

Abstract
BCR-ABL positive (+) acute lymphoblastic leukemia (ALL) accounts for ~30% of cases of ALL. We recently demonstrated that 2-deoxy-D-glucose (2-DG), a dual energy (glycolysis inhibition) and ER-stress (N-linked-glycosylation inhibition) inducer, leads to cell death in ALL via ER-stress/UPR-mediated apoptosis. Among ALL subtypes, BCR-ABL+ ALL cells exhibited the highest sensitivity to 2-DG suggesting BCR-ABL expression may be linked to this increased vulnerability. To confirm the role of BCR-ABL, we constructed a NALM6/BCR-ABL stable cell line and found significant increase in 2-DG-induced apoptosis compared to control. We found that Mcl-1 was downregulated by agents inducing ER-stress and Mcl-1 levels correlated with ALL sensitivity. In addition, we showed that Mcl-1 expression is positively regulated by the MEK/ERK pathway, dependent on BCR-ABL, and further downregulated by combining ER-stressors with TKIs. We determined that energy/ER stressors led to translational repression of Mcl-1 via the AMPK/mTOR and UPR/PERK/eIF2α pathways. Taken together, our data indicate that BCR-ABL+ ALL exhibits heightened sensitivity to induction of energy and ER- stress through inhibition of the MEK/ERK pathway, and translational repression of Mcl-1 expression via AMPK/mTOR and UPR/PERK/eIF2α pathways. This study supports further consideration of strategies combining energy/ER-stress inducers with BCR-ABL TKIs for future clinical translation in BCR-ABL+ ALL patients.
Keywords: Leukemia, BCR-ABL+, 2-deoxy-D-glucose, ER stress, energy stress, UPR, imatinib, dasatinib, ERK, Mcl-1, AMPK, eIF2a, shRNA, apoptosis, tyrosine kinase inhibitors, combination targeted therapy, acute lymphoblastic leukemia
1. INTRODUCTION
Acute Lymphoblastic Leukemia (ALL) is the most common malignancy in children and adolescents [1]. BCR-ABL positive ALL (BCR-ABL+ ALL, t(9;22)) is present in 5% and 25% of pediatric and adult cases of ALL, respectively [2]. Recent strategies combining intensive multi-agent chemotherapy plus a tyrosine kinase inhibitor (TKI) have improved outcomes for BCR-ABL+ ALL patients albeit with significant toxicity [3, 4]. Therefore, finding novel strategies that synergize with TKIs against BCR-ABL+ ALL and are less toxic is desirable.
Tumor metabolism has emerged as a hallmark of cancer [5]. Oncogenes such as BCR-ABL, Akt, and Ras have the ability to both upregulate glucose metabolism and prevent cell death [6]. The BCR-ABL fusion upregulates Glut1 through activation of the PI3K/Akt signaling pathway [7], and inhibition of BCR-ABL with TKIs leads to decreased glucose uptake and cell death [8, 9]. Additionally, increased glucose uptake and metabolism has been reported to protect cells by blocking GSK-3β mediated degradation of Mcl-1, a member of the anti-apoptotic Bcl2 family [10]. Expression of the Mcl-1 protein has been shown to be important for BCR-ABL+ ALL cell survival [11–13], whereas reduced Mcl-1 levels allow the pro-apoptotic proteins (Noxa, Bim, Puma) to promote cell death via Bax and Bak [14]. In CML, BCR-ABL has been shown to promote Mcl-1 expression through the RAS/RAF/MEK/ERK and STAT5 pathways [15].
The dual glycolysis and N-linked glycosylation inhibitor, 2-deoxy-D-glucose (2-DG), induces both energy and ER-stress. As a glucose analog, 2-DG inhibits hexokinase (HK) and phosphoglucose isomerase (PGI), and as a mannose analog it interferes with N-linked glycosylation by incorporating into lipid-linked oligosaccharide (LLO) chains leading to premature termination of LLO synthesis (Figure S1) [16]. We recently demonstrated that 2-DG induces ALL cell death preferentially by inhibition of N-linked glycosylation resulting in ER-stress/unfolded protein response (UPR)-mediated apoptosis, although inhibition of glycolysis contributes to cell death in a phenotype specific-manner [17]. Among ALL subtypes, BCR-ABL+ ALL cell lines exhibited the highest sensitivity to 2-DG, suggesting that BCR-ABL expression may be linked to this phenotype [17]. Our group also demonstrated that in ALL cells, AMPK is a central regulator of the UPR under conditions of metabolic stress [18], acting as a sensor of ER-stress through CaMKKβ, independent of LKB1 signaling [19].
In this study, we investigated the role of BCR-ABL expression in heightening the sensitivity of ALL cells to the induction of energy and ER-stress, and examined the role of Mcl-1 in BCR-ABL+ ALL cells under these conditions of stress.
2. MATERIALS AND METHODS
2.1 Reagents
2-deoxy-D-glucose, D-mannose, thapsigargin, and tunicamycin (Sigma-Aldrich). Imatinib, dasatinib, nilotinib, bortezomib, and PD98059 (Selleck Chemicals), and qVD-OPH (SM Biochemicals). PERK inhibitor GSKA, a kind gift from Drs. Rakesh Kumar and Jeffrey Axten (GlaxoSmithKline, Collegeville, PA) [20].
2.2 Cell culture, cell proliferation and apoptosis assays
CCRF-CEM (T-ALL), NALM6 (Bp-ALL), and K562 (CML, BCR-ABL p210) cell lines were grown in RPMI 1640 medium [17], and BCR-ABL+ ALL SUPB15 and TOM-1 cells were grown in Iscove’s DMEM medium and RPMI 1640, respectively [17]. Cell proliferation and apoptosis were determined as previously described [18]. Data were analyzed by one-way ANOVA or by Student’s t test using GraphPad PRISM (Version 5.0c).
2.3 Plasmids, trasnfection, construction of stable cell lines, and shRNA
To construct pSG5-P190-Neo, a 1.7 kbp fragment containing the neomycin gene from pCI-Neo plasmid was PCR amplified using oligonucleotides designed with BamHI sites, inserted into the BamHI site of pSG5-P190 (Addgene #31285), and characterized by restriction enzyme digestions and DNA sequence. Transfections in ALL cells were carried out using the Nucleofector II device (program C-005) (Lonza). Stable transfectants were selected in presence of geneticin (G418) [500 μg/mL] (Life Technologies). Plasmids: pCI-neo (Promega, #E1841), pCDNA3.1 (Invitrogen, #V790-20), and pCDNA3.1-hMcl-1 (gift from Roger Davis, Addgene plasmid # 25375) [21].
Mcl-1 was downregulated using a pool of lentiviral particles encoding specific shRNAs against Mcl-1 (sc-35877-V; Santa Cruz Biotechnology) or scrambled shRNA sequence (sc-108080) as previously described [17].
2.4 SYBR Green real-time qRT-PCR
Mcl-1 and GAPDH mRNA expression were measured by real-time quantitative RT-PCR using Mcl-1 (Hs Mcl-1_1_SG #QT00094122) and GAPDH (Hs GAPDH_1_SG #QT00079247) Quantitect Primers (Qiagen) as described elsewhere [22].
2.5 Western immunoblots
Immunoblots were done as previously described [18] using antibodies from Active Motif (ATF6, 40962), Santa Cruz Biotechnology (c-ABL, sc131; NOXA, sc-30209) and Cell Signaling: β-actin (4967), p-Akt/S473 (4060), p-AMPK/T172 (2535), Bak (3814), Bax (2772), Bcl-2 (2870), Bcl-xL (2764), Bid (2002), Bim (2933), p-cABL (#5300), CHOP (2895), GRP78 (3177), GRP94 (2104), p-eIF2α (9721), p-ERK1/2 (9101), GSK-3β (9315), p-GSK-3β/S9 (9322), IRE1α (3294), Mcl-1 (4572), p-MEK1/2 (9154), p-mTOR/S2448 (2971), PUMA (4976), p-4EBP1/T70 (9455), p-p70S6K/T389 (9205), p-p70S6K/T421/S424 (9204).
