

Abstract
Human immunodeficiency virus type 1 (HIV-1) acquisition occurs predominantly through mucosal transmission. We hypothesized that greater mucosal immune responses and protective efficacy against mucosal HIV-1 infection may be achieved by prime-boost immunization at mucosal sites. We used a macaque model to determine the safety, immunogenicity, and protective efficacy of orally delivered, replication-competent but attenuated recombinant vaccinia viruses expressing full-length HIV-1 SF162 envelope (Env) or simian immunodeficiency virus (SIV) Gag-Pol proteins. We examined the dose and route that are suitable for oral immunization with recombinant vaccinia viruses. We showed that sublingual inoculation of two vaccinia virus-naive pigtailed macaques with 5 × 108 PFU of recombinant vaccinia viruses was safe. However, sublingual inoculation with a higher dose or tonsillar inoculation resulted in secondary oral lesions, indicating the need to optimize the dose and route for oral immunization with replication-competent vaccinia virus vectors. Oral priming alone elicited antibody responses to vaccinia virus and to the SF162 Env protein. Intramuscular immunization with the SF162 gp120 protein at either 20 or 21 weeks postpriming resulted in a significant boost in antibody responses in both systemic and mucosal compartments. Furthermore, we showed that immune responses induced by recombinant vaccinia virus priming and intramuscular protein boosting provided protection against intrarectal challenge with the simian-human immunodeficiency virus SHIV-SF162-P4.
INTRODUCTION
Human immunodeficiency virus type 1 (HIV-1) is a major public health concern of unprecedented dimensions. Therefore, development of a vaccine to prevent the spread of HIV remains a global public health priority. Our laboratory first demonstrated the protective efficacy of the “prime-boost” immunization strategy in animal models by using recombinant poxvirus priming followed by subunit envelope protein (Env) boosters to augment innate and antigen-specific responses in both the T- and B-cell compartments (1, 2). Using this regimen, protection was demonstrated in macaques challenged through both parenteral and mucosal routes by various viruses, including simian immunodeficiency virus (SIV) and simian-human immunodeficiency virus (SHIV) strains of different tropisms and in vivo pathogenicities (1, 3–7). In humans, the only vaccine regimen that has demonstrated efficacy is the poxvirus prime-protein boost regimen used in the RV144 trial. This regimen consisted of recombinant canarypox virus priming (ALVAC-HIV; Sanofi Pasteur) and a gp120 protein booster (AIDSVAX B/E; VaxGen) and was associated with a 31% reduction in the risk of HIV-1 acquisition (8).
The moderate success observed in the RV144 trial provides proof of concept that protective immunity can be elicited by a poxvirus prime-protein boost regimen and supports further efforts to build on this approach. One strategy for improving efficacy may be to target induction of stronger mucosal immune responses. This is because the majority of HIV-1 infections occur by mucosal transmission at vaginal or rectal sites. Additionally, HIV-1 and SIV preferentially replicate in the gut mucosa and deplete intestinal CD4 T cells in the early stages of infection (9, 10). Protection against mucosal transmission or disseminated infection in the gut-associated lymphoid tissue (GALT) may be better achieved by effective mucosal immune responses. Studies also indicate that generation of immune responses at mucosal sites can be better achieved by mucosal immunization than by parenteral immunization (11–14). It is therefore important to examine mucosal immunization approaches that may generate protective immunity against mucosal acquisition of HIV.
Recombinant vaccinia virus (rVV) strains expressing a variety of foreign antigens have been shown to confer protection in immunized animals against challenges with the respective pathogens, such as respiratory syncytial virus, hepatitis B virus, influenza virus, and rinderpest virus (15–20). Early studies also showed protection against smallpox by oral immunization with attenuated VV (21). Indeed, the first recombinant poxvirus vector approved for immunization of wildlife animals against rabies was developed as an oral vaccine (22–25). Thus, we tested recombinant poxvirus priming at mucosal sites as a strategy to induce mucosal immune responses at potential sites of infection. In a pilot study, we investigated the dose and route that are suitable for oral immunization of macaques with a poxvirus-based HIV-1 vaccine. Our results indicate that oral inoculation primes the immune system and results in induction of immune responses in both systemic and mucosal compartments. Furthermore, our results also indicate that oral priming and intramuscular (i.m.) protein boosting could afford significant protection against mucosal SHIV challenge in some macaques.
MATERIALS AND METHODS
Reagents.
A SIVmac239 Gag peptide pool and complete peptide sets for SIVmac239 Pol and SHIV-SF162-P3 Env were obtained from the NIH AIDS reagent program.
Cells.
TZM-bl cells (NIH AIDS reagent program), African green monkey kidney cells (BSC40; ATCC), and 293T cells (ATCC) were cultured in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM glutamine (complete DMEM). A human osteosarcoma cell line that lacks a functional thymidine kinase (TK) gene (143B; ATCC) was cultured in complete DMEM containing 25 μg/ml of 5-bromodeoxyuridine.
rVV.
A plaque-purified, replication-competent VV (generated from New York City Board of Health strain v-NY) (26, 27) was used as the vector to express either the HIV-1 SF162 full-length Env protein or SIVmac239 Gag-Pol proteins as previously described (28). In brief, transgenes were inserted into the TK gene of the v-NY strain of VV by homologous recombination. Three rounds of plaque purification were performed, the first two of which were under negative-selection conditions, using cell culture medium containing 25 μg/ml of 5-bromodeoxyuridine on 143B cells. The third round of selection was performed under nonselective conditions on BSC40 cells to ensure that a pure recombinant vaccinia virus stock had been obtained. Plaques were screened for the transgene of interest by PCR using primer sets specific to the TK gene and the transgene. Recombinant viruses were then expanded and propagated on BSC40 cells to generate crude virus stocks. Expression of the transgene was verified by Western blotting using antisera specific for the transgene product. Viruses used as immunogens were further purified by sucrose density sedimentation as described by Joklik (29), with the following modifications. rVV-infected BSC40 cell pellets (∼1 billion cells) were resuspended in 14 ml of cold 10 mM Tris, pH 9.0. The resuspended cells were transferred to a 40-ml glass Dounce homogenizer, and cell suspensions were homogenized with 40 strokes of a tight pestle on ice. The material was then centrifuged for 5 min at 1,360 rpm (GH3.8 rotor and Beckman GS-R centrifuge) at 4°C. The supernatant was removed to a sterile tube, and the cell pellet was resuspended in 3 ml of cold 10 mM Tris before again being centrifuged. The supernatant from the second spin was pooled with the supernatant from the first spin, and the material was then sonicated in a cup horn, using a model 550 Sonic Dismembrator machine at an amplitude setting of 8. The material was sonicated at 1-min intervals three times in ice water, with 1- to 3-min resting periods on ice. The material was then gently layered onto a 17-ml solution of 36% sucrose in 10 mM Tris, pH 9.0, and spun at 15,800 rpm (SW28 rotor and Optima XL-100k ultracentrifuge) for 80 min at 4°C. The supernatant was aspirated, and the virus pellet was resuspended in cold 1 mM Tris, pH 9.0, and sonicated as described above before being stored and frozen at −80°C.
Protein immunogen.
