
- 3.Anzelon AN, Wu H, Rickert RC. Pten inactivation alters peripheral B lymphocyte fate and reconstitutes CD19 function. Nature immunology. 2003 Mar;4(3):287–94. doi: 10.1038/ni892. [DOI] [PubMed] [Google Scholar]
- 4.Buhl AM, Cambier JC. Phosphorylation of CD19 Y484 and Y515, and linked activation of phosphatidylinositol 3-kinase, are required for B cell antigen receptor-mediated activation of Bruton’s tyrosine kinase. Journal of immunology. 1999 Apr 15;162(8):4438–46. [PubMed] [Google Scholar]
- 5.Fujimoto M, Fujimoto Y, Poe JC, Jansen PJ, Lowell CA, DeFranco AL, et al. CD19 regulates Src family protein tyrosine kinase activation in B lymphocytes through processive amplification. Immunity. 2000 Jul;13(1):47–57. doi: 10.1016/s1074-7613(00)00007-8. [DOI] [PubMed] [Google Scholar]
- 6.Fujimoto M, Poe JC, Satterthwaite AB, Wahl MI, Witte ON, Tedder TF. Complementary roles for CD19 and Bruton’s tyrosine kinase in B lymphocyte signal transduction. Journal of immunology. 2002 Jun 1;168(11):5465–76. doi: 10.4049/jimmunol.168.11.5465. [DOI] [PubMed] [Google Scholar]
- 7.Engel P, Zhou LJ, Ord DC, Sato S, Koller B, Tedder TF. Abnormal B lymphocyte development, activation, and differentiation in mice that lack or overexpress the CD19 signal transduction molecule. Immunity. 1995 Jul;3(1):39–50. doi: 10.1016/1074-7613(95)90157-4. [DOI] [PubMed] [Google Scholar]
- 8.Rickert RC, Rajewsky K, Roes J. Impairment of T-cell-dependent B-cell responses and B-1 cell development in CD19-deficient mice. Nature. 1995 Jul 27;376(6538):352–5. doi: 10.1038/376352a0. [DOI] [PubMed] [Google Scholar]
- 9.van Zelm MC, Reisli I, van der Burg M, Castano D, van Noesel CJ, van Tol MJ, et al. An antibody-deficiency syndrome due to mutations in the CD19 gene. The New England journal of medicine. 2006 May 4;354(18):1901–12. doi: 10.1056/NEJMoa051568. [DOI] [PubMed] [Google Scholar]
- 10.van Zelm MC, Smet J, van der Burg M, Ferster A, Le PQ, Schandene L, et al. Antibody deficiency due to a missense mutation in CD19 demonstrates the importance of the conserved tryptophan 41 in immunoglobulin superfamily domain formation. Human molecular genetics. 2011 May 1;20(9):1854–63. doi: 10.1093/hmg/ddr068. [DOI] [PubMed] [Google Scholar]
- 11.Vince N, Boutboul D, Mouillot G, Just N, Peralta M, Casanova JL, et al. Defects in the CD19 complex predispose to glomerulonephritis, as well as IgG1 subclass deficiency. The Journal of allergy and clinical immunology. 2011 Feb;127(2):538–41. doi: 10.1016/j.jaci.2010.10.019. [DOI] [PubMed] [Google Scholar]
- 12.Cunningham-Rundles C, Radigan L, Knight AK, Zhang L, Bauer L, Nakazawa A. TLR9 activation is defective in common variable immune deficiency. Journal of immunology. 2006 Feb 1;176(3):1978–87. doi: 10.4049/jimmunol.176.3.1978. [DOI] [PubMed] [Google Scholar]
- 13.Romberg N, Chamberlain N, Saadoun D, Gentile M, Kinnunen T, Ng YS, et al. CVID-associated TACI mutations affect autoreactive B cell selection and activation. The Journal of clinical investigation. 2013 Oct 1;123(10):4283–93. doi: 10.1172/JCI69854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu JE, Knight AK, Radigan L, Marron TU, Zhang L, Sanchez-Ramon S, et al. Toll-like receptor 7 and 9 defects in common variable immunodeficiency. The Journal of allergy and clinical immunology. 2009 Aug;124(2):349–56. 56 e1–3. doi: 10.1016/j.jaci.2009.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Denz A, Eibel H, Illges H, Kienzle G, Schlesier M, Peter HH. Impaired up-regulation of CD86 in B cells of “type A” common variable immunodeficiency patients. European journal of immunology. 2000 Apr;30(4):1069–77. doi: 10.1002/(SICI)1521-4141(200004)30:4<1069::AID-IMMU1069>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 16.Weller S, Bonnet M, Delagreverie H, Israel L, Chrabieh M, Marodi L, et al. IgM+IgD+CD27+ B cells are markedly reduced in IRAK-4-, MyD88-, and TIRAP- but not UNC-93B-deficient patients. Blood. 2012 Dec 13;120(25):4992–5001. doi: 10.1182/blood-2012-07-440776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hornung V, Rothenfusser S, Britsch S, Krug A, Jahrsdorfer B, Giese T, et al. Quantitative expression of toll-like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. Journal of immunology. 2002 May 1;168(9):4531–7. doi: 10.4049/jimmunol.168.9.4531. [DOI] [PubMed] [Google Scholar]