2.6 Fluorophore-Assisted Carbohydrate Electrophoresis (FACE)
LLOs were analyzed as previously described [16].
3. RESULTS
3.1 BCR-ABL expression leads to increased ALL sensitivity to the dual energy/ER-stress inducing agent 2-DG
We previously reported that 2-DG induces cell death in ALL cell lines and primary cells [17]. Among ALL subtypes, BCR-ABL+ ALL SUPB15 and TOM1 cells consistently exhibited the highest sensitivity to 2-DG (Figure 1A). In contrast, the CML cell line K562 also expressing BCR-ABL exhibited the highest resistance to 2-DG (Figure 1A). These findings suggested that expression of the BCR-ABL fusion protein in ALL cells is linked to their sensitivity to energy/ER-stress. To directly investigate the role that BCR-ABL played in heightened sensitivity of ALL cells to 2-DG, we constructed NALM6 stable cell lines expressing the BCR-ABL fusion p190 (NALM6/BCR-ABL), and assessed their sensitivity to 2-DG. Expression of the BCR-ABL p190 fusion in these stable transfectants was confirmed by immunoblot analysis (Figure 1B). More importantly, we found that expression of BCR-ABL in these stable transfectants significantly increased 2-DG-induced apoptosis compared to mock transfected NALM6 cells (NALM6/pCI-Neo) (Figures 1A and 1C), indicating that expression of BCR-ABL sensitizes ALL cells to 2-DG. In addition, the level of 2-DG induced cytotoxicity in NALM6/BCR-ABL was dose dependent (Figure 1C) [17].
Figure 1. BCR-ABL expression leads to increased sensitivity to 2-DG in NALM6 ALL cells.

(A) Cell death in CML (K562), control ALL (NALM6), and BCR-ABL+ ALL (NALM6/BCR-ABL, TOM-1, SUPB15) cells treated with 2-DG (4mM) for 72 h under normoxia (pO2 = 21%). (B) Western blot analysis of BCR-ABL p190 protein in SUPB15, NALM6, and NALM6 stable cell expressing BCR-ABL p190 fusion. (C) NALM6 and stable transfectant NALM6/BCR-ABL were treated with 2-DG (1 mM, 2 mM, and 4 mM) for 48 h under normoxia and assayed for cell death (apoptosis) using the Annexin V/PI staining; * and # denote p<0.001 and p<0.01, respectively, for NALM6 BCR-ABL vs. NALM6 cells treated with 2-DG. Data represents the mean ± SEM (n=3).
3.2 2-DG induces ER-stress/UPR in BCR-ABL + and − ALL via inhibition of Lipid-linked oligosccharides (LLOs) synthesis
Co-treatment of BCR-ABL+ and BCR-ABL− ALL cells with 2-DG and D-mannose demonstrated that D-mannose, known to reverse the cytotoxicity associated with inhibition of N-linked glycosylation but not of glycolysis [23], conferred only partial protection (≈50%) against 2-DG-induced apoptosis (Figure 2A), indicating that inhibition of N-linked glycosylation was not solely responsible for the observed enhanced cell death. In addition, LLOs synthesis (the precursors of N-linked oligosaccharides) was analyzed by FACE in 2-DG-treated ALL cells, and ER-stress/UPR markers assessed by immunoblots. 2-DG inhibited LLOs synthesis in both BCR-ABL+ SUPB15 and BCR-ABL− NALM6 ALL cells (Figure 2B) which correlated with induction of ER stress/UPR markers (GRP78, CHOP, Figure 2C). Co-treatment with D-mannose reduced the inhibitory effects of 2-DG on LLO synthesis which was consistent with the reduction in ER stress/UPR markers indicating a causal relationship between 2-DG’s inhibition of N-linked glycosylation and ER stress/UPR-mediated cell death (Figure 2D).
Figure 2. 2-DG induces ER-stress/UPR in BCR-ABL+ and − ALL cell lines.

(A) Effect of D-mannose in BCR-ABL+ and − ALL cell lines treated with 2-DG. SUPB15 and NALM6 cell lines were treated with 2-DG (2mM or 4mM) ± D-mannose (Man, 1mM or 2mM) for 72 h, and assayed for cell death using the ViCell XR Cell system. Data represents the mean ± SEM (n=3). (B) 2-DG interferes with N-linked glycosylation (inhibits LLO synthesis) in BCR-ABL+ and − ALL cell lines. SUPB15 and NALM6 cell lines were treated with 2-DG (4 mM) for 24 h, and LLOs analyzed by FACE. The standard oligosaccharides are as follows: G4 to G7, glucose oligomers; G3M9, mature oligosaccharide (G3M9Gn2); M5 and M9, oligosaccharides. (C) 2-DG induces UPR signaling factors in BCR-ABL+ and − ALL cell lines. Western blots of UPR signaling factors in SUPB15 and NALM6 cell lines treated with 2-DG (4 mM) for 24 h under normoxia. (D) Western blots of UPR signaling factors in SUPB15 and NALM6 cell lines treated with 2-DG (2 mM or 4 mM] and either with (+) or without (−) D-mannose (1 mM or 2 mM) for 24 h. β-actin was used as a loading control.
3.3 ER-stress inducing agents lead to downregulation of Mcl-1 expression in BCR-ABL+ and − ALL
Mcl-1 was shown to promote survival of BCR-ABL expressing myeloid and lymphoid cells [11–13, 15]. This prompted us to determine the role of Mcl-1 expression in 2-DG-induced cell death in BCR-ABL+ and BCR-ABL− ALL, and compare it to CML cells. Figure 3A shows that constitutive expression of Mcl-1 is lower in BCR-ABL+ ALL (SUPB15, TOM1) than in CML (KU812, K562) cells, which inversely correlates with their sensitivity to 2-DG (Figure 1A). Moreover, we found that Mcl-1 expression was reduced in BCR-ABL+/− ALL cells treated with 2-DG as well as with other ER stressors, i.e. tunicamycin (TUN), or thapsigargin (TG) (Figure 3B), which correlated with apoptotic death (Figure 1A). Analysis of anti-apoptotic and pro-apoptotic factors in 2-DG-treated BCR-ABL+/− ALL cells demonstrated that the only anti-apoptotic factor downregulated was Mcl-1, whereas no discernible differences was seen among pro-apoptotic proteins except for PUMA which was reduced (Figure 3C).
Figure 3. ER-stressors induce down-regulation of Mcl-1 in BCR-ABL+ and − ALL cells.

(A) Basal levels of Mcl-1 protein expression in CML (KU812, K562) and BCR-ABL+ ALL (SUPB15, TOM-1) cell lines. (B) Levels of Mcl-1 protein expression in SUPB15, NALM6, and NALM6 BCR-ABL cells treated with 2-DG (4 mM), tunicamycin (TUN, 1μg/ml), or thapsigargin (TG, 50nM) for 24 h. (C) Levels of anti-apoptotic and pro-apoptotic proteins in SUPB15 and NALM6 cells treated with 2-DG (4 mM) for 24 h. (D) NALM6 cells expressing either scramble shRNAs (shCTRL) or shRNAs against Mcl-1 (shMcl-1) were treated with 2-DG (4 mM) for 72 h and assayed for cell death using the ViCell XR Cell system. Insert shows Western blot analysis of Mcl-1 expression detected in the shCTRL and shMcl-1 expressing NALM6 cells treated with 2-DG for 24 h. Data represents the mean ± SEM (n=3). β-actin was used as a loading control. * denotes p<0.001.
