

Summary
Dementia is the cardinal feature of Alzheimer's disease (AD), yet the clinical symptoms of this disorder also include a marked loss of motor function. Tau abnormal hyperphosphorylation and malfunction are well‐established key events in AD neuropathology but the impact of the loss of normal Tau function in neuronal degeneration and subsequent behavioral deficits is still debated. While Tau reduction has been increasingly suggested as therapeutic strategy against neurodegeneration, particularly in AD, there is controversial evidence about whether loss of Tau progressively impacts on motor function arguing about damage of CNS motor components. Using a variety of motor‐related tests, we herein provide evidence of an age‐dependent motor impairment in Tau−/− animals that is accompanied by ultrastructural and functional impairments of the efferent fibers that convey motor‐related information. Specifically, we show that the sciatic nerve of old (17–22‐months) Tau−/− mice displays increased degenerating myelinated fibers and diminished conduction properties, as compared to age‐matched wild‐type (Tau+/+) littermates and younger (4–6 months) Tau−/− and Tau+/+ mice. In addition, the sciatic nerves of Tau−/− mice exhibit a progressive hypomyelination (assessed by g‐ratio) specifically affecting large‐diameter, motor‐related axons in old animals. These findings suggest that loss of Tau protein may progressively impact on peripheral motor system.
Keywords: Tau, motor deficits, peripheral nerve, myelination, nerve conduction, knockout
Introduction
Clinical presentation of Alzheimer's disease (AD) is complex and extends well beyond the cognitive impairments that characterize this disorder (Duker et al., 2012). Alterations in facial expression, gait and posture, and manifestations of rigidity, bradykinesia, and tremor are found in late AD stages although mounting evidence suggests that motor problems emerge long before any recognizable sign of AD (Wilson et al., 2000; Scarmeas et al., 2004; Buchman & Bennett, 2011). Tau abnormal hyperphosphorylation and subsequent malfunction are postulated as crucial mechanisms in AD neuronal dysfunction where hyperphosphorylated and/or aggregated (insoluble) forms of Tau exhibit neurodegenerative action(s) that also interfere with normal Tau, sequestering and reducing soluble Tau forms (Ksiezak‐Reding et al., 1988; Zhukareva et al., 2003). These lesions are mainly found at different areas of CNS such as hippocampus and cortex although some studies also demonstrate the presence of Tau deficits in peripheral nervous system (PNS; e.g., autonomic ganglia and sciatic nerves Bohl et al., 1997; Holzer et al., 1999). While evidence suggests that Tau reduction can block AD pathology progression (Roberson et al., 2007), indicating that Tau‐targeted strategies might be of interest for AD therapy (Gotz et al., 2012), the safety and/or potential side effect(s) of these approaches is not well studied.
Tau genetic deletion seems to be well tolerated by young animals as the majority of Tau−/− models do not exhibit behavioral or microtubule alterations (Dawson et al., 2001; reviewed by Ke et al., 2012). However, chronic loss of Tau has been described to result in subtle or mild motor deficits in increasingly aged animals (reviewed by Gotz et al., 2013). Indeed, one study revealed loss of substantia nigra (SN) dopaminergic neurons in middle‐aged Tau−/− animals (Lei et al., 2012), while in another study, using aged Tau−/− of the same strain, similar motor deficits were shown but in a SN‐/dopamine‐independent manner (Morris et al., 2013), raising uncertainty on the underlying mechanisms of the motor deficits in Tau−/− animals. Surprisingly, the involvement of the PNS has not been assessed in any of the previous studies given the fact that Tau reduction is found in peripheral nerves (e.g., sciatic nerve) of patients with AD (Holzer et al., 1999). This study aimed therefore to monitor the impact of chronic loss of Tau protein in PNS efferents, primary compartment of motor circuitry, and motor performance using a battery of behavioral tests analyzing motor function in both young (4–6 months) and old (17–22 months) Tau−/− mice combined with a systematic morphofunctional analysis of their sciatic nerve.
Results
Chronic lack of Tau results in motor deficits in old animals
To clarify the discrepancies of previous studies regarding the age‐dependent motor deficits in Tau−/− animals (cf. Lei et al., 2012 and Morris et al., 2013), we performed a battery of motor‐related behavioral tests in adult (4–6 months old) and old (17–22 months old) male Tau−/− and Tau+/+ animals. Motor function/coordination and locomotor activity were assessed in the rotarod and open‐field tests, respectively. As shown in Fig. 1A, the accelerating protocol of rotarod test showed that old Tau−/− animals presented reduced latency to fall, indicating an impairment in motor coordination. Two‐way ANOVA rendered an overall genotype effect [F 1,37 = 4.050, p = 0.021], where old Tau−/− exhibit a motor impairment when compared with old Tau+/+ (p = 0.019). In the open‐field arena, we found a Genotype x Aging interaction of total area travelled by the animals [F 1,31 = 4.339, p = 0.045] (Fig. 1B). Further analysis revealed that old Tau−/− travelled significantly less than their age‐matched Tau+/+ (p = 0.005) as well as than adult Tau−/− animals (p = 0.019). As potential body weight differences among groups could affect the assessment of the above motor‐related parameters, we monitor body weight of these animals (adult Tau+/+: 27.876 ± 0.95; adult Tau−/−: 27.879 ± 0.53; old Tau+/+: 34.943 ± 0.60; old Tau−/−: 36.215 ± 1.67) finding no overall Genotype ([F 1,37 = 0.4187, p = 0.52]), but a significant Aging overall effect ([F 1,37 = 61.15, p < 0.0001]). Further analysis showed that both old Tau+/+ and Tau−/− animals exhibit higher body weight than their younger counterparts (p < 0.0001 for both genotypes). For monitoring muscle strength, we use wire‐hanging test where a Genotype x Aging interaction [F 1,31 = 5.768, p = 0.022] was found; old Tau−/− present significantly less hanging time than old Tau+/+ (P = 0.008) as well as when compared with adult Tau−/− (p = 0.0001) suggesting compromised muscle strength (Fig. 1C). Furthermore, monitoring separately hindlimb and forelimb performance by hindlimb tonus and forelimb grid strength tests, we found a Genotype x Aging interaction (for tonus F 1,38 = 11.97, p = 0.001; for strength F 1,38 = 12.14, p = 0.001) represented by diminished hindlimb tonus resistance and reduced forelimb strength in old Tau−/− animals when compared to old Tau+/+ (ptonus = 0.0003 and pstrength = 0.004, respectively; Fig. 1D,E); note that this difference was not found in adult animals (ptonus = 0.98 and pstrength = 0.80, respectively) in line with the findings of the above tests used in this study. In addition, no significant changes among groups were found in hindlimb clasping score (Fig. 1F). Moreover, for extra monitoring of motor coordination and gait, we performed footprinting‐based analysis showing a Genotype x Aging interaction in the stride length of animals (F 1,23 = 4.69, p = 0.04) where, in contrast to adult animals, old Tau−/− presented a decreased stride length when compared to old Tau+/+ (p = 0.03) (Fig. 1G). In addition, we found an Aging main effect on forepaw base (F 1,23 = 13.01, p = 0.001) where old Tau−/− animals presented an increase in this parameter compared to adult Tau−/− animals (p = 0.04) (Fig. 1H). No significant differences were found in other parameters of gait analysis such as hindpaw base and forepaw–hindpaw overlap.