- 18.Rawlings DJ, Schwartz MA, Jackson SW, Meyer-Bahlburg A. Integration of B cell responses through Toll-like receptors and antigen receptors. Nature reviews Immunology. 2012 Apr;12(4):282–94. doi: 10.1038/nri3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huggins J, Pellegrin T, Felgar RE, Wei C, Brown M, Zheng B, et al. CpG DNA activation and plasma-cell differentiation of CD27- naive human B cells. Blood. 2007 Feb 15;109(4):1611–9. doi: 10.1182/blood-2006-03-008441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Isnardi I, Ng YS, Srdanovic I, Motaghedi R, Rudchenko S, von Bernuth H, et al. IRAK-4-and MyD88-dependent pathways are essential for the removal of developing autoreactive B cells in humans. Immunity. 2008 Nov 14;29(5):746–57. doi: 10.1016/j.immuni.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nickerson KM, Christensen SR, Cullen JL, Meng W, Luning Prak ET, Shlomchik MJ. TLR9 promotes tolerance by restricting survival of anergic anti-DNA B cells, yet is also required for their activation. Journal of immunology. 2013 Feb 15;190(4):1447–56. doi: 10.4049/jimmunol.1202115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006 Sep;25(3):417–28. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 23.Artac H, Reisli I, Kara R, Pico-Knijnenburg I, Adin-Cinar S, Pekcan S, et al. B-cell maturation and antibody responses in individuals carrying a mutated CD19 allele. Genes and immunity. 2010 Oct;11(7):523–30. doi: 10.1038/gene.2010.22. [DOI] [PubMed] [Google Scholar]
- 24.Conley ME, Fitch-Hilgenberg ME, Cleveland JL, Parolini O, Rohrer J. Screening of genomic DNA to identify mutations in the gene for Bruton’s tyrosine kinase. Human molecular genetics. 1994 Oct;3(10):1751–6. doi: 10.1093/hmg/3.10.1751. [DOI] [PubMed] [Google Scholar]
- 25.Conley ME, Broides A, Hernandez-Trujillo V, Howard V, Kanegane H, Miyawaki T, et al. Genetic analysis of patients with defects in early B-cell development. Immunological reviews. 2005 Feb;203:216–34. doi: 10.1111/j.0105-2896.2005.00233.x. [DOI] [PubMed] [Google Scholar]
- 26.Sauer AV, Morbach H, Brigida I, Ng YS, Aiuti A, Meffre E. Defective B cell tolerance in adenosine deaminase deficiency is corrected by gene therapy. The Journal of clinical investigation. 2012 Jun 1;122(6):2141–52. doi: 10.1172/JCI61788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schickel JN, Pasquali JL, Soley A, Knapp AM, Decossas M, Kern A, et al. Carabin deficiency in B cells increases BCR-TLR9 costimulation-induced autoimmunity. EMBO molecular medicine. 2012 Dec;4(12):1261–75. doi: 10.1002/emmm.201201595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zennou V, Petit C, Guetard D, Nerhbass U, Montagnier L, Charneau P. HIV-1 genome nuclear import is mediated by a central DNA flap. Cell. 2000 Apr 14;101(2):173–85. doi: 10.1016/S0092-8674(00)80828-4. [DOI] [PubMed] [Google Scholar]
- 29.Fujimoto M, Poe JC, Inaoki M, Tedder TF. CD19 regulates B lymphocyte responses to transmembrane signals. Seminars in immunology. 1998 Aug;10(4):267–77. doi: 10.1006/smim.1998.9999. [DOI] [PubMed] [Google Scholar]
- 30.Okkenhaug K, Vanhaesebroeck B. PI3K in lymphocyte development, differentiation and activation. Nature reviews Immunology. 2003 Apr;3(4):317–30. doi: 10.1038/nri1056. [DOI] [PubMed] [Google Scholar]
- 31.Aiba Y, Kameyama M, Yamazaki T, Tedder TF, Kurosaki T. Regulation of B-cell development by BCAP and CD19 through their binding to phosphoinositide 3-kinase. Blood. 2008 Feb 1;111(3):1497–503. doi: 10.1182/blood-2007-08-109769. [DOI] [PubMed] [Google Scholar]