To investigate whether there is a causal relationship between Mcl-1 expression and increased vulnerability to 2-DG-induced energy/ER-stress, we used lentiviral-based shRNAs to knock-down Mcl-1 expression in NALM6 cells, and found that Mcl-1 knockdown sensitized NALM6 cells to low dose 2-DG (2 mM) as compared to control NALM6/shCTRL (35% vs. 13% cell death; p < 0.001; Figure 3D). Similar knockdown experiments in SUPB15 cells led to cell death even in the absence of treatment with 2-DG (not shown), highlighting the pro-survival role of Mcl-1 in BCR-ABL+ ALL cells. To further confirm the importance of Mcl-1 expression in the sensitivity to 2-DG, we examined the effect of lowering Mcl-1 expression in the highly 2-DG resistant CML cell line K562 (Figure 1A), which exhibited higher level of Mcl-1 (Figure 3A). We found that down-regulation of Mcl-1 expression through inhibition of the PI3K/Akt/GSK-3β pathway using LY294002 sensitized K562 cells to 2-DG (Figure S2). Conversely, Mcl-1 over-expression in NALM6 cells transfected with pCDNA3.1-hMCL-1, lowered their sensitivity to 2-DG compared to mock transfectants (NALM6/pCDNA3.1) (Figure S3). These data indicate a central role for Mcl-1 expression in the sensitivity to induction of energy and ER-stress by 2-DG, and provide evidence for a causal relationship between Mcl-1 expression and the survival of BCR-ABL+ ALL.
3.4 Energy/ER-stress-induced Mcl-1 downregulation is mediated by decreased phosphorylation of MEK/ERK in BCR-ABL+ ALL
In CML cells, Mcl-1 expression is regulated by the RAF/MEK/ERK pathway in a BCR-ABL-dependent manner [15]. To investigate the BCR-ABL-dependent role of the RAF/MEK/ERK pathway on Mcl-1 expression in BCR-ABL+ ALL cells, we treated SUPB15 cells with TKIs (imatinib and dasatinib) and examined p-MEK1/2, p-ERK1/2, and Mcl-1 expression. Treatment with TKIs downregulated p-MEK1/2 and p-ERK1/2 expression in SUPB15 cells (Figure 4A) which correlated with decreased Mcl-1 expression, indicating constitutive expression of Mcl-1 is BCR-ABL-dependent in ALL. We next examined the effect of energy/ER-stress on MEK/ERK expression in BCR-ABL+ SUPB15 cells, and found that the energy/ER-stress inducer 2-DG and the pure ER-stressor TUN downregulated p-ERK1/2 (Figure 4B) which correlated with decreased Mcl-1 expression. This suggest that energy and/or ER-stress inducing agents downregulate Mcl-1 via the MEK/ERK pathway in ALL. Consistent with these findings, when ER-stress inducing agents were combined with a TKI (imatinib), further downregulation of p-ERK1/2 and Mcl-1 expression was observed (Figure 4B). Similar results were seen following pharmacological inhibition of MEK phosphorylation by PD98059, and further p-ERK1/2 and Mcl-1 downregulation were seen when PD98059 was combined with 2-DG or imatinib (Figure 4C). In addition, we found no correlation between the known regulator of Mcl-1 expression glycogen synthase kinase-3β (GSK-3β) [24] and Mcl-1 levels in BCR-ABL+ (SUPB15 and TOM-1) and BCR-ABL− (NALM6) ALL cells (Figure S4). These data suggest that in contrast to CML (K562), Mcl-1 downregulation in BCR-ABL+ and BCR-ABL− ALL cells treated with 2-DG is independent of the Akt/GSK-3β pathway.
Figure 4. ER-stressors and TKIs decrease Mcl-1 expression via downregulation of the MEK/ERK pathway in BCR-ABL+ ALL cells.

(A) Western blot of p-MEK1/2 (S217/221), p-ERK1/2 (p44/p42, T202/Y204) and Mcl-1 expression in SUPB15 cells treated with TKIs (imatinib, IM, 1 μM; dasatinib, DAS, 5 nM) for 24 h. (B) Western blot of p-ERK1/2 (p44/p42, T202/Y204) and Mcl-1 expression in SUPB15 cells treated with ER-stressors (2-DG, 4mM; tunicamycin, TUN, 1μg/ml), imatinib (IM, 1 μM), or the combination ER-stressors + IM for 24 h. (C) Western blot of p-ERK1/2 (p44/p42, T202/Y204) and Mcl-1 expression in SUPB15 cells treated with 2-DG (4mM), imatinib (IM, 1 μM), MEK inhibitor (PD98059, PD, 50μM), or in combination (2-DG + PD and IM + PD) for 24 h. β-actin was used as a loading control.
3.5 Energy/ER-stress inducing agents downregulate Mcl-1 in BCR-ABL+ ALL through translational mechanisms mediated by the AMPK/mTOR and UPR/PERK/eIF2α pathways
Initial experiments into the mechanism of Mcl-1 downregulation in BCR-ABL+/− ALL cells ruled out transcriptional control (Figure S5), caspase cleavage [25] (Figure S6A), and post-translational (ubiquitin-mediated proteasomal degradation) [26, 27] (Figure S6B) mechanisms.
Translational control mediated by the AMPK/mTOR pathway has been implicated in Mcl-1 downregulation in lymphoid malignancies [28, 29]. As reported for NALM6 [17], we found that energy/ER-stressors led to AMPK activation (p-AMPK/T172) and downstream downregulation of p-mTOR, p-p70S6K and p-4EBP1 in BCR-ABL+ SUPB15 cells [30] (Figures 5A and 5B). These data suggest that energy/ER-stressors could induce translational repression of the Mcl-1 protein via an AMPK/mTOR-dependent manner. To test this, we treated SUPB15 cells with 2-DG ± the mTOR inhibitor rapamycin, a translation inhibitor. We found that rapamycin + 2-DG led to greater p-mTOR/p-4EBP1 downregulation (compared to single agent), lowered Mcl-1 expression (Figure 5C), and led to enhanced cell death (Figure 5D). Therefore, we conclude that Mcl-1 downregulation by energy/ER-stress inducers such as 2-DG, occurs through translational control mediated by AMPK/mTOR.
Figure 5. Mcl-1 expression is controlled by a translational mechanism mediated by the AMPK/mTOR pathway.

Western blots of AMPK (T172), p-mTOR (S2448), p-p70S6K (T389), and p-4EBP1 (T70) proteins in SUPB15 cells treated with (A) 2-DG (4 mM) or (B) TUN (2 μg/ml) or TG (200 mM) for 24 h. (C) Effects of mTOR inhibition on Mcl-1 expression. Western blots of p-mTOR (S2448), p-4EBP1 (T70) and Mcl-1 expression in SUPB15 cells treated with 2-DG (4 mM) ± rapamycin (1μg/ml) for 24 h. β-actin was used as a loading control. (D) Cell death in SUPB15 BCR-ABL+ ALL cells treated with 2-DG (2 mM) ± rapamycin (1μg/ml) for 72 h. Data are representative of at least two independent experiments.
Based on the known function of the ER-stress-induced UPR/PERK pathway in shutting down protein synthesis via p-eIF2α, we use the PERK inhibitor GSKA to assess the role of the UPR/PERK/eIF2α pathway on Mcl-1 expression [20]. SUPB15 cells were co-treated with 2-DG or TUN ± GSKA (100 μM), and Mcl-1 expression assessed. As expected, 2DG and TUN induced the UPR as evidenced by upregulation of GRP78 and CHOP (Figure 6). More importantly, PERK inhibition by GSKA decreased p-eIF2α expression, known to relieve inhibition of general protein synthesis, which correlated with increased Mcl-1 expression. These data suggest that Mcl-1 expression is downregulated through a translational mechanism mediated by the UPR/PERK/p-eIF2α pathway in BCR-ABL+ ALL cells under conditions of ER-stress.