Figure 1.
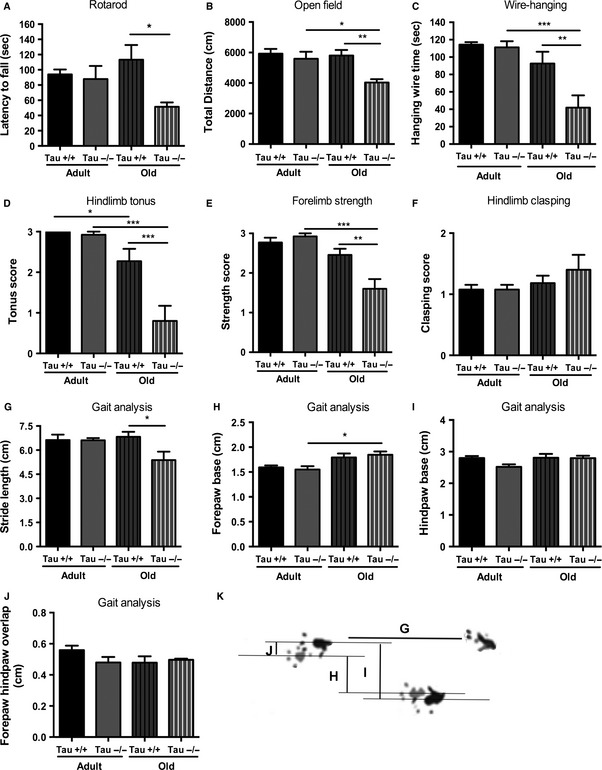
Impaired motor behavior of old Tau−/− animals. Behavioral screening of motor functions revealed that lack of Tau protein led to an age‐dependent motor dysfunction. (A) Rotarod test showed that old, but not adult, Tau−/− animals exhibited reduced latency to fall when compared with Tau+/+ reflecting motor impairment. (B) Total distance travelled in open field was reduced in old Tau−/− when compared with old Tau+/+ and adult Tau−/− animals indicating reduced locomotor activity. (C) Wire‐hanging test assessments showed that old Tau−/− animals differed from old Tau+/+ and adult Tau−/− presenting significantly less hanging time. D‐E) Hindlimb tonus resistance (D) and forelimb strength (E) were severely reduced in old Tau−/− animals compared with old and adult Tau+/+. (F) No changes in hindlimb clasping score between animals of both ages and genotypes. (G–J) Footprinting‐based gait analysis showed that, in contrast to adult animals, old Tau−/− present reduced stride length when compared to old Tau+/+ (G), while no other changes were found between animals of two genotypes. (K) Representative image of animal footprints which were used to calculate the different gait parameters, for example, stride length *(G), forepaw base (H), hindpaw base (I), and forepaw/hindpaw overlap (J). All numeric data are presented as mean ± SEM; *p < 0.05; **p < 0.01; ***p < 0.001.
Tau ablation alters sciatic nerve structure and function by aging
As Tau−/− mice are shown to exhibit motor deficits, it was of interest to monitor the impact of Tau ablation on the peripheral component of motor circuit; for that purpose, we studied the sciatic nerve, in the same mouse line and background (C57BL/6; Dawson et al., 2001) used in previous studies describing motor deficits after Tau ablation (Morris et al., 2013; Lei et al., 2014). It is important to note that the sciatic nerve is known to exhibit important morphological and functional changes with aging (Melcangi et al., 2000). Thus, we monitored myelinated (A‐fibers) density in p‐phenylene diamine (PPD)‐stained sections. In agreement with previous studies (Ceballos et al., 1999), two‐way ANOVA showed an aging effect [F 1,16 = 11.71, p = 0.0038] with aged animals exhibiting reduced density of myelinated fibers independently of genotype (p < 0.05 for both genotypes) (Fig. 2A,B). Furthermore, electron micrographs of the sciatic nerves revealed, in addition to normal myelinated axons (Fig. 2Ci), the presence of dysmorphic myelin sheaths as shown in Fig. 2Cii–iv. Thus, we also evaluated the percentage of myelinated fibers that exhibit the above abnormalities in their myelin sheath finding an overall aging effect [F 1,16 = 7.41, p = 0.015] and an overall genotype effect [F 1,16 = 5.58, p = 0.031]. Additional analysis showed a significant increase in the percentage of degenerating myelinated fibers in old Tau−/− animals when compared with old Tau+/+ animals (p = 0.049) and adult Tau−/− (p = 0.0289) (Fig. 2D). In the next step of our analysis, we compared myelin sheath thickness of fibers of sciatic nerves by electron microscopy determining the g‐ratio, an index of myelination independent of axonal diameter (Michailov et al., 2004). A total of 2885 axons were sampled and classified according to axon diameter. Tau−/− axons exhibited increased g‐ratio indicating reduced myelination while two‐way ANOVA per category exhibited an overall Genotype effect in all categories [F 1,270 = 23.891, p < 0.001 (0–2 μm); F 1,1390 = 99.885, p < 0.001 (2–4 μm); F 1,705 = 132.891, p < 0.001 (4–6 μm); F 1,372 = 39.379, p < 0.001 (6–8 μm); F 1,143 = 9.060, p = 0.02 (8–10 μm)] and an age effect [F 1,1390 = 31.672 (2–4 μm); F 1,705 = 74.302 (4–6 μm); F 1,372 = 100.001 (6–8 μm); F 1,143 = 30.694 (8–10 μm); p < 0.001] in all categories except 0–2 μm class of axons (F 1,270 = 3.256; p = 0.072) (Fig. 2E,F). Large‐diameter (8–10 μm) axons g‐ratio (motor‐related) was different between old (p < 0.001), but not adult, Tau+/+ and Tau−/−. This g‐ratio variation in large‐diameter axons between Tau−/− and Tau+/+ of the two ages is also depicted in the regression lines of g‐ratio growth in Tau−/− (grey) and Tau+/+ (black) of adult and old animals (inserted micrographs in Fig. 2E and 2F, respectively), where the regression lines for adult, but not old, animals are converging at the large‐diameter axons indicating no difference between g‐ratios of Tau−/− and Tau+/+ of this fiber category. The above ultrastructural hypomyelination findings in sciatic nerves of Tau−/− animals were further confirmed by reduced protein levels of myelin basic protein (MBP) (Fig. 2G).