- 32.Jabara HH, McDonald DR, Janssen E, Massaad MJ, Ramesh N, Borzutzky A, et al. DOCK8 functions as an adaptor that links TLR-MyD88 signaling to B cell activation. Nature immunology. 2012 Jun;13(6):612–20. doi: 10.1038/ni.2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Doyle SL, Jefferies CA, Feighery C, O’Neill LA. Signaling by Toll-like receptors 8 and 9 requires Bruton’s tyrosine kinase. The Journal of biological chemistry. 2007 Dec 21;282(51):36953–60. doi: 10.1074/jbc.M707682200. [DOI] [PubMed] [Google Scholar]
- 34.Yang G, Zhou Y, Liu X, Xu L, Cao Y, Manning RJ, et al. A mutation in MYD88 (L265P) supports the survival of lymphoplasmacytic cells by activation of Bruton’s tyrosine kinase in Waldenstrom’s macroglobulinemia. Blood. 2013 Jul 8; doi: 10.1182/blood-2012-12-475111. [DOI] [PubMed] [Google Scholar]
- 35.Conley ME, Dobbs AK, Farmer DM, Kilic S, Paris K, Grigoriadou S, et al. Primary B cell immunodeficiencies: comparisons and contrasts. Annual review of immunology. 2009;27:199–227. doi: 10.1146/annurev.immunol.021908.132649. [DOI] [PubMed] [Google Scholar]
- 36.Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nature reviews Immunology. 2006 Nov;6(11):823–35. doi: 10.1038/nri1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Buhl AM, Pleiman CM, Rickert RC, Cambier JC. Qualitative regulation of B cell antigen receptor signaling by CD19: selective requirement for PI3-kinase activation, inositol-1,4,5-trisphosphate production and Ca2+ mobilization. The Journal of experimental medicine. 1997 Dec 1;186(11):1897–910. doi: 10.1084/jem.186.11.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hasegawa M, Fujimoto M, Poe JC, Steeber DA, Lowell CA, Tedder TF. A CD19-dependent signaling pathway regulates autoimmunity in Lyn-deficient mice. Journal of immunology. 2001 Sep 1;167(5):2469–78. doi: 10.4049/jimmunol.167.5.2469. [DOI] [PubMed] [Google Scholar]
- 39.Suzuki H, Matsuda S, Terauchi Y, Fujiwara M, Ohteki T, Asano T, et al. PI3K and Btk differentially regulate B cell antigen receptor-mediated signal transduction. Nature immunology. 2003 Mar;4(3):280–6. doi: 10.1038/ni890. [DOI] [PubMed] [Google Scholar]
- 40.Castello A, Gaya M, Tucholski J, Oellerich T, Lu KH, Tafuri A, et al. Nck-mediated recruitment of BCAP to the BCR regulates the PI(3)K-Akt pathway in B cells. Nature immunology. 2013 Sep;14(9):966–75. doi: 10.1038/ni.2685. [DOI] [PubMed] [Google Scholar]
- 41.Ni M, MacFarlane AWt, Toft M, Lowell CA, Campbell KS, Hamerman JA. B-cell adaptor for PI3K (BCAP) negatively regulates Toll-like receptor signaling through activation of PI3K. Proceedings of the National Academy of Sciences of the United States of America. 2012 Jan 3;109(1):267–72. doi: 10.1073/pnas.1111957108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Troutman TD, Hu W, Fulenchek S, Yamazaki T, Kurosaki T, Bazan JF, et al. Role for B-cell adapter for PI3K (BCAP) as a signaling adapter linking Toll-like receptors (TLRs) to serine/threonine kinases PI3K/Akt. Proceedings of the National Academy of Sciences of the United States of America. 2012 Jan 3;109(1):273–8. doi: 10.1073/pnas.1118579109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baumann CL, Aspalter IM, Sharif O, Pichlmair A, Bluml S, Grebien F, et al. CD14 is a coreceptor of Toll-like receptors 7 and 9. The Journal of experimental medicine. 2010 Nov 22;207(12):2689–701. doi: 10.1084/jem.20101111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Han C, Jin J, Xu S, Liu H, Li N, Cao X. Integrin CD11b negatively regulates TLR-triggered inflammatory responses by activating Syk and promoting degradation of MyD88 and TRIF via Cbl-b. Nature immunology. 2010 Aug;11(8):734–42. doi: 10.1038/ni.1908. [DOI] [PubMed] [Google Scholar]