Figure 6. ER-stress-induced downregulation of Mcl-1 expression is controlled by a translational mechanism mediated by the UPR PERK/p-eIF2α pathway.
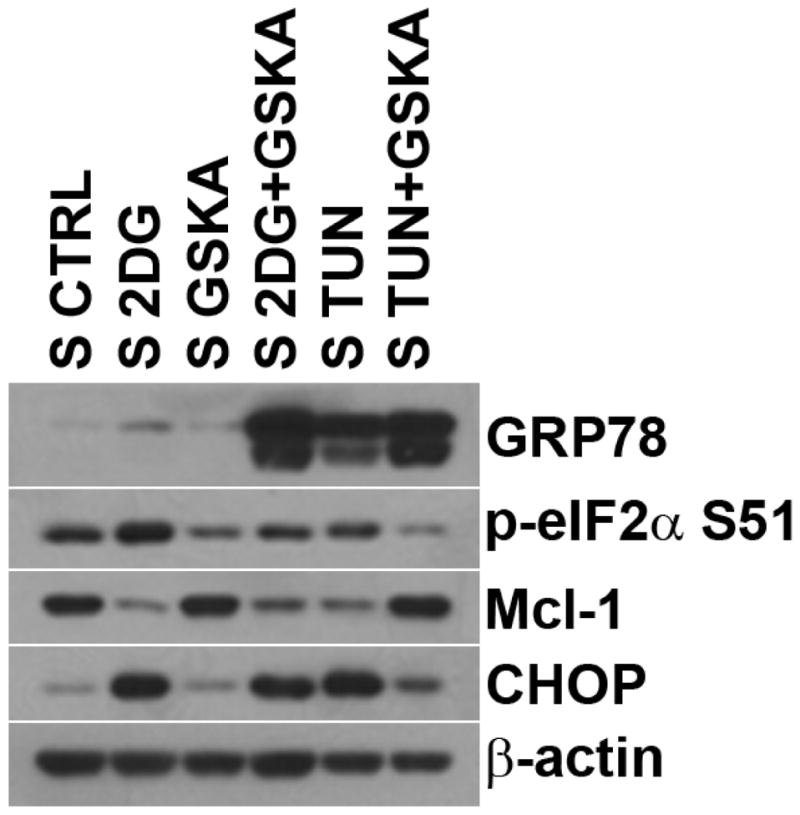
Western blots of Mcl-1 and UPR proteins (GRP78, p-eIF2α (S51), and CHOP) in SUPB15 cells treated with ER-stressors (2-DG, 4 mM; TUN, 2μg/ml) ± PERK inhibitor (GSKA, 200nM) for 24 h. β-actin was used as a loading control.
3.6 Combining BCR-ABL TKIs with energy/ER-stress inducing agents heightens cell death in BCR-ABL+ ALL
Similar to the effect in myeloid leukemia [9, 31–33], TKIs lowered ATP (~ 50%) and induced energy stress in BCR-ABL+ ALL cells, and the combination 2-DG + TKIs further decreased ATP production (Table I). Therefore, we examined the cytotoxicicity of combining TKIs (imatinib, dasatinib, nilotinib) with ER-stressors (2-DG, TUN, TG) in SUPB15 cells, and found these combinations induced greater cell death in SUPB15 cells than either drug alone (Figure 7A). As expected, Mcl-1 expression was decreased by each of these agents alone and further downregulated in the combinations TKIs + ER-stressors (Figure 7B). Under all conditions, greater Mcl-1 downregulation correlated with increased cell death (Figure 7A) confirming that decreased Mcl-1 expression is responsible for the heightened cell death exhibited by BCR-ABL+ ALL cells treated with TKIs ± ER-stress inducing agents. Imatinib was shown to downregulate the UPR in HepG2 hepatoma cells undergoing ER-stress [6]. We found a similar effect in SUPB15 ALL cells (lower GRP78) treated with TKIs ± ER-stressors (Figure 7B). On this basis, we propose that downregulation of the MEK/ERK/Mcl-1 pathway coupled with the inability of BCR-ABL+ ALL cells to effectively engage the UPR synergize to induce cell death in BCR-ABL+ ALL undergoing energy/ER-stress.
Table I.
Levels of ATP production (ATP/total number of cells) in SUPB15 cells treated with 2-DG and ER stressors relative to untreated cells.
| Time | 2-DG | IM | DAS | 2-DG + IM | 2-DG + DAS |
|---|---|---|---|---|---|
| 6 h | 73.3 ± 6.6 | 74.7 ± 5.1 | 70.6 ± 6.2 | 58.2 ± 6.4 | 52.6 ± 6.1 |
| 24 h | 54.6 ± 8.3 | 57.8 ± 7.6 | 43.4 ± 6.7 | 41.0 ± 6.5 | 32.2 ± 4.6 |
SUPB15 cells were treated with 2-DG (4 mM) or TKIs (imatinib, IM 1μM; dasatinib, DAS, 0.5μM) for 24 h and assayed for ATP production (ATP/total number of cells) as described in Materials and methods. Data are from at least to independent experiments assayed in triplicate and normalized to control values (%).
Figure 7. Targeting BCR-ABL+ ALL cells with TKIs and energy/ER-stressors significantly increase cell death.

(A) SUPB15 cells were treated with TKIs (imatinib, 1 μM; dasatinib, 5 nM; nilotinib, 250nM), ER-stressors (2-DG, 2 mM; TUN, 1 μg/ml; TG, 50 nM), or the combination TKIs + ER-stressors, and assayed for cell death at 72 h using the ViCell XR Cell system. Data represents the mean ± SEM (n=3). (B) Levels of Mcl-1 and UPR (IRE1α and GRP78) protein expression in SUPB15 cell line treated with TKIs and ER-stressors as described in (A) for 24 h. β-actin was used as a loading control.
4. DISCUSSION
We reported for the first time that ALL cell models and primary cells are vulnerable to ER-stress leading to UPR-mediated apoptotic cell death [17]. We also demonstrated that among ALL subtypes, BCR-ABL+ ALL cells exhibited the highest sensitivity to 2-DG-induced energy/ER-stress [17]. Our original findings were recently corroborated by a report indicating that normal and leukemic lymphoid cells are uniquely vulnerable to agents that block the UPR pathway and induce ER stress [34]. Herein, we investigated the role of the BCR-ABL fusion in heightening the sensitivity of ALL cells to perturbations in energy and ER-stress. To this effect we focused on the role, the regulation, and the contribution of the anti-apoptotic protein Mcl-1.
Using genetic models, we demonstrated that BCR-ABL expression was sufficient to sensitize ALL cells to the dual energy/ER-stressing agent 2-DG, suggesting BCR-ABL expression regulates signaling pathways involved in energy (ATP) and/or ER homeostasis in ALL. We showed that the increased sensitivity of BCR-ABL+ ALL cells to 2-DG was not due to the induction of ER-stress alone because treatment with D-mannose only partially rescued ALL cells, suggesting that simultaneous induction of energy and ER-stress are required for maximal induction of apoptotic death in ALL cells harboring the BCR-ABL fusion.
Recently, the antiapoptotic protein Mcl-1 was shown to be required for cell survival in BCR-ABL+ leukemia [11], and to play a role in the survival of ALL [12, 13], CML [15], hematopoietic stem cells [35], T-lymphocytes [36], and plasma cells [37]. Herein, we demonstrate that Mcl-1 plays a crucial role in the survival of BCR-ABL+ ALL cells undergoing energy/ER-stress. We found that treatment with energy/ER-stressors (2-DG), pure ER-stressors (TUN, TG), or energy stressors (TKIs) decreased the level of Mcl-1 expression which correlated with apoptotic death. More importantly, when BCR-ABL+ ALL cells were simultaneously challenged with energy and ER-stress, further downregulation of Mcl-1 leading to increased cell death was observed. Similar observations were reported in alveolar rhabdomyosarcoma cell lines treated with 2-DG [38].