The HIV-1 SF162 gp120 protein used for boosting in this study was purified from the cell culture medium of BSC40 cells infected with rVV as described previously (30). Briefly, BSC40 cells were infected with rVV expressing the SF162 gp120 protein at a multiplicity of infection (MOI) of 3 for 48 h. The culture medium was then collected, and the cells were removed by centrifugation at 2,645 × g for 20 min at 4°C. After addition of Empigen BB (Sigma) to a final concentration of 0.25%, the sample was used directly for purification, without any further treatment. All purification steps were handled at 4°C, using an Äkta 10/100 purifier (GE Life Sciences). The sample was loaded at 1 ml/min onto a 10-ml GNA column (Galanthus nivalis lectin-coupled agarose; Vector Laboratories) preequilibrated with binding buffer (150 mM NaCl, 20 mM Tris-HCl, 0.25% Empigen BB, pH 7.5). After washing with 10 column volumes (CV) of high-salt wash buffer (500 mM NaCl, 20 mM Tris-HCl, 0.25% Empigen BB, pH 7.5) followed by 10 CV of binding buffer, the bound protein was eluted with GNA elution buffer containing methyl-α-d-mannopyranoside (MMP; Sigma) (150 mM NaCl, 20 mM Tris-HCl, 0.25% Empigen BB, 1 M MMP, pH 7.5). Peak fractions were pooled and dialyzed overnight against DEAE binding buffer (100 mM NaCl, 20 mM Tris-HCl, pH 8.0), followed by one additional buffer exchange for another 3 h. The dialyzed sample was loaded at 1 ml/min onto a prepacked 5-ml DEAE column (GE Healthcare), and the flowthrough was collected. After concentration with an Amicon centrifugal concentrator (Millipore), the sample was loaded onto a HighLoad 26/600 Superdex 200 column (GE Life Sciences) for size exclusion chromatography and run at 1.2 ml/min in phosphate-buffered saline (PBS; 10 mM sodium phosphate, 150 mM NaCl, pH 7.4). Peak fractions were pooled and concentrated. The protein concentration was determined by bicinchoninic acid (BCA) assay (Pierce).
Animals, immunization plan, and challenge study.
A total of 6 juvenile pigtailed macaques (PTMs) that were negative for SIV, simian retrovirus type D, and simian T-cell leukemia virus were enrolled in this study. They were housed and maintained according to the standards of the American Association for Assessment and Accreditation of Laboratory Animal Care. In stage I, two PTMs (R10106 and R10131) were immunized by sublingual inoculation at weeks 0 and 8 with an equal-part mixture of two rVV, one expressing full-length SF162 Env (gp160) (2.5 × 108 PFU) and the other expressing SIV Gag-Pol proteins (2.5 × 108 PFU), in a total volume of 0.62 ml of 1 mM Tris, pH 9.0. The macaques were boosted intramuscularly 21 weeks after primary immunization, using 100 μg/animal of SF162 gp120 protein formulated in 2% alum adjuvant (Alhydrogel; InvivoGen). In stage II, two macaques (Z09183 and L10124) were immunized by sublingual inoculation with a higher dose of the same mixture of the two rVV (1 × 109 PFU total in 0.788 ml of 1 mM Tris, pH 9.0), and two additional macaques (R10090 and L10141) were immunized by tonsillar inoculation (5 × 108 PFU total in 0.394 ml of 1 mM Tris, pH 9.0). The macaques were then boosted by i.m. injection with the SF162 gp120 protein 20 weeks after the primary immunization. Four weeks after the gp120 boost, these 4 macaques were challenged intrarectally with a standard challenge stock of strain SHIV-SF162-P4 (Advance BioScience Laboratory). The in vivo infectivity of SHIV-SF162-P4 in PTMs was previously determined in our lab (31), and the same stock was used for the present challenge study. The challenge virus was administered nontraumatically in 1 ml of PBS per animal (640 50% tissue culture infective doses [TCID50]/ml) by intrarectal inoculation.
Sample collection.
Plasma, peripheral blood mononuclear cell (PBMC), and serum samples were collected as previously described (32). Briefly, peripheral blood was drawn by venipuncture into EDTA-containing tubes or serum-separating tubes for extraction of plasma and PBMCs or serum, respectively. PBMCs were isolated from EDTA-treated blood by using 95% Lymphoprep (Axis Shield). Saliva was collected from ketamine-sedated animals by keeping the macaques in a facedown position and allowing saliva to flow freely into a petri dish. Rectal lavage fluid was collected by gently flushing 3 ml of PBS through the rectum.
Antibody assays.
The antibody response against VV was measured by an enzyme-linked immunosorbent assay (ELISA) using VV-infected cell lysate as the capture antigen (100 ng total protein per well of a 96-well microtiter plate). HIV Env-specific antibody titers in serum (IgG and IgA) were determined by an ELISA using the SF162 gp120 protein as the capture antigen (100 ng gp120 per well). ELISA procedures were performed as described previously (33). The endpoint ELISA titer of binding antibodies was defined as the reciprocal of the serum dilution that resulted in optical density (OD) readings higher than the mean OD reading for the preimmune sera (1:100 dilution) plus 3 times the standard deviation. The detection limit of the ELISA was considered to be the starting dilution (1:100) of the test sera. Antibody responses in serum, rectal lavage fluid, and saliva against the SF162 gp120 purified protein were determined by Western blotting. Briefly, the purified SF162 gp120 protein was run in Criterion precast gels (Bio-Rad) and transferred to either polyvinylidene difluoride (PVDF) or nylon membranes. Transferred membranes were then cut into small strips and blotted in 5% blocking solution prepared in 1× PBS with 0.05% Tween 20 (PBST) for 30 min to 1 h. Membrane strips were then incubated in serum (1:200 dilution), rectal lavage fluid (1:7.5 dilution), or saliva (1:7.5 dilution) overnight at 4°C. After 3 washes in 1× PBST, membranes were incubated with goat anti-human IgG(H+L) conjugated with horseradish peroxidase (1:5,000 dilution; Thermo Scientific) for 1 h at room temperature. After 3 washes in 1× PBST, membranes were developed with Super Signal West Pico chemiluminescence substrate (Thermo Scientific), and signals of bound antibodies were detected by autoradiography.
An assay of neutralization, measured by a reduction of luciferase gene expression, was performed as previously described (3), using SF162 Env-pseudotyped HIV-1. Briefly, an indicator virus containing 150 TCID50 was incubated with either a single dilution (1:20) or serial dilutions of serum samples in duplicate in a total volume of 60 μl for 90 min at 37°C in 96-well plates. At the same time, TZM-bl cells, plated 1 day before at 3,000 cells/well in 96-well white assay plates with clear flat bottoms (Costar), were treated with serum-free DMEM containing 2 μg/ml Polybrene for 30 min at 37°C. After the incubation step, 50 μl of virus-serum was added to the TZM-bl cells and incubated at 37°C and 5% CO2. After 3 days of incubation, virus was removed, and luciferase activity (in relative luciferase units [RLU]) was measured by using BrightGlo substrate solution (Promega) according to the manufacturer's instructions. Neutralization activity was expressed either as the percent reduction of RLU, for experiments with single (1:20) serum dilutions, or as the 50% inhibitory dilution (ID50), which was defined as the reciprocal serum dilution that resulted in a 50% reduction of RLU in serial dilution experiments compared to that for virus control wells which contained preimmune sera (week 0).
ELISPOT assay.