Figure 2.
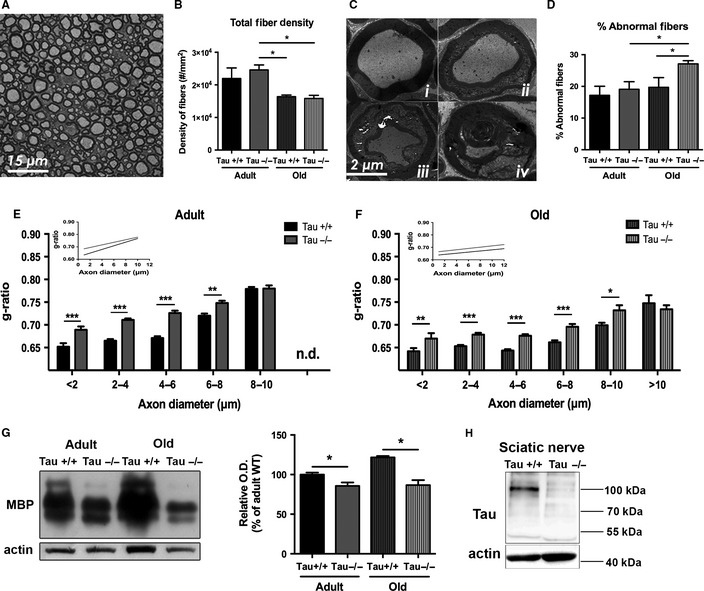
Ultrastructural analysis of Tau−/− and Tau+/+ sciatic nerves. (A, B) Representative light microscopy photograph of ppd staining of sciatic nerve (A) and quantification of fibers density (B) showing an age‐dependent reduction in both Tau−/− and Tau+/+. (C) TEM analysis images of sciatic nerve showing normal (i) and degenerating (ii, iii) myelinated fibers with obvious myelin anomalies while severely damaged axons (iv) were very rarely found in all groups. (D) Old Tau−/− animals present a significant increase in percentage of degenerating fibers when compared with old Tau+/+ animals. (E, F) Detailed evaluation of g‐ratio [axon diameter/(axon diameter + myelin thickness)] per axon diameter category in adult (E) and old (F) animals. There is an age‐dependent g‐ratio reduction (hypomyelination) in Tau−/− vs. Tau+/+ starting from axons of small and middle diameter (nonmotor related) in adult animals (E) and extending to large‐diameter (8–10 μm), motor‐related axons in old Tau−/− (F). Specifically, compared to adult Tau+/+ ones, adult Tau−/− animals exhibited increased g‐ratio in all fiber categories except that of 8–10 μm, while in old Tau−/−, this category g‐ratio was also affected (E). Insets represent regression lines of g‐ratio growth in Tau−/− (red) and Tau+/+ (black) of adult (D) and old (E) animals where the regression lines for adult, but not old, animals are converging at the large‐diameter axons, indicating no difference between g‐ratio of Tau−/− and Tau+/+ of this fiber category. G‐H) Western blot analysis showed reduced myelin basic protein (MBP) levels in Tau−/− sciatic nerves (G) in line with the g‐ratio analysis as well as absence of the characteristic high molecular weight Tau (HMW‐Tau; 110 kDa) (H) in sciatic nerves of Tau−/− animals. Data are presented as mean ± SEM; *p < 0.05; **p < 0.01; ***p < 0.001.
Next, we monitored in vivo compound muscle (M) and reflex (H) nerve action potentials in hindpaw of anesthetized animals (Fig. 3A–E). As shown in Fig. 3B,C, there were no differences between amplitudes or total response area of direct muscle action potentials—M‐wave—between Tau+/+ and Tau−/− on both ages. Regarding the H‐waves which represent the reflex response that travels via the sciatic nerve (sensitive to lidocaine local sciatic application; Fig. 3A), two‐way ANOVA showed an Aging main effect [F 1,17 = 6.701, p = 0.018] on the amplitude but no changes between Tau−/− and Tau+/+ animals of both ages (Fig. 3D). Moreover, assessment of the total response area exhibited a significant reduction in old Tau−/− animals when compared to old Tau+/+ (p = 0.02; Fig. 3E); note that this difference was not found in adult animals (p = 0.64). We also found a Genotype x Aging interaction effect [F 1,17 = 9.40, p = 0.007] as well as an aging main effect [F 1,17 = 49.78, p < 0.0001]. Based on the morphological alterations of sciatic nerve described above, we next monitored nerve function by ex vivo recordings in isolated sciatic nerves, as previously described (Pinto et al., 2008) (Fig. 3F). Two‐way ANOVA revealed no differences in conduction velocity of A‐fibers among both ages and genotypes (Fig. 3G). However, assessment of the total response area revealed a significant Genotype x Aging interaction [F 1,31 = 6.715, P = 0.0144]. Particularly, we observed that sciatic nerves from old Tau−/− animals presented a decreased conduction capacity when compared with old Tau+/+ animals (P = 0.03) (Fig. 3H), indicating diminished overall response ability for old Tau−/− sciatic nerves; the same was not valid for adult animals again providing further evidence about the age‐dependent impact of Tau loss on sciatic nerve.
Figure 3.