In agreement with reports in B-cell lymphoma [39–41], T-ALL [28, 41], rhabdomyosarcoma [38], and non-small cell lung cancer cells [29], our data also supports a translational mechanism via AMPK/mTOR as responsible for Mcl-1 downregulation under energy/ER-stress. In addition to the AMPK/mTOR pathway, we uncovered that the UPR/PERK/p-eIF2α pathway also contributes to the translational downregulation of Mcl-1 expression in BCR-ABL+ ALL cells undergoing ER-stress. Interestingly, ER-stress-induced phosphorylation of eIF2α was recently found to be coupled with mitochondrial control of apoptosis via translational repression of Mcl-1 [42]. Based on the new function we recently reported for AMPK in sensing ER-stress in addition to sensing energy stress [17, 19], these findings suggest that Mcl-1 expression is translationally downregulated by energy stress via AMPK/mTOR pathway, and by ER-stress via both the UPR/PERK/eIF2α and AMPK/mTOR pathways. Therefore, in BCR-ABL+ ALL cells treated with energy and/or ER-stress inducing agents, AMPK/mTOR and UPR/PERK/eIF2α cooperate to downregulate Mcl-1 via inhibition of protein translation.
BCR-ABL promotes Mcl-1 expression mediated by the RAS/RAF/MEK/ERK and STAT5 pathways [15], and MEK/ERK positively regulates Mcl-1 expression [43, 44]. Consistent with this, we demonstrated that BCR-ABL upregulated MEK/ERK to promote Mcl-1 expression in BCR-ABL+ ALL cells, and that these pathways play a crucial role in the survival in BCR-ABL+ ALL via Mcl-1. Further, our data suggest that ER-stressors and TKIs regulate MEK/ERK expression via alterations of the same pathway(s). Recently, we reported a novel role for AMPK as a molecular switch regulating Akt and RAS signaling in ALL cells [45]. We found that AMPK could inhibit RAF (phosphorylation at S621) and downstream MEK/ERK signaling under metabolic stress, suggesting AMPK could also downregulate Mcl-1 expression via decreased RAS/MEK/ERK pathway. Taken together, we propose that energy/ER-stress-induced AMPK activation leads to Mcl-1 downregulation via three pathways: i) AMPK/RAS/MEK/ERK, ii) AMPK/mTOR/p70S6K, and iii) AMPK/UPR/eIF2α pathways (Figure 8).
Figure 8. Proposed model for the effects of energy and/or ER-stressors (2DG, tunicamycin), and TKIs (imatinib) on UPR/eIF2α, AMPK/mTOR, MEK/ERK, and Mcl-1 expression in BCR-ABL+ ALL cells.

Our data indicate that treatment with energy/ER-stressors (2-DG, tunicamycin) and TKIs (imatinib, dasatinib) lead to Mcl-1 downregulation via AMPK activation regulating RAS/MEK/ERK, mTOR/p70S6K, and UPR/PERK/eIF2α pathways. In addition to AMPK signaling, ER-stress alone can also inhibit protein synthesis through activation of the UPR/PERK and subsequent phosphorylation of the initiator of translation eIF2α.
We and others have shown that TKIs lower ATP and induce energy stress (Table I) [9, 31, 32]. In BCR-ABL+ ALL cells, treatment with TKIs alone lead to AMPK activation (data not shown) but induced minimal cell death. Interestingly, it was only when we combined TKIs with ER-stressors that cell death ensued. Our interpretation is that BCR-ABL kinase activity maintains energy levels in BCR-ABL+ ALL via stimulation of glycolysis, and when BCR-ABL kinase is blocked with a TKI, energy stress is induced (in the form of lower ATP) leading to cell growth inhibition, whereas when combined with ER-stress it leads to increased cell death as evidenced by further downregulation of MEK/ERK pathway and Mcl-1 expression. As both TKIs and ER-stressors lead to downregulation of the MEK/ERK pathway and Mcl-1 expression in BCR-ABL+ ALL cells, and Mcl-1 is critical for their survival, the enhanced downregulation of Mcl-1 expression by these agents in combination is central to explain their heightened vulnerability when these forms of stress are induced simultaneously. Exploiting these molecular vulnerabilities of BCR-ABL+ ALL through targeted approaches in combination with TKIs is highly desirable, highlighting the translational relevance of our findings.
Recently, small-inhibitors of Mcl-1 have been identified and shown to effectively suppress tumor growth in vivo using xenograft models [46, 47]. However, to our knowledge, there is no report of Mcl-1 downregulation in BCR-ABL-positive ALL cells in vivo using these pharmacological inhibitors. Recently, energy and/or ER-stress inducing agents have been used in humans. For instance, the proteasome inhibitor bortezomib, known to induce ER-stress, has been approved in the U.S. for treating relapsed multiple myeloma and mantle cell lymphoma [48] whereas a phase I clinical trial of 2-DG in combination with docetaxel has been reported in patients with advanced malignancies, showing tolerable toxicity [49]. Based on these published findings and our data, we propose that the combination of small-inhibitors of Mcl-1 plus energy and/or ER-stress inducing agents should lead to further downregulation or inhibition of Mcl-1 and induce greater cell death in cancer cells. Clinical trials using these combinations, or alike, will be required to established the safety and tolerability of the same.
In conclusion, we established that the BCR-ABL fusion is responsible for the increased vulnerability of BCR-ABL+ ALL cells to induction of energy and ER-stress via downregulation of Mcl-1 expression. We identified the antiapoptotic protein Mcl-1 as a crucial factor for the survival of BCR-ABL+ ALL cells treated with energy/ER-stressors ± TKIs. Our findings reveal that under energy/ER-stress conditions, Mcl-1 expression is downregulated via i) translational mechanisms mediated by AMPK/mTOR and UPR/PERK/eIF2α and ii) by the MEK/ERK pathway in a BCR-ABL-dependent manner. Finally, our study demonstrates that strategies with agents that induce ER-stress plus BCR-ABL TKIs warrant further consideration as a novel approach for future clinical translation for BCR-ABL+ ALL patients.
Supplementary Material
Figure S1. Glucose analogues 2-DG and 2-FDG act as inhibitors of glycolysis and N-linked glycosylation. 2-DG and 2-FDG inhibit hexokinase, phosphoglucose isomerase (PGI), and mannose-1-phosphate guanylyltransferase (GDP-Man synthase). Due to the resemblance of 2-DG with mannose, it not only competes with mannose metabolism in this pathway but also incorporates fraudulently into dolichol-pyrophosphate (lipid)–linked oligosaccharides (LLOs), which are the precursors for N-linked glycosylation. 2-FDG is a more potent glycolysis inhibitor than does 2-DG. In contrast, 2-DG is more potent than 2-FDG in interfering with N-linked glycosylation. (▲, glucose; ●, mannose; and ■, N-acetyl-glucosamine).
Figure S2. Mcl-1 downregulation sensitized CML cells to 2-DG. K562 cells were treated with the PI3K inhibitor LY294002 (25 mM) and 2-DG (10 mM) for 72 h and assayed for cell death using the ViCell XR Cell system Data represents the mean ± SEM (n=3); * denotes p<0.001. Right insert shows Western blot analysis of Mcl-1 expression in K562 cells treated with the PI3K inhibitor LY294002 (25 mM and 50 mM) ± 2-DG (10 mM) for 24 h. β-actin was used as a loading control.
Figure S3. Mcl-1 expression increases NALM6 resistance to 2-DG. Mcl-1 overexpression alleviates 2-DG cytotoxicity in NALM6 cells. NALM6 cells were transfected with either pCDNA3.1 or pCDNA3.1-hMCL-1 vector (addgene # 25375) [21], and 24 h post transfection, cells were treated with 2-DG (4 mM) for 72 h and assayed for cell death using the ViCell XR Cell system. Data represents the mean ± SEM (n=3); * denotes p<0.001.