HIV Env- and SIV Gag-Pol-specific T-cell responses were measured by a gamma interferon (IFN-γ)-specific enzyme-linked immunosorbent spot (ELISPOT) assay as previously described (34). Approximately 1.5 × 105 or 2 × 105 PBMCs in duplicate wells were stimulated in the presence of Env, Gag, or Pol 15-mer peptides overlapping by 11 amino acids, at a final concentration of 1 μg/ml/peptide, for 24 h at 37°C. Concanavalin A (5 μg/ml) and dimethyl sulfoxide (DMSO) (at a concentration equivalent to or greater than that used for the peptide stimulations) were used as the positive and negative controls, respectively. The spots were counted using a CTL-ImmunoSpot S6 microanalyzer (Cellular Technology Ltd.). The number of spots observed for the DMSO-stimulated PBMCs was subtracted from those for peptide-stimulated samples before converting the results to numbers of spot-forming cells (SFC) per million PBMCs.
Viral load quantitation and CD4 T-cell counts.
Plasma viral loads were determined by real-time reverse transcription-PCR (RT-PCR) as described previously (3, 4, 35). The lower limit for quantification was 30 viral RNA copies/ml plasma. Absolute numbers of peripheral blood CD3+ CD4+ cells were determined by flow cytometry as previously described (4).
RESULTS
Safety and immunogenicity of sublingual immunization with recombinant poxviruses.
To assess safety and immunogenicity, we inoculated two juvenile PTMs (R10106 and R10131) with 5 × 108 PFU of rVV. Animals were monitored for a panel of parameters related to general health, such as body weight, body temperature, and food intake. Clinical parameters included hematology and blood chemistry parameters. All hematology parameters, such as white blood cell count, red blood cell count, hemoglobin concentration, percent hematocrit (Hct; relative volume of erythrocytes), percent granulocytes, and percentages or absolute numbers of lymphocyte subsets, were within normal limits. We also measured blood chemistry parameters, such as kidney function (concentrations of creatinine and blood urea nitrogen), blood glucose, lipid concentration profile (concentrations of cholesterol, low-density lipoprotein, and high-density lipoprotein), liver function (concentrations of alkaline phosphatase, alanine aminotransferase, aspartate aminotransferase, and bilirubin), and electrolytes (concentrations of sodium, potassium, calcium, and chloride). All were within normal limits (data not shown). We also monitored vaccinated macaques for oral lesions. Importantly, no signs of lesions in the oral cavity or adverse effects on the overall health of the macaques were observed. We measured rVV loads in cytobrush samples collected from sublingual, inner cheek, and tonsil sites, but we detected virus only at 1 week postinoculation, mainly in samples collected from tonsils (Table 1), suggesting tonsils as the main site of VV replication as observed in other studies (36). rVV loads were undetectable at later time points, even after the second priming. This may have been due to the generation of antibody responses against VV by 2 weeks after the first rVV inoculation (Table 2).
TABLE 1.
Vaccinia virus loads
| Sample type | Viral load (PFU/ml) at indicated time (wk) postinoculationa |
|||||||
|---|---|---|---|---|---|---|---|---|
|
R10106 |
R10131 |
|||||||
| 1 | 2 | 9 | 10 | 1 | 2 | 9 | 10 | |
| Sublingual supernatant | — | — | — | — | — | — | — | — |
| Inner cheek supernatant | — | — | — | — | 24 | — | — | — |
| Tonsil supernatant | 64 | — | — | — | 4,000 | — | — | — |
—, not detected.
TABLE 2.
Serum antibody titers against vaccinia virus as measured by ELISA
| Study stage | Animal no. | Serum antibody titer after primary immunizationa |
||
|---|---|---|---|---|
| Wk 1 | Wk 2 | Wk 4 | ||
| I | R10106 | 200 | 1,600 | 3,200 |
| R10131 | 100 | 800 | 3,200 | |
| II | Z09183 | 100 | 3,200 | 1,600 |
| L10124 | 100 | 1,600 | 800 | |
| R10090 | — | 1,600 | 1,600 | |
| L10141 | — | 800 | 1,600 | |
Reciprocal serum dilution that resulted in a positive OD reading in ELISA (see Materials and Methods for details). —, below the detection limit at the starting serum dilution of 1:100.
Next, we evaluated antibody responses against the SF162 Env protein. Sublingual immunization induced Env-specific IgG in serum (Fig. 1A and B), but Gag-specific antibodies were not detectable (data not shown). Following i.m. boosting with the SF162 gp120 protein at 21 weeks postpriming, approximately 250- to 500-fold increases in serum IgG titer were observed (Fig. 1A). An increase in the Env-specific IgA titer was also observed in serum following boosting (Fig. 1C). Interestingly, following boosting, Env-specific IgG responses were detected in mucosal samples, including saliva and rectal lavage samples, by Western blotting (Fig. 1D and E). However, Env-specific IgA was not detected in mucosal samples by Western blotting (data not shown), which may have been due to low concentrations of IgA in the mucosal samples. HIV-1 neutralizing antibody (NAb) responses were observed in sera collected at 2 weeks postboosting. These sera showed neutralizing activities against autologous SF162-pseudotyped HIV-1 in a TZM-bl cell assay, with NAb titers (ID50) of ∼20,000 and 4,400 in macaques R10106 and R10131, respectively (Table 3; see Fig. S1 in the supplemental material), compared to the low NAb titers observed in sera collected on the day of boosting. As expected, no neutralization activity was observed against murine leukemia virus (MLV) envelope-pseudotyped HIV-1 (ID50 of <10), suggesting that neutralization activity is specific to HIV-1 Env-pseudotyped viruses (data not shown). We also measured T-cell responses against Gag and Env peptides. However, the Gag-specific T-cell response was low. Interestingly, one macaque (R10131) showed an Env-specific T-cell response (>50 spots/million PBMCs) after i.m. boosting with the SF162 gp120 protein (see Fig. 3A).
FIG 1.

Antibody responses to the SF162 gp120 protein in immunized macaques in stage I of this study. Env-specific antibody titers in serum samples were measured by ELISA (A and C) and Western blotting (B). Antibody responses in mucosal samples, such as rectal lavage fluid (D) and saliva (E), were detected by Western blotting. For Western blotting, a 1:200 dilution of sera and a 1:7.5 dilution of rectal lavage and saliva samples were used. The starting dilution of serum used for ELISA was 1:100.
TABLE 3.
HIV-1 NAb titers and outcomes of SHIV challenge
| Study stage | Dose (PFU)/route | Animal no. | HIV-1 NAb titera |
Outcome of SHIV-SF162-P4 challenge |
|||
|---|---|---|---|---|---|---|---|
| Day of boost | 2 wk postboost | 4 wk postboost | 2 wk postchallenge | ||||
| I | 5 × 108/sublingual | R10106 | 17 | 20,698 | 11,924 | — | — |
| 5 × 108/sublingual | R10131 | <10 | 4,440 | 2,883b | — | — | |
| II | 1 × 109/sublingual | Z09183 | <10 | 17 | 35c | 191,743 | Infected |
| 1 × 109/sublingual | L10124 | 26 | 8,633 | 2,353c | 1,033 | Delayed acquisition | |
| 5 × 108/tonsillar | R10090 | 31 | 13,659 | 9,711c | 4,103 | Protected | |
| 5 × 108/tonsillar | L10141 | 10 | 8,429 | 4,476c | 3,866 | Protected | |
Defined as the reciprocal serum dilution that resulted in a 50% reduction of viral infectivity as determined in the TZM-bl cell assay, using HIV-1 SF162 Env-pseudotyped virus as the indicator (see Materials and Methods). HIV-1 pseudotyped with Env from murine leukemia virus (MLV) was used as a control for nonspecific activities. NAb titers (ID50) against MLV-pseudotyped virus were <10 for all serum samples tested. —, animals were not challenged.