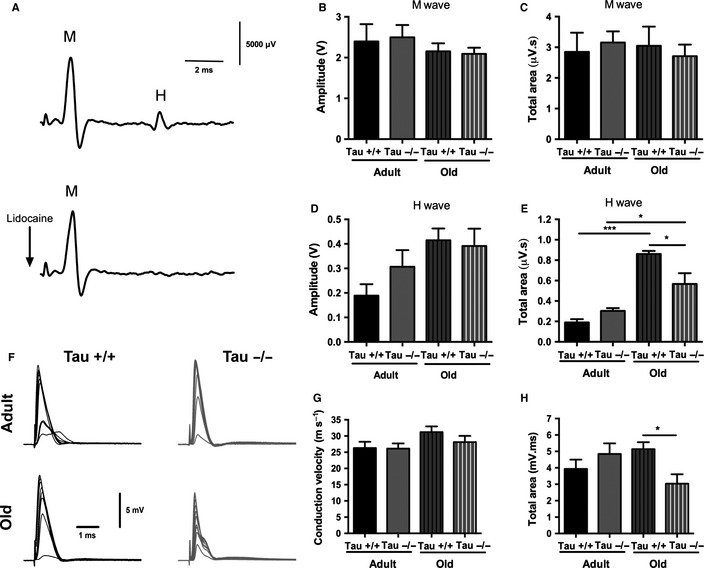
Tau ablation results in age‐dependent sciatic nerve malfunction. (A) Representative traces of in vivo electrophysiological recordings of compound muscle (M) and reflex (H) nerve action potentials (upper panel); lidocaine injection locally at the sciatic resulted in loss of H‐wave without affecting the muscle M‐wave (lower panel). (B, C) No differences in both amplitude (B) and total response area (C) of M‐waves between animals of both genotypes and ages. (D) Age‐dependent increase in amplitude of H‐waves (Aging main effect, see also results), but no difference between Tau−/− and Tau+/+ animals. (E) Old Tau−/− exhibited significantly reduced total response area compared to old Tau+/+ animals, while this difference was not found in adult animals. In addition, there was an age‐dependent difference in both Tau+/+ and Tau−/− animals. (F) Representative traces of current‐clamp recordings of sciatic nerve compound action potentials in adult and old Tau+/+ and Tau−/− mice. Sciatic nerves received repeated stimuli with increasing amplitudes pulses until saturation. Stimulation artifacts were truncated (conduction distance: adult Tau+/+: 5.5 mm; adult Tau−/−: 9.6 mm; old Tau+/+: 8.0 mm; old Tau−/−: 9.8 mm); traces are averages of five consecutive recordings. (G, H) While no differences in conduction velocity (m s−1) between Tau+/+ and Tau−/− were found (G), total area under the curve (mV.ms) of old Tau−/− was significantly reduced compared to old Tau+/+ animals indicating sciatic nerve malfunction (H). Data presented as mean ± SEM; p < 0.05.
Discussion
Tau is a soluble cytoskeletal protein that is expressed mainly in neurons and in glial cells, of both CNS and PNS; it is implicated in various cellular processes such as microtubule stabilization, axonal maintenance, and transport. Tau malfunction is also causally related to AD pathology in where aberrant, hyperphosphorylated and/or aggregated, forms of Tau are found in the CNS and in the PNS (Holzer et al., 1999). Accordingly, recent evidence suggests Tau protein as the final executor of AD neuronal dysfunction (Roberson et al., 2007; Ittner & Gotz, 2011) and, therefore, anti‐Tau therapies emerged as potential strategies against AD (Frost et al., 2015); however, their safety or potential risks have not been defined yet.
Evidence from different Tau−/− models suggests that absence of Tau is well tolerated by young/adult animals probably due to the occurrence of compensatory mechanisms (e.g., increase in MAP1A, another microtubule‐associated protein) (Dawson et al., 2001; Ma et al., 2014). In contrast, aged Tau−/− seem to exhibit mild motor deficits albeit the effect of Tau depletion on motor performance by aging remains controversial and unclear. A study from Ikegami et al. (2000) reports signs of muscle weakness (tested by wire‐hanging) and reduced balance coordination in 2‐month‐old Tau−/−, but Morris et al. (2011) showed normal motor function of young (3–5 months old) and adult animals of another Tau−/− mouse line in a variety of motor tests (B6 background; Dawson et al. (2001)) in agreement with this study findings. Moreover, the same team found mild motor deficits in middle‐aged (12–15 months old) Tau−/− animals using rotarod and pole tests but, unexpectedly, these motor deficits were not clearly evident in old (21–22 months old) Tau−/− (Morris et al., 2013). In contrast, we observed clear motor deficits in old (17–22 months old) Tau−/− using the rotarod, wire‐hanging, and open‐field tests and these deficits were not correlated with body weight. In addition, our study offers additional evidence of motor dysfunction showing that, in line with our wire‐hanging data, old Tau−/− animals also exhibit reduced forelimb (grip) strength and diminished hindlimb tonus resistance compared to old wild‐types but they exhibit no deficits in gross aspects of motor function/balance as assessed by hindlimb clasping tests (see Fig. 1) and negative geotaxis (data not shown). Interestingly, reduced motor strength and coordination was also shown in 13‐ to 17‐month‐old animals lacking 4R‐Tau, suggesting the potential role of 4RTau in the age‐dependent development of motor deficits (Gumucio et al., 2013).
The mechanisms underlying the impaired motor behavior in the absence of Tau in aged individuals are not entirely understood with contradictory findings being reported. Lei et al. (2012), for instance, reported an age‐related loss of SN dopaminergic neurons in middle‐aged Tau−/− animals. In contrast, a recent study on the same Tau−/− line failed to observe any major dopaminergic loss in different CNS motor components of old Tau−/− mice (Morris et al., 2013). While both studies showed motor deficits, Lei et al. (2012) showed nigrostriatal loss in middle‐aged Tau−/− mice of C57BL/6/SV129 background, while our study and the study of Morris et al. (2013) found no nigrostriatal neuronal loss in old mice with C57BL/6 background (data not shown). Furthermore, in a later study, Lei et al. (2014) showed that motor deficits of old Tau−/− animals do not depend on background or have a sex‐dependent profile. In line with it, despite the fact that our study has used male animals and Morris study (2013) used a mixed (male and female) cohort, both studies exhibit motor deficits providing further support of no gender influence in the old Tau−/− motor deficits.