Figure S4. Mcl-1 downregulation is independent of the PI3K/GSK-3β pathway in BCR-ABL+/− ALL cells treated with 2-DG. Levels of p-Akt (S473), p-GSK-3β (S9), GSK-3β and Mcl-1 protein expression in BCR-ABL+ (SUPB15 and TOM1) and control ALL cells (NALM6) treated with 2-DG [4 mM] for 24 h. β-actin was used as a loading control.
Figure S5. Mcl-1 downregulation in BCR-ABL+ and − ALL cells treated with energy/ER-stress inducers is not due to transcriptional mechanism. To explore other potential mechanisms by which Mcl-1 is downregulated in BCR-ABL+ and − ALL cells treated with energy/ER-stressing agents, we analyzed the level of Mcl-1 mRNA expression in NALM6, SUPB15, and NALM6/BCR-ABL cells treated with or without 2-DG (4 mM) for 24 h. Mcl-1 mRNA expression was detected by real-time qRT-PCR and normalized to GAPDH mRNA levels. Each experiment was performed at least two times in tetraplicate. Data represents the mean ± SEM (n=4). No significant differences were detected in BCR-ABL+ or BCR-ABL− ALL cells treated with 2-DG compared to untreated controls, indicating that 2-DG-induced Mcl-1 downregulation is not mediated by transcriptional mechanisms.
Figure S6. Mcl-1 downregulation in BCR-ABL+ and − ALL cells treated with energy/ER-stress inducers is not due to posttranslational modification mechanisms. (A) To rule out that the decrease in Mcl-1 expression was due to caspase dependent cleavage during apoptosis, we examined the level of Mcl-1 protein expression in SUPB15 and NALM6 cells treated with or without 2-DG (4 mM) in presence or absence of the pan-caspase inhibitor qVD-OPH (20 μM) for 24 h. qVD-OPH was added 1 hr prior treatment. In NALM6 cells, staurosporin (STP, 3 μM for 4 h) was used as positive control on Mcl-1 cleavage. We found that qVD-OPH did not prevent Mcl-1 downregulation in ALL cells treated with 2-DG but could inhibit Mcl-1 cleavage in NALM6 cells treated with staurosporin (STP)-induced caspase activation, indicating that 2-DG-induced Mcl-1 downregulation is not mediated by caspase cleavage activity during apoptosis. (B) To investigate whether Mcl-1 was regulated by ubiquitin-mediated proteasomal degradation in ALL, we determined the level of Mcl-1 protein expression in SUPB15 and NALM6 cells treated with or without 2-DG (4 mM) in presence or absence of the proteasome inhibitor Bortezomib (BTZ, 5 nM) for 24 h. β-actin was used as a loading control. We found that the decreased levels of Mcl-1 expression in SUPB15 and NALM6 cells treated with 2-DG were unchanged in the presence of bortezomib, ruling out an ubiquitin-mediated proteasomal degradation mechanism to downregulate Mcl-1 in our experimental system. Taken together, our results indicate that Mcl-1 downregulation in ALL cells treated with energy/ER-stressors appears not to be mediated through post-translational mechanisms.
Acknowledgments
Supported in part by the National Institute of Health (NIH) grant GM038545 (to M.A. Lehrman)
Footnotes
Contribution: GJL, JCB, TJL, GML conceived the study, participated in its design and coordination, and drafted the manuscript. GJL carried out the molecular cloning, generated NALM6 stable transfectants, performed transfection (nucleofection), shRNA experiments, RNA extraction, and real-time qRT-PCR. GJL, JD, JD, GML carried out cell proliferation and apoptosis assays, Western immunoblots, and performed the statistical analysis. NG, MAL performed FACE analysis. All authors have read and approved the final manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gelinas C, White E. BH3-only proteins in control: specificity regulates MCL-1 and BAK-mediated apoptosis. Genes Dev. 2005;19:1263–8. doi: 10.1101/gad.1326205. [DOI] [PubMed] [Google Scholar]
- 2.Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006;354:166–78. doi: 10.1056/NEJMra052603. [DOI] [PubMed] [Google Scholar]
- 3.Yung HW, Charnock-Jones DS, Burton GJ. Regulation of AKT phosphorylation at Ser473 and Thr308 by endoplasmic reticulum stress modulates substrate specificity in a severity dependent manner. PLoS One. 2011;6:e17894. doi: 10.1371/journal.pone.0017894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schonthal AH, Chen TC, Hofman FM, Louie SG, Petasis NA. Preclinical development of novel anti-glioma drugs targeting the endoplasmic reticulum stress response. Current pharmaceutical design. 2011;17:2428–38. doi: 10.2174/138161211797249242. [DOI] [PubMed] [Google Scholar]
- 5.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 6.Thomas LW, Lam C, Edwards SW. Mcl-1; the molecular regulation of protein function. FEBS Lett. 2010;584:2981–9. doi: 10.1016/j.febslet.2010.05.061. [DOI] [PubMed] [Google Scholar]
- 7.Yamani L, Latreille M, Larose L. Interaction of Nck1 and PERK phosphorylated at Y(5)(6)(1) negatively modulates PERK activity and PERK regulation of pancreatic beta-cell proinsulin content. Molecular biology of the cell. 2014;25:702–11. doi: 10.1091/mbc.E13-09-0511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barnes K, McIntosh E, Whetton AD, Daley GQ, Bentley J, Baldwin SA. Chronic myeloid leukaemia: an investigation into the role of Bcr-Abl-induced abnormalities in glucose transport regulation. Oncogene. 2005;24:3257–67. doi: 10.1038/sj.onc.1208461. [DOI] [PubMed] [Google Scholar]
- 9.Gottschalk S, Anderson N, Hainz C, Eckhardt SG, Serkova NJ. Imatinib (STI571)-mediated changes in glucose metabolism in human leukemia BCR-ABL-positive cells. Clin Cancer Res. 2004;10:6661–8. doi: 10.1158/1078-0432.CCR-04-0039. [DOI] [PubMed] [Google Scholar]
- 10.van Huizen R, Martindale JL, Gorospe M, Holbrook NJ. P58IPK, a novel endoplasmic reticulum stress-inducible protein and potential negative regulator of eIF2alpha signaling. J Biol Chem. 2003;278:15558–64. doi: 10.1074/jbc.M212074200. [DOI] [PubMed] [Google Scholar]
- 11.Bates SE, Robey RW, Piekarz RL. CCR 20th Anniversary Commentary: Expanding the Epigenetic Therapeutic Portfolio. Clin Cancer Res. 2015;21:2195–7. doi: 10.1158/1078-0432.CCR-14-2555. [DOI] [PubMed] [Google Scholar]
- 12.Aries IM, Hansen BR, Koch T, van den Dungen R, Evans WE, Pieters R, et al. The synergism of MCL1 and glycolysis on pediatric acute lymphoblastic leukemia cell survival and prednisolone resistance. Haematologica. 2013;98:1905–11. doi: 10.3324/haematol.2013.093823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC, Korsmeyer SJ. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature. 2003;426:671–6. doi: 10.1038/nature02067. [DOI] [PubMed] [Google Scholar]
- 14.Domina AM, Vrana JA, Gregory MA, Hann SR, Craig RW. MCL1 is phosphorylated in the PEST region and stabilized upon ERK activation in viable cells, and at additional sites with cytotoxic okadaic acid or taxol. Oncogene. 2004;23:5301–15. doi: 10.1038/sj.onc.1207692. [DOI] [PubMed] [Google Scholar]