Measured at 6 weeks postboosting.
Day of challenge for stage II animals.
FIG 3.
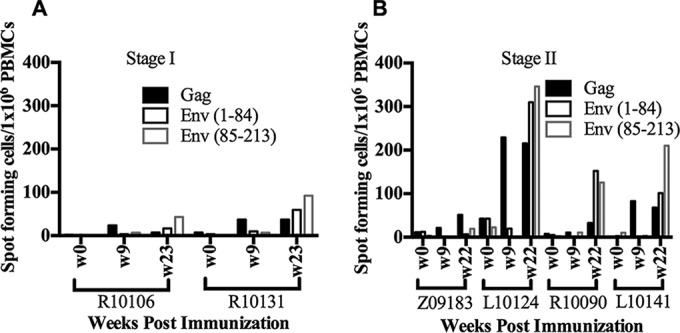
T-cell responses in macaques primed with recombinant vaccinia viruses by sublingual or tonsillar inoculation. PBMCs were stimulated with SIVmac239 Gag, SIVmac239 Pol, and SHIV-SF162-P3 Env peptide pools (1 μg/ml/peptide [final concentration]). IFN-γ released by the peptide-stimulated PBMCs was detected by ELISPOT assay. Numbers of spot-forming cells per million PBMCs were plotted for immunized macaques in stage I (A) and stage II (B).
These results indicate that sublingual immunization with rVV is safe, induces HIV Env-specific antibody responses, and results in immunological memory, which can be boosted by subsequent i.m. injection with a gp120 protein immunogen.
Effects of higher dose and tonsillar inoculation on safety and immunogenicity.
Because the previous set of experiments suggested that sublingual immunization with 5 × 108 PFU of rVV was safe, we next assessed the tolerability of a higher sublingual dose (total of 1 × 109 PFU per dose) in two macaques (Z09183 and L10124). Additionally, because vaccinia virus replication was observed primarily in the tonsils, we also tested the effects of direct tonsillar inoculation of rVV (5 × 108 PFU per dose) in two additional macaques (R10090 and L10141). All clinical parameters related to hematology and blood chemistry were within normal limits for all macaques (data not shown). Animal L10124 showed a temporary decrease in hematocrit, which was most likely a response to the blood draws and was resolved with iron supplementation. Both immunization approaches also resulted in secondary oral lesions (data not shown), indicating the need to optimize the dose and route for oral immunization with rVV vectors. However, as expected of vaccinia virus inoculation, these lesions healed and resolved by 2 weeks postinoculation. Both routes of immunization induced antibody responses to VV (Table 2). Env-specific IgG antibodies were observed in two of the macaques that received inoculation directly into the tonsils. In one of the sublingually inoculated macaques (Z09183), Env-specific IgG antibodies were below the detection limit (Fig. 2A). As observed in the previous experiment, increases in Env-specific IgG and IgA antibody titers were observed following the gp120 booster (Fig. 2A and B). Env-specific IgG antibodies were also detected in saliva samples from 3 of 4 macaques (L10124, R10090, and L10141) and in rectal lavage samples from 2 of 4 macaques (Z09183 and R10090) following boosting (Fig. 2C). Sera from 3 of 4 macaques showed autologous HIV-1-neutralizing activity, with ID50 values in the ranges of 4,000 to 20,000 and 2,000 to 12,000 at 2 and 4 weeks postboosting, respectively (Table 3; see Fig. S1 in the supplemental material), compared to the low NAb titer observed in sera collected on the day of boosting. The low NAb titer observed for macaque Z09183 (Table 3) might have been due to the low Env-specific antibody response (Fig. 2A). We also observed no neutralization activity against HIV-1 pseudotyped with MLV Env (ID50 of <10). To determine the neutralization breadth in vaccinated macaques, we measured the percent inhibition of virus infection at a single serum dilution (1:20), using a panel of pseudotyped viruses generated with Env proteins from different HIV-1 primary isolates. The neutralization activity assay was considered positive if the reduction of virus infectivity was 50% or higher. Sera collected at 2 weeks postboosting from all the macaques except animal Z09183 showed >90% inhibition of SF162 Env-pseudotyped HIV-1 infection. Interestingly, serum collected from macaque L10124 after boosting showed 50% or more inhibition of HIV-1 strains pseudotyped with heterologous Envs of BAL.26, DJ263.8, and SS1196.1 in TZM-bl cells (see Table S1 in the supplemental material). However, heterologous neutralization activity was not observed in sera collected from the other 3 macaques against the pseudotyped viruses tested (see Table S1). In regard to cellular immune responses, Env-specific T-cell responses were observed in peripheral blood following boosting for 3 of 4 macaques (Fig. 3). One animal (L10124) that received a higher sublingual dose also showed Gag-specific T-cell responses at 9 and 22 weeks postpriming. However, we did not detect Pol-specific T-cell responses in PBMCs (data not shown). Overall, these results indicate that both systemic and mucosal immune responses were induced by the oral rVV prime-intramuscular protein boost regimen.
FIG 2.
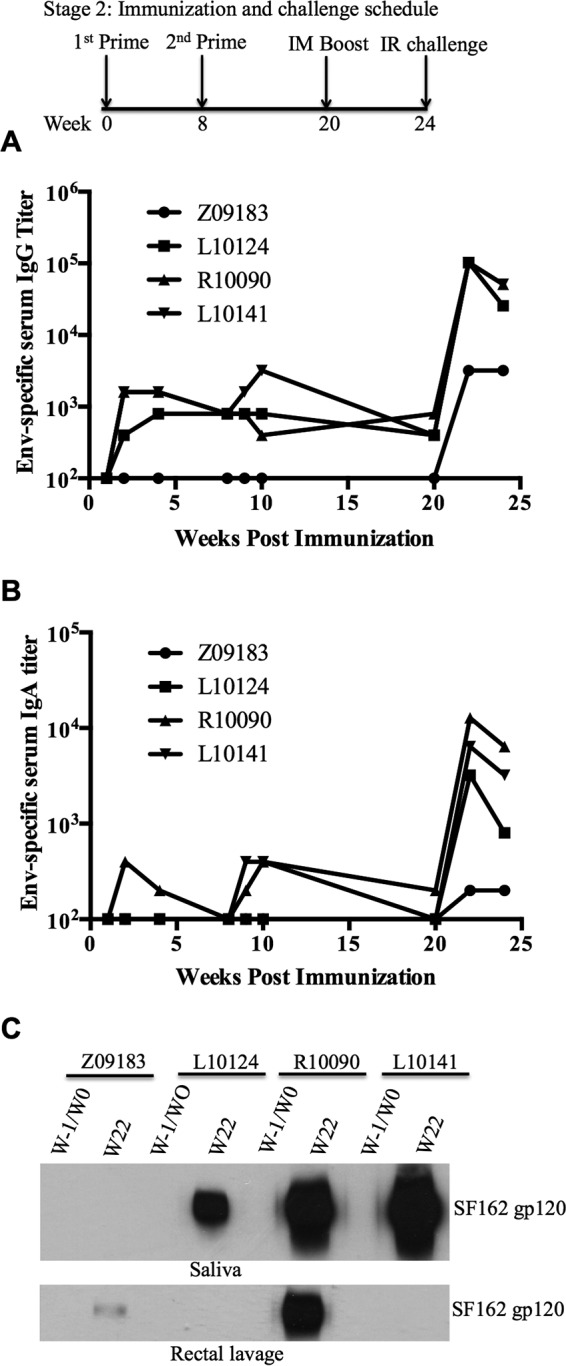
Antibody responses to the SF162 gp120 protein in immunized macaques in stage II of this study. (A and B) Antibody titers in serum samples were measured by ELISA. (C) Antibody responses in saliva and rectal lavage fluid were detected by Western blotting. For Western blotting, a 1:7.5 dilution of rectal lavage and saliva samples was used. The starting dilution of serum used for ELISA was 1:100. IR, intrarectal.