While a number of CNS areas have been implicated in the emergence of AD‐associated motor deficits, little attention has been devoted to PNS primary efferents. Previous evidence suggests that the sciatic nerves of patients with AD, but not of age‐matched healthy individuals, exhibit reduced Tau levels (Holzer et al., 1999). In our study, we demonstrate that chronic loss of Tau protein results in sciatic nerve morphofunctional deficits which includes increased percentage of degenerating fibers, hypomyelination of large‐diameter, motor‐related fibers, and diminished conduction properties in old, but not young, Tau−/− sciatic nerve. While other mechanisms cannot be excluded, the aforementioned sciatic nerve deficits may critically contribute to motor deficits found in old Tau−/− as fine‐tuning of myelin sheath thickness and formation is important for maintenance and proper function of motor fibers. In addition, while no amplitude differences in muscle action potentials of Tau−/− were found, further studies are needed to clarify the impact of loss of Tau on neuromuscular junction by aging as Tau‐related pathology in motor neurons has been shown to have neuromuscular junction malfunction and motor deficits in AD Tg models (Zhang et al., 2005; Ubhi et al., 2007).
Despite the fact that axon–Schwann cell interactions are critical for nerve function and maintenance, the underlying mechanisms of maintaining normal nerve and Schwann cell structure and function remain poorly understood. Tau is expressed in both CNS and PNS, but the low molecular weight isoforms of Tau protein that are expressed in adult CNS differ from high molecular weight (HMW) Tau isoforms mainly found in PNS (e.g., sciatic nerves) but also in optical nerves (Sato‐Yoshitake et al., 1989; Georgieff et al., 1991; Nothias et al., 1995). The expression of HMW tau isoforms may confer increased stabilization and spacing of microtubules (Frappier et al., 1994; Boyne et al., 1995) but yet, our knowledge about Tau function in PNS is very limited. While previous cell‐based evidence suggests the involvement of Tau in myelination of oligodendrocytes, the current study provides ultrastructural and molecular support of Tau role in myelination of PNS. Neuronal axon–Schwann cell interaction(s) are essential for myelination, and myelin maintenance is a dynamic process tightly controlled by axon‐dependent transcription factors such as Krox20 and Oct6 (Murphy et al., 1996; Decker et al., 2006). In line with previous studies demonstrating an absence of axonal deficits in neurons of Tau−/− animals (Yuan et al., 2008; Vossel et al., 2010), there were no differences in mRNA levels of transcription factors that critically regulate myelination process and/or maintenance, for example, Krox20 and Oct6 (data not shown), suggesting no gross changes in myelin gene regulation of Tau−/− Schwann cells. Furthermore, supporting the involvement of Tau and other cytoskeletal elements in myelination process, previous evidence suggests that Tau strongly colocalizes with MBP in distal tips of oligodendrocytes (LoPresti et al., 1995; Muller et al., 1997), suggesting that transportation and/or local MBP translation may require microtubule cytoskeleton and might be controlled by Tau–Fyn interaction (Klein et al., 2002). As recent evidence suggests that Tau is responsible to locate Fyn in spines and Fyn has a well‐described role in myelination of both CNS and PNS (Kramer‐Albers & White, 2011), the above Tau–Fyn–MBP complex could be disrupted in the absence of Tau protein, affecting myelination signaling and process. Indeed, Taiep myelin‐mutant animals exhibit irregular microtubule polarity and display abnormal Tau accumulation and intracellular accumulation of myelin proteins in oligodendrocytes (Song et al., 2001), while PNS myelin‐deficient mice also exhibited reduction in Tau and other cytoskeletal protein in their sciatic nerves (Kirkpatrick & Brady, 1994). The above findings suggest that the absence of Tau may impact on Schwann cells’ myelination process/signaling, opening a novel window for further research on role of Tau protein in PNS. Furthermore, in light of the suggested therapeutic potential of Tau reduction against AD, a better understanding of the impact of Tau ablation in both CNS and PNS is of great importance. Our findings highlight the morphological and functional implication of peripheral nerves induced by Tau loss in the precipitation of motor deficits with increasing aging and should be taken into account in the development of future AD therapies.
Experimental procedures
Animals
In this study, 4‐ to 6‐ and 17‐ to 22‐month‐old male Tau+/+ and Tau−/− (Dawson et al. (2001); C57BL/6 background) were used. Mice were housed 4–5 animals per cage under standard environmental conditions with ad libitum access to food and water. All experimental procedures were approved by the local ethical committee and national authority for animal experimentation and were in accordance with the guidelines for the care and handling of laboratory animals, as described in the Directive 2010/63/EU.
Behavioral tests
Rotarod, wire‐hanging, and open‐field tests were performed. Animals were acclimated to the testing conditions for 60 min before testing. For rotarod test (TSE systems, Bad Homburg, Germany), mice were first trained a constant speed (15 rpm) for 3 days (4 trials per day; 60‐s max trial time; 10‐min resting interval among trials). At fourth day, mice (N = 9–11 for each genotype) were tested on an accelerating rod (4–40 rpm, 5 min; four trials); in this case, latency to fall was automatically recorded. For wire‐hanging test, animals were placed on a standard wire cage grid, which was then inverted and at 30 cm height from the table surface. The latency to animal fall was manually recorded (two trials; maximum time of each trial: 120 s). The open field was performed in a square arena (43.2 × 43.2 cm) surrounded by tall perspex walls (Med Associates Inc., St. Albans, VT, USA). Mice were placed in the center of the arena allowed to explore during 10 min. Infrared beams were used to automatically register animals’ movements. We next monitored forelimb strength, hindlimb tonus, hindlimb clasping, negative geotaxis, and gait analysis as previously described (Silva‐Fernandes et al., 2014). For all tests, scoring was carried out manually by an experimenter blind to the genotype and age of the animals. Briefly, for the assessment of forelimb (grid) strength, animal was positioned on a metal grid and gently pulled off by its tail (hindlimbs were suspended slightly above the grid), while the animal was gripping a wire using its forelimbs. A scale of four different scores was used by the experimenter evaluating forelimb strength of the animal: 0 (no strength), 1 (light/semi‐effective), 2 (moderate/effective), and 3 (active/effective). Hindlimb tonus was tested by pressing animals’ hindlimb paw (by experimenter index finger) evaluating muscular resistance and classifying into four different categories: 0 (no resistance), 1 (slight resistance), 2 (moderate resistance), and 3 (pronounced resistance). Assessment of hindlimb clasping was performed suspending animals by the tail during 30 s. The hindlimbs were observed, and each mouse was given a score for each trial. Hindlimb clasping was rated based on two classes: 1 (hindlimb splayed outward and away of the abdomen) and 2 (hindlimbs retracted inward, toward the abdomen for at least more than 50% of the testing time). Next, we also performed gait analysis based on footprinting. Briefly, the hind‐ and forepaws of the mice were coated with black and red nontoxic paints, respectively. The animals were allowed to walk along 100‐cm‐long × 4.2‐cm‐width × 10‐cm‐height corridor, with ordinary white paper on the floor of the runway for each run. Each animal was tested to achieve one valid trial. The footprint patterns were analyzed manually for four step parameters (measured in cm): the front‐ and hind‐base width, the footstep uniformity, and the stride length. For the analysis of each step parameter, three values were used based on three consecutive steps.