- 15.Bates SE. Quit early, quit often. Clin Cancer Res. 2015;21:2212. doi: 10.1158/1078-0432.CCR-14-2749. [DOI] [PubMed] [Google Scholar]
- 16.Kurtoglu M, Gao N, Shang J, Maher JC, Lehrman MA, Wangpaichitr M, et al. Under normoxia, 2-deoxy-D-glucose elicits cell death in select tumor types not by inhibition of glycolysis but by interfering with N-linked glycosylation. Mol Cancer Ther. 2007;6:3049–58. doi: 10.1158/1535-7163.MCT-07-0310. [DOI] [PubMed] [Google Scholar]
- 17.DeSalvo J, Kuznetsov JN, Du J, Leclerc GM, Leclerc GJ, Lampidis TJ, et al. Inhibition of Akt potentiates 2-DG-induced apoptosis via downregulation of UPR in acute lymphoblastic leukemia. Mol Cancer Res. 2012;10:969–78. doi: 10.1158/1541-7786.MCR-12-0125. [DOI] [PubMed] [Google Scholar]
- 18.Kuznetsov JN, Leclerc GJ, Leclerc GM, Barredo JC. AMPK and Akt Determine Apoptotic Cell Death following Perturbations of One-Carbon Metabolism by Regulating ER Stress in Acute Lymphoblastic Leukemia. Mol Cancer Ther. 2011;10:437–47. doi: 10.1158/1535-7163.MCT-10-0777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wolf D, Wolf AM. CCR 20th Anniversary Commentary: From Regulatory T Cells to Checkpoint Monoclonal Antibodies-Immuno-oncology Advances Clinical Cancer Research. Clin Cancer Res. 2015;21:2657–9. doi: 10.1158/1078-0432.CCR-14-2558. [DOI] [PubMed] [Google Scholar]
- 20.Sledge GW, Pegram MD. “Vertical” Inhibition of HER2 Yields Horizontal Gains in the Clinic. Clin Cancer Res. 2015;21:2663–5. doi: 10.1158/1078-0432.CCR-14-3183. [DOI] [PubMed] [Google Scholar]
- 21.Cox AD, Der CJ, Philips MR. Targeting RAS Membrane Association: Back to the Future for Anti-RAS Drug Discovery? Clin Cancer Res. 2015;21:1819–27. doi: 10.1158/1078-0432.CCR-14-3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leclerc GJ, Sanderson C, Hunger S, Devidas M, Barredo JC. Folylpolyglutamate synthetase gene transcription is regulated by a multiprotein complex that binds the TEL-AML1 fusion in acute lymphoblastic leukemia. Leuk Res. 2010;34:1601–9. doi: 10.1016/j.leukres.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Datema R, Schwarz RT. Interference with glycosylation of glycoproteins. Inhibition of formation of lipid-linked oligosaccharides in vivo. Biochem J. 1979;184:113–23. doi: 10.1042/bj1840113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hieke S, Kleber M, Konig C, Engelhardt M, Schumacher M. Conditional Survival: A Useful Concept to Provide Information on How Prognosis Evolves over Time. Clin Cancer Res. 2015;21:1530–6. doi: 10.1158/1078-0432.CCR-14-2154. [DOI] [PubMed] [Google Scholar]
- 25.Hicklin DJ. CCR 20th Anniversary Commentary: In Search of Cetuximab’s First Indication-Combination Therapy with Irinotecan in Colorectal Cancer. Clin Cancer Res. 2015;21:1505–7. doi: 10.1158/1078-0432.CCR-14-2553. [DOI] [PubMed] [Google Scholar]
- 26.Zhong Q, Gao W, Du F, Wang X. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell. 2005;121:1085–95. doi: 10.1016/j.cell.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 27.Kharabi Masouleh B, Geng H, Hurtz C, Chan LN, Logan AC, Chang MS, et al. Mechanistic rationale for targeting the unfolded protein response in pre-B acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2014;111:E2219–28. doi: 10.1073/pnas.1400958111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pradelli LA, Beneteau M, Chauvin C, Jacquin MA, Marchetti S, Munoz-Pinedo C, et al. Glycolysis inhibition sensitizes tumor cells to death receptors-induced apoptosis by AMP kinase activation leading to Mcl-1 block in translation. Oncogene. 2010;29:1641–52. doi: 10.1038/onc.2009.448. [DOI] [PubMed] [Google Scholar]
- 29.Blank JL, Liu XJ, Cosmopoulos K, Bouck DC, Garcia K, Bernard H, et al. Novel DNA damage checkpoints mediating cell death induced by the NEDD8-activating enzyme inhibitor MLN4924. Cancer Res. 2013;73:225–34. doi: 10.1158/0008-5472.CAN-12-1729. [DOI] [PubMed] [Google Scholar]
- 30.Gingras AC, Kennedy SG, O’Leary MA, Sonenberg N, Hay N. 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt(PKB) signaling pathway. Genes Dev. 1998;12:502–13. doi: 10.1101/gad.12.4.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boren J, Cascante M, Marin S, Comin-Anduix B, Centelles JJ, Lim S, et al. Gleevec (STI571) influences metabolic enzyme activities and glucose carbon flow toward nucleic acid and fatty acid synthesis in myeloid tumor cells. J Biol Chem. 2001;276:37747–53. doi: 10.1074/jbc.M105796200. [DOI] [PubMed] [Google Scholar]
- 32.Serkova N, Boros LG. Detection of resistance to imatinib by metabolic profiling: clinical and drug development implications. American journal of pharmacogenomics: genomics-related research in drug development and clinical practice. 2005;5:293–302. doi: 10.2165/00129785-200505050-00002. [DOI] [PubMed] [Google Scholar]
- 33.Masaki A, Ishida T, Maeda Y, Suzuki S, Ito A, Takino H, et al. Prognostic Significance of Tryptophan Catabolism in Adult T-cell Leukemia/Lymphoma. Clin Cancer Res. 2015;21:2830–9. doi: 10.1158/1078-0432.CCR-14-2275. [DOI] [PubMed] [Google Scholar]
- 34.Vellinga TT, Borovski T, de Boer VC, Fatrai S, van Schelven S, Trumpi K, et al. SIRT1/PGC1alpha-Dependent Increase in Oxidative Phosphorylation Supports Chemotherapy Resistance of Colon Cancer. Clin Cancer Res. 2015;21:2870–9. doi: 10.1158/1078-0432.CCR-14-2290. [DOI] [PubMed] [Google Scholar]
- 35.Paoletti C, Li Y, Muniz MC, Kidwell KM, Aung K, Thomas DG, et al. Significance of Circulating Tumor Cells in Metastatic Triple-Negative Breast Cancer Patients within a Randomized, Phase II Trial: TBCRC 019. Clin Cancer Res. 2015;21:2771–9. doi: 10.1158/1078-0432.CCR-14-2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eisele SC, Gill CM, Shankar GM, Brastianos PK. PLEKHA5: A Key to Unlock the Blood-Brain Barrier? Clin Cancer Res. 2015;21:1978–80. doi: 10.1158/1078-0432.CCR-14-2604. [DOI] [PubMed] [Google Scholar]
- 37.Kang MH, Wang J, Makena MR, Lee JS, Paz N, Hall CP, et al. Activity of MM-398, nanoliposomal irinotecan (nal-IRI), in Ewing’s family tumor xenografts is associated with high exposure of tumor to drug and high SLFN11 expression. Clin Cancer Res. 2015;21:1139–50. doi: 10.1158/1078-0432.CCR-14-1882. [DOI] [PubMed] [Google Scholar]
- 38.Touat M, Ileana E, Postel-Vinay S, Andre F, Soria JC. Targeting FGFR Signaling in Cancer. Clin Cancer Res. 2015;21:2684–94. doi: 10.1158/1078-0432.CCR-14-2329. [DOI] [PubMed] [Google Scholar]
- 39.Jamal-Hanjani M, Quezada SA, Larkin J, Swanton C. Translational implications of tumor heterogeneity. Clin Cancer Res. 2015;21:1258–66. doi: 10.1158/1078-0432.CCR-14-1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pabla N, Sparreboom A. CCR 20th anniversary commentary: BMS-247550-microtubule stabilization as successful targeted therapy. Clin Cancer Res. 2015;21:1237–9. doi: 10.1158/1078-0432.CCR-14-2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paiva B, Puig N, Garcia-Sanz R, San Miguel JF. Is This the Time to Introduce Minimal Residual Disease in Multiple Myeloma Clinical Practice? Clin Cancer Res. 2015;21:2001–8. doi: 10.1158/1078-0432.CCR-14-2841. [DOI] [PubMed] [Google Scholar]