Oral rVV prime-intramuscular protein boost regimen provides protection against intrarectal SHIV-SF162-P4 challenge.
We showed previously that PTMs can be infected intrarectally with doses of SHIV-SF162-P4 ranging from 360 to 1,800 TCID50/ml (31). To determine the protective efficacy of the immune responses induced by an oral rVV prime-intramuscular protein boost regimen, macaques were challenged intrarectally with 640 TCID50/ml of the same stock of SHIV-SF162-P4. Interestingly, two macaques that were primed by direct inoculation into tonsils did not acquire infection. However, of the two macaques that were primed sublingually with 1 × 109 PFU, one (Z09183) acquired infection and the other (L10124) showed a delayed acquisition of infection (Fig. 4A). Plasma viral loads peaked at 1 week postinfection in the infected animal and gradually declined thereafter, indicating effective control of virus replication after infection. The macaque that showed delayed acquisition of infection also controlled viral replication. However, both sublingually vaccinated macaques showed small rebounds in plasma viral load at later time points, indicating viral persistence. Although CD4+ T-cell counts decreased immediately following infection, they stabilized at later time points, coinciding with decreases in plasma viral loads (Fig. 4B). Antibody titers increased rapidly at 2 weeks postinfection in one of the sublingually vaccinated macaques (Z09183) (Fig. 4C) compared to the naive animals (the historic controls) that were inoculated with 1,800 TCID50/ml SHIV-SF162-P4 (Fig. 4D), consistent with an anamnestic immune response in the vaccinated macaques. Serum collected postinfection from animal Z09183 also showed strong autologous neutralization activity against SF162 Env-pseudotyped HIV-1 (Table 3), as well as cross-neutralization activity against HIV-1 pseudotyped with BAL.26 or SS1196.1 Env (see Table S1 in the supplemental material). Due to the small size of this pilot study, it is difficult to ascertain the significance of the differences in immune responses between the experimental groups and their potential role in protection. However, our observation is consistent with the notion that a higher NAb titer at the time of challenge may provide protection against acquisition of infection (Table 3).
FIG 4.

Plasma viral loads, CD4 T-cell counts, and Env-specific antibody responses following intrarectal challenge with SHIV-SF162-P4. (A) Plasma viral loads were measured by real-time PCR, using an Applied Biosystems universal master mix II kit. (B) Peripheral blood CD4+ T-cell counts were measured by flow cytometry. Env-specific serum antibody titers in vaccinated animals (C) and historic controls (D) were measured by ELISA. The starting dilution of serum used for ELISA was 1:100.
Furthermore, we performed Fisher's two-tailed exact test to determine the P value for the outcome of the SHIV challenge. If we combine the results for 12 historic control animals (31) that received either a 3-fold higher inoculum (1,800 TCID50; 6/6 animals infected) or a 2-fold lower inoculum (360 TCID50; 5/6 animals infected) of the same challenge stock by the same challenge route used in the current study, the protection observed here is statistically significant: 11/12 control animals were infected, in contrast to 0/2 animals primed with recombinant vaccinia virus by tonsillar inoculations and boosted with gp120 by intramuscular injections (P ≤ 0.033). This difference is still statistically significant (P ≤ 0.027) if we combine the outcomes for both immunization groups: 3/4 immunized animals showed protection (two were completely protected and one showed delayed acquisition and control of viremia), while 11/12 naive control animals were infected. Overall, these results suggest that oral rVV priming and intramuscular protein boosting could provide significant protection against mucosal SHIV challenge in some immunized macaques.
DISCUSSION
In this study, we examined the safety and immunogenicity of oral immunization with rVV in macaques. This is the first study that shows the feasibility of using replication-competent rVV in sublingual and tonsillar immunization regimens in macaques. We show that sublingual immunization with 5 × 108 PFU of rVV is safe. However, macaques that were inoculated with a higher dose or directly inoculated in the tonsils showed self-healing secondary oral lesions, suggesting that further studies are required to optimize the dose and route for oral immunization with live VV vectors. Importantly, we showed that oral immunization followed by intramuscular boosting with a protein immunogen afforded protection against intrarectal challenge with pathogenic SHIV-SF162-P4.
The oral cavity includes sublingual, buccal, pharyngeal, and tonsillar regions. Different vectors and protein immunogens have been applied to individual regions as a way to induce mucosal immune responses (reviewed in references 37 and 38). However, the ability of live attenuated vaccinia virus vectors to induce an immune response by sublingual or tonsillar immunization in macaques has not been reported. Thus, we used an attenuated, replication-competent VV (v-NY), developed in our lab, as a vector for oral vaccination in a prime-boost regimen. We also showed that sublingual or tonsillar inoculation of v-NY expressing the HIV SF162 full-length Env protein induces antibody responses to both the VV vector and the SF162 Env protein. However, in one of the macaques, for reasons that are not clear, the antibody response to the SF162 Env protein was below the level of detection, although it generated an antibody response to VV. Due to the small number of animals used in this study, it was difficult to determine a correlation between antibody responses to the vector and to the foreign antigen expressed by the vector. It is possible that a strong antibody response to VV might limit its replication and the immune response against the foreign antigen expressed by the vector.
Mucosal antibodies have been correlated with protection against SIV or SHIV infection in macaques (12, 39, 40). One of the goals of our study was also to induce an antibody response in the mucosa. Although we did not detect Env-specific IgA responses, we did detect an Env-specific IgG antibody response in mucosal samples, such as saliva and rectal lavage fluid, by Western blotting. These samples were also positive for Env-specific IgG by ELISA, although the antibody titers were very low (data not shown). The NAb titers in rectal lavage samples were low, with ID50 values of <30, consistent with the low binding antibody titers in these samples. The low-titer Env-specific responses could have been due to the dilution factor of the rectal lavage fluid collected. In fact, the IgG concentrations in the mucosal samples, measured by using Easy-Tier IgG assay kits, were either very low or not detected, which could have been due to sampling errors (data not shown). Thus, further improvements in sample collection strategies are required to better detect Env antibody in mucosal samples.