Sciatic nerve ultrastructure analysis
Under deep anesthesia [ketamine hydrochloride (75 mg kg−1) and medetomidine (1 mg kg−1)], sciatic nerves were collected and immediately fixed in 4% glutaraldehyde (in 0.1 m sodium cacodylate buffer, pH 7.4) for 7 days and room temperature and then postfixed in 1% OsO4, dehydrated. Finally, the tissue was embedded in epon resin (Electron Microscopy Sciences) and sectioned according to the objective (see below). One‐micrometer transverse sections covering the complete cross‐sectional nerve area were stained with 1% p‐phenylene diamine and mounted on Entellan (Merk). Light microscope (Olympus DP70, Hamburg, Germany) images were then mounted on Photoshop and used for manual calculation of number and density of myelinated fibers per transverse section (4–5 animals per group). For the assessment of degenerating fibers, 16 nonoverlapping TEM photographs (3000×) of counterstained ultrathin sections (60 nm) were used (obtained by JEM‐1400 TEM). The same TEM images were also used for g‐ratio [axon diameter/(axon diameter + myelin thickness)] calculation; more than one hundred fibers were measured per animal. Morphometric analysis was performed by an experimenter blind to the samples provenience.
Ex vivo measurement of compound action potentials
Acutely isolated sciatic nerves from each group (6–8 animals per group) were used for the assessment of A‐ and C‐fibers conduction velocity and compound action potentials as previously described (Pinto et al., 2008). Briefly, sciatic nerves were dissected and cleaned from the connective tissue sheath in artificial cerebrospinal fluid. Compound action potentials recordings were made with a Multiclamp 700B amplifier in CC mode and digitized with the Digidata 1440a digitizer using PCLAMP 10 software (Axon Instruments, Sunnyvale, CA, USA). Signals were low‐pass‐filtered at an effective corner frequency of 16 KHz and sampled at 50 KHz. Fibers were stimulated at 60 μs, and conduction velocities were calculated for the first compound action potentials peak; total areas were calculated using CLAMPFIT software (Axon Instruments). Electrophysiological recordings and analyses were performed by an experimenter blind to the provenience of the tissue.
In vivo electrophysiological measurements
The in vivo approach allowed the acquisition of electroneuromyographic recordings in a noninvasive manner as described previously (Petit et al., 2014). Briefly, 5–6 mice per group were anesthetized with a mixture of ketamine (75 mg kg−1; Imalgene®, Merial, France) and medetomidine (1 mg kg−1; Dormitor®, Pfizer, New York, NY, USA) i.p. administered. Temperature was monitored and maintained at 37 °C by a homeothermic blanket (Stoelting, Dublin, Ireland). A concentric bipolar platinum/iridium–stainless steel stimulation electrode (WPI, Worcester, MA, USA) was used to deliver 0.1 ms square pulses to the tibial nerve at the ankle, through a small skin incision. The ground stainless steel electrode was inserted at the base of the tail. Compound muscle action potentials (CMAPs) were recorded from the plantar muscles of both hindpaws individually through stainless steel electrodes (diameter: 0.28 mm; Science Products, Hofheim, Germany) inserted in the plantar muscle and subcutaneously in the third toe. Signals evoked by 1‐mA electrical stimulation at 0.1 Hz were amplified, filtered (3–3000 Hz, LP511 Grass Amplifier, Astro‐Med, Rodgau, Germany), acquired (Micro 1401 mkII, CED, Cambridge, UK), and recorded using SIGNAL Software (CED). This setup allowed the recording of CMAPs containing (1) stimulus artifact; (2) early nerve action potential; (3) direct muscle response (M‐wave); and (4) monosynaptic reflex response (H‐wave; Fig. 3A, upper panel); the latest was specifically abolished by lidocaine (2%; B Brown, Germany) injected in the vicinity of the sciatic nerve in the mouse thigh in a control experiment (Fig. 3A, lower panel). Averages of five consecutive responses were analyzed to measure amplitudes and area under curve of M‐ and H‐waves to the baseline. The values obtained for each paw were averaged per animal.
Western blot analysis
Tau−/− and Tau+/+ sciatic nerves were homogenized [10 mm HEPES pH 7.9, 150 mm NaCl, 1 mm EGTA, 1 mm EDTA, 10% glycerol, 1% NP‐40, Complete Protease Inhibitor (Roche, Mannheim, Germany) and Phosphatase Inhibitor Cocktails II and III (Sigma, St Louis, MO, USA)]. After sonication and centrifugation (15 000 g; 10 min; 4 °C), protein contents were estimated by Bradford assay and lysates were electrophoresed on 10% acrylamide gels, and transferred onto nitrocellulose membranes (BIORAD Turbo, Munich, Germany). For detecting MBP levels, membranes were blocked in Tris‐buffered saline containing 5% nonfat milk in TBS‐T before incubation with antibodies against MBP (Serotec, Oxford, UK; 1:500), Tau (abcam, Cambridge, UK; 1:1000), and actin (DSHB, University of Iowa, IA, USA; 1:2000). Antigens were revealed by enhanced chemiluminescence (BIORAD) after incubation with appropriate horseradish peroxidase–immunoglobulin G conjugates (BIORAD). Blots were scanned and quantified using tina 3.0 bioimaging software (Raytest, Straubenhardt, Germany). All values were normalized against actin.
Statistical analysis
Unless otherwise specified, two‐way ANOVA was used having genotype (Tau+/+ vs. Tau−/−) and aging (adult vs. old) as factors and followed by Tukey post hoc analysis (SPSS, Aspire Software, Armonk, NY, USA). Differences were considered to be significant if p < 0.05.