- 45.Yazawa N, Fujimoto M, Sato S, Miyake K, Asano N, Nagai Y, et al. CD19 regulates innate immunity by the toll-like receptor RP105 signaling in B lymphocytes. Blood. 2003 Aug 15;102(4):1374–80. doi: 10.1182/blood-2002-11-3573. [DOI] [PubMed] [Google Scholar]
- 46.Kubo T, Uchida Y, Watanabe Y, Abe M, Nakamura A, Ono M, et al. Augmented TLR9-induced Btk activation in PIR-B-deficient B-1 cells provokes excessive autoantibody production and autoimmunity. The Journal of experimental medicine. 2009 Aug 31;206(9):1971–82. doi: 10.1084/jem.20082392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Janssen E, Morbach H, Ullas S, Bannock JM, Massad C, Menard L, et al. Dedicator of cytokinesis 8-deficient patients have a breakdown in peripheral B-cell tolerance and defective regulatory T cells. The Journal of allergy and clinical immunology. 2014 Sep 10; doi: 10.1016/j.jaci.2014.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.He B, Santamaria R, Xu W, Cols M, Chen K, Puga I, et al. The transmembrane activator TACI triggers immunoglobulin class switching by activating B cells through the adaptor MyD88. Nature immunology. 2010 Sep;11(9):836–45. doi: 10.1038/ni.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures
FIG E1. Defective TLR9-induced B-cell activation in CD19-deficient patients is B-cell specific and not caused by altered TLR9 expression levels. A, Immunoblot analysis of lysates from EBV-immortalized B-cell lines derived from a CD19-deficient patient and two unrelated healthy controls. B, Surface IgM expression levels on CD20+ CD10-CD21+CD27− mature naïve B cells from a CD19-def. patient and a healthy control were analyzed by flow cytometry. C, Surface expression of CD80 and CD86 on purified CD303+CD4+ plasmacytoid dendritic cells of a healthy control and a CD19-deficient patient after no stimulation or in vitro stimulation with the TLR9 ligand CpG or the TLR7 ligand Gardiquimod for two days was analyzed by flow cytometry.
FIG E2. Defective TLR7-induced B-cell activation in CD19-deficient patients. Surface expression of TACI, CD23, CD86 and CD80 on CD20+CD27- naïve B cells of a healthy family member (CD19-non carrier), a CD19-heterozygous carrier (CD19-het.) and a CD19-deficient patient (CD19-def.) after no stimulation or in vitro stimulation with the TLR7 ligand Loxorobine for two days.
FIG E3. TLR7 stimulation induces phosphorylation of CD19, BTK and AKT in human B cells. Phosphorylation of CD19, BTK, AKT and SYK was assessed by immunoblot in lysates from purified healthy control peripheral blood B cells stimulated or not for the indicated time with TLR7 ligand Gardiquimod.
FIG E4. CD19 knock-down in Ramos B cells. Ramos B cells were transduced with lentiviral constructs containing no shRNA (pTRIP control) or an shRNA targeting CD19 (pTRIP CD19 shRNA) and GFP. GFP+CD19+ (control) or GFP+CD19− (CD19 knockdown) Ramos B cells were sorted and expanded in culture. CD19 expression was determined by flow cytometry, dashed lines display the isotype control staining (MFI, mean fluorescence intensity). CD19 expression was also determined by immunoblot analysis of lysates from control or CD19 shRNA transduced Ramos B-cell lines.
FIG E5. Inhibition of PI3K, AKT or BTK mimics TLR7 induced B-cell activation defects observed in CD19-deficient B cells. Surface expression of TACI, CD23, CD86 and CD69 on purified CD19+CD27− naive B cells of healthy individuals after in vitro stimulation with the TLR7 ligand Gardiquimod for two days with or without addition of PI3K-inhibitor (CAL-101), BTK-inhibitor (PCI32765) or AKT inhibitor (AKT-IV inhibitor) was analyzed by flow cytometry. Dot blots of a representative experiments are shown in A and the data of five independent experiments is summarized in B. Each bar represents the mean ± SEM frequency, horizontal dashed lines represent the mean of the unstimulated samples. (* p<0.05; ** p<0.01; *** p<0.001).