In our previous study, we reported that PTMs can be infected by intrarectal inoculation with SHIV-SF162-P4. We showed that 6 of 6 and 5 of 6 macaques were infected with 1,800 TCID50/ml and 360 TCID50/ml SHIV-SF162-P4, respectively (31). In a recent study, Eugene et al. also showed infection of 4 of 4 unvaccinated rhesus macaques challenged rectally with 640 TCID50/ml of SHIV-SF162-P4 (41). Based on these observations, we used 640 TCID50/ml of the same stock of SHIV-SF162-P4 that was used in our previous study to intrarectally challenge vaccinated macaques. Interestingly, two macaques immunized by direct tonsillar inoculation did not acquire infection. The lack of an anamnestic response and the gradual decrease in Env-specific antibody titer following challenge in these two macaques also suggest that the animals did not acquire infection. The reason for the protection observed in macaques immunized by tonsillar inoculation is not clear. However, previous studies have shown that vaccine administration to the tonsils can elicit mucosal immune responses at distal sites, such as the genital and gastrointestinal tracts (8, 39, 41–43). Moreover, strong NAb titers in sera have also been found to correlate with protection against intrarectal challenge with SHIV-SF162-P4 (44). In this study, we also observed strong NAb responses in the macaques that received tonsillar inoculation followed by intramuscular protein boosting. Considering that SHIV-SF162-P4 is a neutralization-sensitive virus (45), the strong NAb titer at the time of challenge might have played a role in prevention of acquisition of infection. Interestingly, in another study, protection against acquisition of infection was not observed in macaques challenged intrarectally with SHIV-SF162-P4, although vaccination induced protective levels of NAb (46). Therefore, it is possible that additional immune responses induced by tonsillar immunization with live rVV may have played a role in prevention of infection. Thus, future studies are required to assess the efficacy of tonsillar immunization in a large group of macaques.
In the case of sublingually immunized macaques, one acquired infection and the other showed delayed acquisition of infection. The macaque that became infected had a low NAb titer compared to the macaques that showed protection. However, the infected macaque generated a NAb response following infection. Previous studies have suggested that NAb responses generated after challenge in vaccinated macaques may play a role in the control of virus infection (47–50). Thus, the strong NAb response (Table 3) generated in macaque Z09183 following challenge might have played a role in control of viral loads at later time points during infection. Interestingly, the macaque that showed delayed acquisition had a NAb titer (ID50) in the range of 2,300 and also showed strong T-cell responses to Gag and Env proteins. The absence of detectable plasma viremia in the first 2 weeks postinfection suggests that the immune response generated in this vaccinated macaque was able to control viral replication during this period. Interestingly, viral loads were detected at 4 weeks postinfection, suggesting either a waning of immune responses or selection for escape viruses.
Conclusions.
Our study shows the feasibility of using live attenuated VV as a vector for vaccination at oral sites. Our results also indicate that oral vaccination with rVV expressing HIV-1 Env followed by a protein boost with gp120 could provide significant protection against mucosal SHIV challenge in macaques. It is believed that live vectors whose biodistribution includes mucosal inductive sites will elicit mucosal immune responses. However, the quality and quantity of mucosal immune responses generated by mucosal immunization could be different from those seen with systemic immunization. Thus, it would be interesting to compare the immunogenicity and protective efficacy of oral vaccination versus parenteral vaccination against mucosal SHIV challenge in macaque models.
Supplementary Material
ACKNOWLEDGMENTS
We thank David Plotnik for critically reading through the manuscript, Samantha Townsley for providing pseudotyped HIV-1 and helping with the neutralization assay, Deb Diamond for assistance with regulatory issues, Paul Munson and Deb Fuller for help with the ELISPOT assay, the veterinary staff of the Washington National Primate Research Center (WaNPRC) for assistance with animal studies, and virology core members (WaNPRC) for assistance with vial load and lymphocyte subset analyses.
This research work was funded by the National Institute of Dental and Craniofacial Research (grant R01 DE021223 to Shiu-Lok Hu) and the National Institutes of Health (grant P51 OD010425 to WaNPRC).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/CVI.00597-15.
REFERENCES
- 1.Hu SL, Abrams K, Barber GN, Moran P, Zarling JM, Langlois AJ, Kuller L, Morton WR, Benveniste RE. 1992. Protection of macaques against SIV infection by subunit vaccines of SIV envelope glycoprotein gp160. Science 255:456–459. doi: 10.1126/science.1531159. [DOI] [PubMed] [Google Scholar]
- 2.Hu SL, Klaniecki J, Dykers T, Sridhar P, Travis BM. 1991. Neutralizing antibodies against HIV-1 BRU and SF2 isolates generated in mice immunized with recombinant vaccinia virus expressing HIV-1 (BRU) envelope glycoproteins and boosted with homologous gp160. AIDS Res Hum Retroviruses 7:615–620. doi: 10.1089/aid.1991.7.615. [DOI] [PubMed] [Google Scholar]
- 3.Li Y, Cleveland B, Klots I, Travis B, Richardson BA, Anderson D, Montefiori D, Polacino P, Hu SL. 2008. Removal of a single N-linked glycan in human immunodeficiency virus type 1 gp120 results in an enhanced ability to induce neutralizing antibody responses. J Virol 82:638–651. doi: 10.1128/JVI.01691-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Polacino P, Cleveland B, Zhu Y, Kimata JT, Overbaugh J, Anderson D, Hu SL. 2007. Immunogenicity and protective efficacy of Gag/Pol/Env vaccines derived from temporal isolates of SIVmne against cognate virus challenge. J Med Primatol 36:254–265. doi: 10.1111/j.1600-0684.2007.00243.x. [DOI] [PubMed] [Google Scholar]
- 5.Polacino P, Stallard V, Montefiori DC, Brown CR, Richardson BA, Morton WR, Benveniste RE, Hu SL. 1999. Protection of macaques against intrarectal infection by a combination immunization regimen with recombinant simian immunodeficiency virus SIVmne gp160 vaccines. J Virol 73:3134–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Polacino PS, Stallard V, Klaniecki JE, Pennathur S, Montefiori DC, Langlois AJ, Richardson BA, Morton WR, Benveniste RE, Hu SL. 1999. Role of immune responses against the envelope and the core antigens of simian immunodeficiency virus SIVmne in protection against homologous cloned and uncloned virus challenge in macaques. J Virol 73:8201–8215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Polacino P, Stallard V, Klaniecki JE, Montefiori DC, Langlois AJ, Richardson BA, Overbaugh J, Morton WR, Benveniste RE, Hu SL. 1999. Limited breadth of the protective immunity elicited by simian immunodeficiency virus SIVmne gp160 vaccines in a combination immunization regimen. J Virol 73:618–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, Kaewkungwal J, Chiu J, Paris R, Premsri N, Namwat C, de Souza M, Adams E, Benenson M, Gurunathan S, Tartaglia J, McNeil JG, Francis DP, Stablein D, Birx DL, Chunsuttiwat S, Khamboonruang C, Thongcharoen P, Robb ML, Michael NL, Kunasol P, Kim JH. 2009. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N Engl J Med 361:2209–2220. doi: 10.1056/NEJMoa0908492. [DOI] [PubMed] [Google Scholar]