Authors’ contribution
SL, AL, VP, VMS, SP, JP, JFO, HLA, and IS performed experiments; SL, AL, MG, VMS, JFO, HLA, and IS analyzed data and contributed with reagents; and IS, NS, and HLA designed the overall study and wrote the manuscript.
Conflict of interests
None of the authors report competing interests.
Funding
The work was supported by grants ‘PTDC/SAU‐NMC/113934/2009,’ ‘PTDC/SAU‐NSC/118194/2010,’ ‘SFRH/BPD/97281/2013,’ PTDC/SAU‐NSC/118194/2010,’ ‘SFRH/BPD/80118/2011,’ ‘SFRH/BD/89714/2012’ funded by FCT—Portuguese Foundation for Science and Technology and project DoIT—Desenvolvimento e Operacionalização da Investigação de Translação (N° do projeto 13853), funded by Fundo Europeu de Desenvolvimento Regional (FEDER) throughout the Programa Operacional Fatores de Competitividade (POFC). In addition, this work was also co‐financed by European Union FP7 project SwitchBox (NS) and the Portuguese North Regional Operational Program (ON.2 ‐ O Novo Norte) under the National Strategic Reference Framework (QREN), through the European Regional Development Fund (FEDER).
Acknowledgments
We would like to thank Rui Fernandes for his excellent TEM technical support and João Relvas for the MBP antibody.
References
- Bohl J, Ulbricht D, Steinmetz H (1997) Neurofibrillary tangles in peripheral autonomic ganglion cells In Alzheimer's Disease: Biology, Diagnosis and Therapeutics (Iqbal K, Winblad B, Nishimura T, Takeda M, Wisniewski HM, eds). Chichester: John Wiley & Sons, pp. 281–287. [Google Scholar]
- Boyne LJ, Tessler A, Murray M, Fischer I (1995) Distribution of Big tau in the central nervous system of the adult and developing rat. J. Comp. Neurol. 358, 279–293. [DOI] [PubMed] [Google Scholar]
- Buchman AS, Bennett DA (2011) Loss of motor function in preclinical Alzheimer's disease. Expert Rev. Neurother. 11, 665–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceballos D, Cuadras J, Verdu E, Navarro X (1999) Morphometric and ultrastructural changes with ageing in mouse peripheral nerve. J. Anat. 195(Pt 4), 563–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson HN, Ferreira A, Eyster MV, Ghoshal N, Binder LI, Vitek MP (2001) Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J. Cell Sci. 114, 1179–1187. [DOI] [PubMed] [Google Scholar]
- Decker L, Desmarquet‐Trin‐Dinh C, Taillebourg E, Ghislain J, Vallat JM, Charnay P (2006) Peripheral myelin maintenance is a dynamic process requiring constant Krox20 expression. J. Neurosci. 26, 9771–9779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duker AP, Espay AJ, Wszolek ZK, Rademakers R, Dickson DW, Kelley BJ (2012) Atypical motor and behavioral presentations of Alzheimer disease: a case‐based approach. Neurologist 18, 266–272. [DOI] [PubMed] [Google Scholar]
- Frappier TF, Georgieff IS, Brown K, Shelanski ML (1994) Tau regulation of microtubule‐microtubule spacing and bundling. J. Neurochem. 63, 2288–2294. [DOI] [PubMed] [Google Scholar]
- Frost B, Gotz J, Feany MB (2015) Connecting the dots between tau dysfunction and neurodegeneration. Trends Cell Biol. 25, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgieff IS, Liem RK, Mellado W, Nunez J, Shelanski ML (1991) High molecular weight tau: preferential localization in the peripheral nervous system. J. Cell Sci. 100(Pt 1), 55–60. [DOI] [PubMed] [Google Scholar]
- Gotz J, Ittner A, Ittner LM (2012) Tau‐targeted treatment strategies in Alzheimer's disease. Br. J. Pharmacol. 165, 1246–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotz J, Xia D, Leinenga G, Chew YL, Nicholas H (2013) What renders TAU toxic. Front Neurol. 4, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumucio A, Lannfelt L, Nilsson LN (2013) Lack of exon 10 in the murine tau gene results in mild sensorimotor defects with aging. BMC Neurosci. 14, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzer M, Holzapfel HP, Krohn K, Gertz HJ, Arendt T (1999) Alterations in content and phosphorylation state of cytoskeletal proteins in the sciatic nerve during ageing and in Alzheimer's disease. J. Neural. Transm. 106, 743–755. [DOI] [PubMed] [Google Scholar]
- Ikegami S, Harada A, Hirokawa N (2000) Muscle weakness, hyperactivity, and impairment in fear conditioning in tau‐deficient mice. Neurosci. Lett. 279, 129–132. [DOI] [PubMed] [Google Scholar]
- Ittner LM, Gotz J (2011) Amyloid‐beta and tau–a toxic pas de deux in Alzheimer's disease. Nat. Rev. Neurosci. 12, 65–72. [DOI] [PubMed] [Google Scholar]
- Ke YD, Suchowerska AK, van der Hoven J, De Silva DM, Wu CW, van Eersel J, Ittner A, Ittner LM (2012) Lessons from tau‐deficient mice. Int. J. Alzheimers Dis. 2012, 873270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick LL, Brady ST (1994) Modulation of the axonal microtubule cytoskeleton by myelinating Schwann cells. J. Neurosci. 14, 7440–7450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein C, Kramer EM, Cardine AM, Schraven B, Brandt R, Trotter J (2002) Process outgrowth of oligodendrocytes is promoted by interaction of fyn kinase with the cytoskeletal protein tau. J. Neurosci. 22, 698–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer‐Albers EM, White R (2011) From axon‐glial signalling to myelination: the integrating role of oligodendroglial Fyn kinase. Cell. Mol. Life Sci. 68, 2003–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ksiezak‐Reding H, Binder LI, Yen SH (1988) Immunochemical and biochemical characterization of tau proteins in normal and Alzheimer's disease brains with Alz 50 and Tau‐1. J. Biol. Chem. 263, 7948–7953. [PubMed] [Google Scholar]
- Lei P, Ayton S, Finkelstein DI, Spoerri L, Ciccotosto GD, Wright DK, Wong BX, Adlard PA, Cherny RA, Lam LQ, Roberts BR, Volitakis I, Egan GF, McLean CA, Cappai R, Duce JA, Bush AI (2012) Tau deficiency induces parkinsonism with dementia by impairing APP‐mediated iron export. Nat. Med. 18, 291–295. [DOI] [PubMed] [Google Scholar]