- 42.Hoffman-Luca CG, Ziazadeh D, McEachern D, Zhao Y, Sun W, Debussche L, et al. Elucidation of Acquired Resistance to Bcl-2 and MDM2 Inhibitors in Acute Leukemia In Vitro and In Vivo. Clin Cancer Res. 2015;21:2558–68. doi: 10.1158/1078-0432.CCR-14-2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lolkema MP, Bohets HH, Arkenau HT, Lampo A, Barale E, de Jonge MJ, et al. The c-Met Tyrosine Kinase Inhibitor JNJ-38877605 Causes Renal Toxicity through Species-Specific Insoluble Metabolite Formation. Clin Cancer Res. 2015;21:2297–304. doi: 10.1158/1078-0432.CCR-14-3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Steelman LS, Franklin RA, Abrams SL, Chappell W, Kempf CR, Basecke J, et al. Roles of the Ras/Raf/MEK/ERK pathway in leukemia therapy. Leukemia. 2011;25:1080–94. doi: 10.1038/leu.2011.66. [DOI] [PubMed] [Google Scholar]
- 45.Leclerc GM, Leclerc GJ, Kuznetsov JN, Barredo JC. A novel role for AMPK as a molecular switch regulating the activation of Akt and RAS signaling in acute lymphoblastic leukemia. Proceedings of the 102nd AACR Annual Meeting; 2011. p. Abstract # 605. [Google Scholar]
- 46.Abulwerdi F, Liao C, Liu M, Azmi AS, Aboukameel A, Mady AS, et al. A novel small-molecule inhibitor of mcl-1 blocks pancreatic cancer growth in vitro and in vivo. Mol Cancer Ther. 2014;13:565–75. doi: 10.1158/1535-7163.MCT-12-0767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun JG, Li H, Li X, Zeng X, Wu P, Fung KP, et al. Clitocine targets Mcl-1 to induce drug-resistant human cancer cell apoptosis in vitro and tumor growth inhibition in vivo. Apoptosis. 2014;19:871–82. doi: 10.1007/s10495-014-0969-0. [DOI] [PubMed] [Google Scholar]
- 48.Chen D, Frezza M, Schmitt S, Kanwar J, Dou QP. Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Current cancer drug targets. 2011;11:239–53. doi: 10.2174/156800911794519752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Raez LE, Papadopoulos K, Ricart AD, Chiorean EG, Dipaola RS, Stein MN, et al. A phase I dose-escalation trial of 2-deoxy-D-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2013;71:523–30. doi: 10.1007/s00280-012-2045-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Glucose analogues 2-DG and 2-FDG act as inhibitors of glycolysis and N-linked glycosylation. 2-DG and 2-FDG inhibit hexokinase, phosphoglucose isomerase (PGI), and mannose-1-phosphate guanylyltransferase (GDP-Man synthase). Due to the resemblance of 2-DG with mannose, it not only competes with mannose metabolism in this pathway but also incorporates fraudulently into dolichol-pyrophosphate (lipid)–linked oligosaccharides (LLOs), which are the precursors for N-linked glycosylation. 2-FDG is a more potent glycolysis inhibitor than does 2-DG. In contrast, 2-DG is more potent than 2-FDG in interfering with N-linked glycosylation. (▲, glucose; ●, mannose; and ■, N-acetyl-glucosamine).
Figure S2. Mcl-1 downregulation sensitized CML cells to 2-DG. K562 cells were treated with the PI3K inhibitor LY294002 (25 mM) and 2-DG (10 mM) for 72 h and assayed for cell death using the ViCell XR Cell system Data represents the mean ± SEM (n=3); * denotes p<0.001. Right insert shows Western blot analysis of Mcl-1 expression in K562 cells treated with the PI3K inhibitor LY294002 (25 mM and 50 mM) ± 2-DG (10 mM) for 24 h. β-actin was used as a loading control.
Figure S3. Mcl-1 expression increases NALM6 resistance to 2-DG. Mcl-1 overexpression alleviates 2-DG cytotoxicity in NALM6 cells. NALM6 cells were transfected with either pCDNA3.1 or pCDNA3.1-hMCL-1 vector (addgene # 25375) [21], and 24 h post transfection, cells were treated with 2-DG (4 mM) for 72 h and assayed for cell death using the ViCell XR Cell system. Data represents the mean ± SEM (n=3); * denotes p<0.001.
Figure S4. Mcl-1 downregulation is independent of the PI3K/GSK-3β pathway in BCR-ABL+/− ALL cells treated with 2-DG. Levels of p-Akt (S473), p-GSK-3β (S9), GSK-3β and Mcl-1 protein expression in BCR-ABL+ (SUPB15 and TOM1) and control ALL cells (NALM6) treated with 2-DG [4 mM] for 24 h. β-actin was used as a loading control.
Figure S5. Mcl-1 downregulation in BCR-ABL+ and − ALL cells treated with energy/ER-stress inducers is not due to transcriptional mechanism. To explore other potential mechanisms by which Mcl-1 is downregulated in BCR-ABL+ and − ALL cells treated with energy/ER-stressing agents, we analyzed the level of Mcl-1 mRNA expression in NALM6, SUPB15, and NALM6/BCR-ABL cells treated with or without 2-DG (4 mM) for 24 h. Mcl-1 mRNA expression was detected by real-time qRT-PCR and normalized to GAPDH mRNA levels. Each experiment was performed at least two times in tetraplicate. Data represents the mean ± SEM (n=4). No significant differences were detected in BCR-ABL+ or BCR-ABL− ALL cells treated with 2-DG compared to untreated controls, indicating that 2-DG-induced Mcl-1 downregulation is not mediated by transcriptional mechanisms.
Figure S6. Mcl-1 downregulation in BCR-ABL+ and − ALL cells treated with energy/ER-stress inducers is not due to posttranslational modification mechanisms. (A) To rule out that the decrease in Mcl-1 expression was due to caspase dependent cleavage during apoptosis, we examined the level of Mcl-1 protein expression in SUPB15 and NALM6 cells treated with or without 2-DG (4 mM) in presence or absence of the pan-caspase inhibitor qVD-OPH (20 μM) for 24 h. qVD-OPH was added 1 hr prior treatment. In NALM6 cells, staurosporin (STP, 3 μM for 4 h) was used as positive control on Mcl-1 cleavage. We found that qVD-OPH did not prevent Mcl-1 downregulation in ALL cells treated with 2-DG but could inhibit Mcl-1 cleavage in NALM6 cells treated with staurosporin (STP)-induced caspase activation, indicating that 2-DG-induced Mcl-1 downregulation is not mediated by caspase cleavage activity during apoptosis. (B) To investigate whether Mcl-1 was regulated by ubiquitin-mediated proteasomal degradation in ALL, we determined the level of Mcl-1 protein expression in SUPB15 and NALM6 cells treated with or without 2-DG (4 mM) in presence or absence of the proteasome inhibitor Bortezomib (BTZ, 5 nM) for 24 h. β-actin was used as a loading control. We found that the decreased levels of Mcl-1 expression in SUPB15 and NALM6 cells treated with 2-DG were unchanged in the presence of bortezomib, ruling out an ubiquitin-mediated proteasomal degradation mechanism to downregulate Mcl-1 in our experimental system. Taken together, our results indicate that Mcl-1 downregulation in ALL cells treated with energy/ER-stressors appears not to be mediated through post-translational mechanisms.