- 9.Veazey RS, Lackner AA. 2004. Getting to the guts of HIV pathogenesis. J Exp Med 200:697–700. doi: 10.1084/jem.20041464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brenchley JM, Schacker TW, Ruff LE, Price DA, Taylor JH, Beilman GJ, Nguyen PL, Khoruts A, Larson M, Haase AT, Douek DC. 2004. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J Exp Med 200:749–759. doi: 10.1084/jem.20040874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gallichan WS, Rosenthal KL. 1996. Long-lived cytotoxic T lymphocyte memory in mucosal tissues after mucosal but not systemic immunization. J Exp Med 184:1879–1890. doi: 10.1084/jem.184.5.1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bomsel M, Tudor D, Drillet AS, Alfsen A, Ganor Y, Roger MG, Mouz N, Amacker M, Chalifour A, Diomede L, Devillier G, Cong Z, Wei Q, Gao H, Qin C, Yang GB, Zurbriggen R, Lopalco L, Fleury S. 2011. Immunization with HIV-1 gp41 subunit virosomes induces mucosal antibodies protecting nonhuman primates against vaginal SHIV challenges. Immunity 34:269–280. doi: 10.1016/j.immuni.2011.01.015. [DOI] [PubMed] [Google Scholar]
- 13.Holmgren J, Czerkinsky C. 2005. Mucosal immunity and vaccines. Nat Med 11:S45–S53. doi: 10.1038/nm1213. [DOI] [PubMed] [Google Scholar]
- 14.Neutra MR, Kozlowski PA. 2006. Mucosal vaccines: the promise and the challenge. Nat Rev Immunol 6:148–158. doi: 10.1038/nri1777. [DOI] [PubMed] [Google Scholar]
- 15.Kanesaki T, Murphy BR, Collins PL, Ogra PL. 1991. Effectiveness of enteric immunization in the development of secretory immunoglobulin A response and the outcome of infection with respiratory syncytial virus. J Virol 65:657–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meitin CA, Bender BS, Small PA Jr. 1994. Enteric immunization of mice against influenza with recombinant vaccinia. Proc Natl Acad Sci U S A 91:11187–11191. doi: 10.1073/pnas.91.23.11187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moss B. 1996. Genetically engineered poxviruses for recombinant gene expression, vaccination, and safety. Proc Natl Acad Sci U S A 93:11341–11348. doi: 10.1073/pnas.93.21.11341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moss B, Smith GL, Gerin JL, Purcell RH. 1984. Live recombinant vaccinia virus protects chimpanzees against hepatitis B. Nature 311:67–69. doi: 10.1038/311067a0. [DOI] [PubMed] [Google Scholar]
- 19.Smith GL, Murphy BR, Moss B. 1983. Construction and characterization of an infectious vaccinia virus recombinant that expresses the influenza hemagglutinin gene and induces resistance to influenza virus infection in hamsters. Proc Natl Acad Sci U S A 80:7155–7159. doi: 10.1073/pnas.80.23.7155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giavedoni L, Jones L, Mebus C, Yilma T. 1991. A vaccinia virus double recombinant expressing the F and H genes of rinderpest virus protects cattle against rinderpest and causes no spock lesions. Proc Natl Acad Sci U S A 88:8011–8015. doi: 10.1073/pnas.88.18.8011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hochstein-Mintzel V, Stickl H, Huber HC. 1976. Oral immunization against smallpox. Dev Biol Stand 33:260–266. [PubMed] [Google Scholar]
- 22.Blancou J, Kieny MP, Lathe R, Lecocq JP, Pastoret PP, Soulebot JP, Desmettre P. 1986. Oral vaccination of the fox against rabies using a live recombinant vaccinia virus. Nature 322:373–375. doi: 10.1038/322373a0. [DOI] [PubMed] [Google Scholar]
- 23.Rupprecht CE, Wiktor TJ, Johnston DH, Hamir AN, Dietzschold B, Wunner WH, Glickman LT, Koprowski H. 1986. Oral immunization and protection of raccoons (Procyon lotor) with a vaccinia-rabies glycoprotein recombinant virus vaccine. Proc Natl Acad Sci U S A 83:7947–7950. doi: 10.1073/pnas.83.20.7947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brochier B, Boulanger D, Costy F, Pastoret PP. 1994. Towards rabies elimination in Belgium by fox vaccination using a vaccinia-rabies glycoprotein recombinant virus. Vaccine 12:1368–1371. doi: 10.1016/0264-410X(94)90143-0. [DOI] [PubMed] [Google Scholar]
- 25.Brochier B, Blancou J, Thomas I, Languet B, Artois M, Kieny MP, Lecocq JP, Costy F, Desmettre P, Chappuis G, Pastoret PP. 1989. Use of recombinant vaccinia-rabies glycoprotein virus for oral vaccination of wildlife against rabies: innocuity to several non-target bait consuming species. J Wildl Dis 25:540–547. doi: 10.7589/0090-3558-25.4.540. [DOI] [PubMed] [Google Scholar]
- 26.Cooney EL, McElrath MJ, Corey L, Hu SL, Collier AC, Arditti D, Hoffman M, Coombs RW, Smith GE, Greenberg PD. 1993. Enhanced immunity to human immunodeficiency virus (HIV) envelope elicited by a combined vaccine regimen consisting of priming with a vaccinia recombinant expressing HIV envelope and boosting with gp160 protein. Proc Natl Acad Sci U S A 90:1882–1886. doi: 10.1073/pnas.90.5.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Graham BS, Matthews TJ, Belshe RB, Clements ML, Dolin R, Wright PF, Gorse GJ, Schwartz DH, Keefer MC, Bolognesi DP, Corey L, Stablein DM, Esterlitz JR, Hu SL, Smith GE, Fast PE, Koff WC, the NIAID AIDS Vaccine Clinical Trials Network . 1993. Augmentation of human immunodeficiency virus type 1 neutralizing antibody by priming with gp160 recombinant vaccinia and boosting with gp160 in vaccinia-naive adults. J Infect Dis 167:533–537. doi: 10.1093/infdis/167.3.533. [DOI] [PubMed] [Google Scholar]
- 28.Hu SL, Kosowski SG, Dalrymple JM. 1986. Expression of AIDS virus envelope gene in recombinant vaccinia viruses. Nature 320:537–540. doi: 10.1038/320537a0. [DOI] [PubMed] [Google Scholar]
- 29.Joklik WK. 1962. The purification of four strains of poxvirus. Virology 18:9–18. doi: 10.1016/0042-6822(62)90172-1. [DOI] [PubMed] [Google Scholar]