- Lei P, Ayton S, Moon S, Zhang Q, Volitakis I, Finkelstein DI, Bush AI (2014) Motor and cognitive deficits in aged tau knockout mice in two background strains. Mol. Neurodegener. 9, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoPresti P, Szuchet S, Papasozomenos SC, Zinkowski RP, Binder LI (1995) Functional implications for the microtubule‐associated protein tau: localization in oligodendrocytes. Proc. Natl Acad. Sci. USA 92, 10369–10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma QL, Zuo X, Yang F, Ubeda OJ, Gant DJ, Alaverdyan M, Kiosea NC, Nazari S, Chen PP, Nothias F, Chan P, Teng E, Frautschy SA, Cole GM (2014) Loss of MAP function leads to hippocampal synapse loss and deficits in the morris water maze with aging. J. Neurosci. 34, 7124–7136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melcangi RC, Magnaghi V, Martini L (2000) Aging in peripheral nerves: regulation of myelin protein genes by steroid hormones. Prog. Neurobiol. 60, 291–308. [DOI] [PubMed] [Google Scholar]
- Michailov GV, Sereda MW, Brinkmann BG, Fischer TM, Haug B, Birchmeier C, Role L, Lai C, Schwab MH, Nave KA (2004) Axonal neuregulin‐1 regulates myelin sheath thickness. Science 304, 700–703. [DOI] [PubMed] [Google Scholar]
- Morris M, Koyama A, Masliah E, Mucke L (2011) Tau reduction does not prevent motor deficits in two mouse models of Parkinson's disease. PLoS ONE 6, e29257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris M, Hamto P, Adame A, Devidze N, Masliah E, Mucke L (2013) Age‐appropriate cognition and subtle dopamine‐independent motor deficits in aged tau knockout mice. Neurobiol. Aging 34, 1523–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller R, Heinrich M, Heck S, Blohm D, Richter‐Landsberg C (1997) Expression of microtubule‐associated proteins MAP2 and tau in cultured rat brain oligodendrocytes. Cell Tissue Res. 288, 239–249. [DOI] [PubMed] [Google Scholar]
- Murphy P, Topilko P, Schneider‐Maunoury S, Seitanidou T, Baron‐Van Evercooren A, Charnay P (1996) The regulation of Krox‐20 expression reveals important steps in the control of peripheral glial cell development. Development 122, 2847–2857. [DOI] [PubMed] [Google Scholar]
- Nothias F, Boyne L, Murray M, Tessler A, Fischer I (1995) The expression and distribution of tau proteins and messenger RNA in rat dorsal root ganglion neurons during development and regeneration. Neuroscience 66, 707–719. [DOI] [PubMed] [Google Scholar]
- Petit B, Giraudet F, Bechon C, Bardin L, Avan P, Boespflug‐Tanguy O, Begou M (2014) Mice with a deletion of the major central myelin protein exhibit hypersensitivity to noxious thermal stimuli: involvement of central sensitization. Neurobiol. Dis. 65, 55–68. [DOI] [PubMed] [Google Scholar]
- Pinto V, Szucs P, Derkach VA, Safronov BV (2008) Monosynaptic convergence of C‐ and Adelta‐afferent fibres from different segmental dorsal roots on to single substantia gelatinosa neurones in the rat spinal cord. J. Physiol. 586, 4165–4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Scearce‐Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L (2007) Reducing endogenous tau ameliorates amyloid beta‐induced deficits in an Alzheimer's disease mouse model. Science 316, 750–754. [DOI] [PubMed] [Google Scholar]
- Sato‐Yoshitake R, Shiomura Y, Miyasaka H, Hirokawa N (1989) Microtubule‐associated protein 1B: molecular structure, localization, and phosphorylation‐dependent expression in developing neurons. Neuron 3, 229–238. [DOI] [PubMed] [Google Scholar]
- Scarmeas N, Hadjigeorgiou GM, Papadimitriou A, Dubois B, Sarazin M, Brandt J, Albert M, Marder K, Bell K, Honig LS, Wegesin D, Stern Y (2004) Motor signs during the course of Alzheimer disease. Neurology 63, 975–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva‐Fernandes A, Duarte‐Silva S, Neves‐Carvalho A, Amorim M, Soares‐Cunha C, Oliveira P, Thirstrup K, Teixeira‐Castro A, Maciel P (2014) Chronic treatment with 17‐DMAG improves balance and coordination in a new mouse model of Machado‐Joseph disease. Neurotherapeutics 11, 433–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Goetz BD, Kirvell SL, Butt AM, Duncan ID (2001) Selective myelin defects in the anterior medullary velum of the taiep mutant rat. Glia 33, 1–11. [PubMed] [Google Scholar]
- Ubhi KK, Shaibah H, Newman TA, Shepherd D, Mudher A (2007) A comparison of the neuronal dysfunction caused by Drosophila tau and human tau in a Drosophila model of tauopathies. Invert. Neurosci. 7, 165–171. [DOI] [PubMed] [Google Scholar]
- Vossel KA, Zhang K, Brodbeck J, Daub AC, Sharma P, Finkbeiner S, Cui B, Mucke L (2010) Tau reduction prevents Abeta‐induced defects in axonal transport. Science 330, 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RS, Bennett DA, Gilley DW, Beckett LA, Schneider JA, Evans DA (2000) Progression of parkinsonian signs in Alzheimer's disease. Neurology 54, 1284–1289. [DOI] [PubMed] [Google Scholar]
- Yuan A, Kumar A, Peterhoff C, Duff K, Nixon RA (2008) Axonal transport rates in vivo are unaffected by tau deletion or overexpression in mice. J. Neurosci. 28, 1682–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Maiti A, Shively S, Lakhani F, McDonald‐Jones G, Bruce J, Lee EB, Xie SX, Joyce S, Li C, Toleikis PM, Lee VM, Trojanowski JQ (2005) Microtubule‐binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc. Natl Acad. Sci. USA 102, 227–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhukareva V, Sundarraj S, Mann D, Sjogren M, Blenow K, Clark CM, McKeel DW, Goate A, Lippa CF, Vonsattel JP, Growdon JH, Trojanowski JQ, Lee VM (2003) Selective reduction of soluble tau proteins in sporadic and familial frontotemporal dementias: an international follow‐up study. Acta Neuropathol. 105, 469–476. [DOI] [PubMed] [Google Scholar]