- 30.Guo W, Cleveland B, Davenport TM, Lee KK, Hu SL. 2013. Purification of recombinant vaccinia virus-expressed monomeric HIV-1 gp120 to apparent homogeneity. Protein Expr Purif 90:34–39. doi: 10.1016/j.pep.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Polacino P, Larsen K, Galmin L, Suschak J, Kraft Z, Stamatatos L, Anderson D, Barnett SW, Pal R, Bost K, Bandivdekar AH, Miller CJ, Hu SL. 2008. Differential pathogenicity of SHIV infection in pig-tailed and rhesus macaques. J Med Primatol 37(Suppl 2):S13–S23. doi: 10.1111/j.1600-0684.2008.00325.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ho O, Larsen K, Polacino P, Li Y, Anderson D, Song R, Ruprecht RM, Hu SL. 2009. Pathogenic infection of Macaca nemestrina with a CCR5-tropic subtype-C simian-human immunodeficiency virus. Retrovirology 6:65. doi: 10.1186/1742-4690-6-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu SL, Zarling JM, Chinn J, Travis BM, Moran PA, Sias J, Kuller L, Morton WR, Heidecker G, Benveniste RE. 1989. Protection of macaques against simian AIDS by immunization with a recombinant vaccinia virus expressing the envelope glycoproteins of simian type D retrovirus. Proc Natl Acad Sci U S A 86:7213–7217. doi: 10.1073/pnas.86.18.7213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fuller DH, Rajakumar P, Che JW, Narendran A, Nyaundi J, Michael H, Yager EJ, Stagnar C, Wahlberg B, Taber R, Haynes JR, Cook FC, Ertl P, Tite J, Amedee AM, Murphey-Corb M. 2012. Therapeutic DNA vaccine induces broad T cell responses in the gut and sustained protection from viral rebound and AIDS in SIV-infected rhesus macaques. PLoS One 7:e33715. doi: 10.1371/journal.pone.0033715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suryanarayana K, Wiltrout TA, Vasquez GM, Hirsch VM, Lifson JD. 1998. Plasma SIV RNA viral load determination by real-time quantification of product generation in reverse transcriptase-polymerase chain reaction. AIDS Res Hum Retroviruses 14:183–189. doi: 10.1089/aid.1998.14.183. [DOI] [PubMed] [Google Scholar]
- 36.Thomas I, Brochier B, Languet B, Blancou J, Peharpre D, Kieny MP, Desmettre P, Chappuis G, Pastoret PP. 1990. Primary multiplication site of the vaccinia-rabies glycoprotein recombinant virus administered to foxes by the oral route. J Gen Virol 71:37–42. doi: 10.1099/0022-1317-71-1-37. [DOI] [PubMed] [Google Scholar]
- 37.Tuero I, Robert-Guroff M. 2014. Challenges in mucosal HIV vaccine development: lessons from non-human primate models. Viruses 6:3129–3158. doi: 10.3390/v6083129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poles J, Alvarez Y, Hioe CE. 2014. Induction of intestinal immunity by mucosal vaccines as a means of controlling HIV infection. AIDS Res Hum Retroviruses 30:1027–1040. doi: 10.1089/aid.2014.0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hidajat R, Xiao P, Zhou Q, Venzon D, Summers LE, Kalyanaraman VS, Montefiori DC, Robert-Guroff M. 2009. Correlation of vaccine-elicited systemic and mucosal nonneutralizing antibody activities with reduced acute viremia following intrarectal simian immunodeficiency virus SIVmac251 challenge of rhesus macaques. J Virol 83:791–801. doi: 10.1128/JVI.01672-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xiao P, Patterson LJ, Kuate S, Brocca-Cofano E, Thomas MA, Venzon D, Zhao J, DiPasquale J, Fenizia C, Lee EM, Kalisz I, Kalyanaraman VS, Pal R, Montefiori D, Keele BF, Robert-Guroff M. 2012. Replicating adenovirus-simian immunodeficiency virus (SIV) recombinant priming and envelope protein boosting elicits localized, mucosal IgA immunity in rhesus macaques correlated with delayed acquisition following a repeated low-dose rectal SIV(mac251) challenge. J Virol 86:4644–4657. doi: 10.1128/JVI.06812-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eugene HS, Pierce-Paul BR, Cragio JK, Ross TM. 2013. Rhesus macaques vaccinated with consensus envelopes elicit partially protective immune responses against SHIV SF162p4 challenge. Virol J 10:102. doi: 10.1186/1743-422X-10-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duerr A. 2010. Update on mucosal HIV vaccine vectors. Curr Opin HIV AIDS 5:397–403. doi: 10.1097/COH.0b013e32833d2e39. [DOI] [PubMed] [Google Scholar]
- 43.Stahl-Hennig C, Kuate S, Franz M, Suh YS, Stoiber H, Sauermann U, Tenner-Racz K, Norley S, Park KS, Sung YC, Steinman R, Racz P, Uberla K. 2007. Atraumatic oral spray immunization with replication-deficient viral vector vaccines. J Virol 81:13180–13190. doi: 10.1128/JVI.01400-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bogers WM, Davis D, Baak I, Kan E, Hofman S, Sun Y, Mortier D, Lian Y, Oostermeijer H, Fagrouch Z, Dubbes R, van der Maas M, Mooij P, Koopman G, Verschoor E, Langedijk JP, Zhao J, Brocca-Cofano E, Robert-Guroff M, Srivastava I, Barnett S, Heeney JL. 2008. Systemic neutralizing antibodies induced by long interval mucosally primed systemically boosted immunization correlate with protection from mucosal SHIV challenge. Virology 382:217–225. doi: 10.1016/j.virol.2008.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seaman MS, Janes H, Hawkins N, Grandpre LE, Devoy C, Giri A, Coffey RT, Harris L, Wood B, Daniels MG, Bhattacharya T, Lapedes A, Polonis VR, McCutchan FE, Gilbert PB, Self SG, Korber BT, Montefiori DC, Mascola JR. 2010. Tiered categorization of a diverse panel of HIV-1 Env pseudoviruses for assessment of neutralizing antibodies. J Virol 84:1439–1452. doi: 10.1128/JVI.02108-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thomas MA, Tuero I, Demberg T, Vargas-Inchaustegui DA, Musich T, Xiao P, Venzon D, LaBranche C, Montefiori DC, DiPasquale J, Reed SG, DeVico A, Fouts T, Lewis GK, Gallo RC, Robert-Guroff M. 2014. HIV-1 CD4-induced (CD4i) gp120 epitope vaccines promote B and T-cell responses that contribute to reduced viral loads in rhesus macaques. Virology 471–473:81–92. doi: 10.1016/j.virol.2014.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Doria-Rose NA, Ohlen C, Polacino P, Pierce CC, Hensel MT, Kuller L, Mulvania T, Anderson D, Greenberg PD, Hu SL, Haigwood NL. 2003. Multigene DNA priming-boosting vaccines protect macaques from acute CD4+-T-cell depletion after simian-human immunodeficiency virus SHIV89.6P mucosal challenge. J Virol 77:11563–11577. doi: 10.1128/JVI.77.21.11563-11577.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Earl PL, Wyatt LS, Montefiori DC, Bilska M, Woodward R, Markham PD, Malley JD, Vogel TU, Allen TM, Watkins DI, Miller N, Moss B. 2002. Comparison of vaccine strategies using recombinant env-gag-pol MVA with or without an oligomeric Env protein boost in the SHIV rhesus macaque model. Virology 294:270–281. doi: 10.1006/viro.2001.1345. [DOI] [PubMed] [Google Scholar]
- 49.Rasmussen RA, Hofmann-Lehman R, Montefiori DC, Li PL, Liska V, Vlasak J, Baba TW, Schmitz JE, Kuroda MJ, Robinson HL, McClure HM, Lu S, Hu SL, Rizvi TA, Ruprecht RM. 2002. DNA prime/protein boost vaccine strategy in neonatal macaques against simian human immunodeficiency virus. J Med Primatol 31:40–60. doi: 10.1034/j.1600-0684.2002.1o019.x. [DOI] [PubMed] [Google Scholar]
- 50.Xu R, Srivastava IK, Kuller L, Zarkikh I, Kraft Z, Fagrouch Z, Letvin NL, Heeney JL, Barnett SW, Stamatatos L. 2006. Immunization with HIV-1 SF162-derived envelope gp140 proteins does not protect macaques from heterologous simian-human immunodeficiency virus SHIV89.6P infection. Virology 349:276–289. doi: 10.1016/j.virol.2006.01.043. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.