

Abstract
Upregulation of β2 subunit-containing (β2*) nicotinic acetylcholine receptors (nAChRs) is implicated in several aspects of nicotine addiction, and menthol cigarette smokers tend to upregulate β2* nAChRs more than nonmenthol cigarette smokers. We investigated the effect of long-term menthol alone on midbrain neurons containing nAChRs. In midbrain dopaminergic (DA) neurons from mice containing fluorescent nAChR subunits, menthol alone increased the number of α4 and α6 nAChR subunits, but this upregulation did not occur in midbrain GABAergic neurons. Thus, chronic menthol produces a cell-type-selective upregulation of α4* nAChRs, complementing that of chronic nicotine alone, which upregulates α4 subunit-containing (α4*) nAChRs in GABAergic but not DA neurons. In mouse brain slices and cultured midbrain neurons, menthol reduced DA neuron firing frequency and altered DA neuron excitability following nAChR activation. Furthermore, menthol exposure before nicotine abolished nicotine reward-related behavior in mice. In neuroblastoma cells transfected with fluorescent nAChR subunits, exposure to 500 nm menthol alone also increased nAChR number and favored the formation of (α4)3(β2)2 nAChRs; this contrasts with the action of nicotine itself, which favors (α4)2(β2)3 nAChRs. Menthol alone also increases the number of α6β2 receptors that exclude the β3 subunit. Thus, menthol stabilizes lower-sensitivity α4* and α6 subunit-containing nAChRs, possibly by acting as a chemical chaperone. The abolition of nicotine reward-related behavior may be mediated through menthol's ability to stabilize lower-sensitivity nAChRs and alter DA neuron excitability. We conclude that menthol is more than a tobacco flavorant: administered alone chronically, it alters midbrain DA neurons of the nicotine reward-related pathway.
SIGNIFICANCE STATEMENT Menthol, the most popular flavorant for tobacco products, has been considered simply a benign flavor additive. However, as we show here, menthol alone exerts several neurobiological changes. We are among the first to show that menthol, by itself, increases the number of nicotinic acetylcholine receptors (nAChRs) in the mouse brain. It does so at a dose that matches nicotine in its ability to increase nAChR number. At this same dose, menthol also alters midbrain dopamine neuron function and prevents nicotine reward-related behavior. Together, our data show that menthol is more than an “inert” flavor additive and is able to change the function of midbrain dopamine neurons that are part of the mesolimbic reward pathway.
Keywords: dopamine neuron, electrophysiology, menthol, nicotine, nicotinic receptor, reward
Introduction
Tobacco use is the leading preventable cause of death in the United States (World Health Organization, 2008). Nicotine, the primary addictive component of tobacco products, upregulates and changes the subunit stoichiometry of nicotinic acetylcholine receptors (nAChRs) containing the β2* subunit (β2* nAChRs) in humans, nonhuman primates, and rodents, as well as in primary cultures and cell lines derived from these species (Breese et al., 1997; Nashmi et al., 2007; Mukhin et al., 2008; Lester et al., 2009; Govind et al., 2012; Srinivasan et al., 2012; Brody et al., 2013; Henderson and Lester, 2015). Although midbrain dopaminergic (DA) neurons dominate the nicotine reward-related pathway, chronic nicotine does not change the α4 subunit-containing (α4*) nAChR number in DA neurons of the ventral tegmental area (VTA) or substantia nigra pars compacta (SNc) but upregulates α4* nAChRs in the GABAergic neurons of the substantia nigra pars reticulata (SNr; Nashmi et al., 2007; Xiao et al., 2009). α6 Subunit-containing (α6*) nAChRs in DA neurons of the VTA and SNc are upregulated by chronic nicotine (Henderson et al., 2014). These changes result in increased inhibitory tone from SNr GABAergic neurons and decreased firing frequency of VTA and SNc DA neurons (Nashmi et al., 2007; Xiao et al., 2009). In consequence, disinhibition and enhanced excitation lead to enhanced DA neuron burst firing and enhanced DA release (Sulzer, 2011; Faure et al., 2014). These changes are likely a primary contributor to nicotine dependence.
Menthol is the sole remaining flavor additive that is allowed in cigarettes, and “menthol cigarettes” are used by a third of all smokers and a majority of African-American smokers (∼84%; McCarthy et al., 1995). Recently, it was revealed that almost all cigarettes contain menthol even if they are not explicitly labeled as such (Ai et al., 2015). Smokers of menthol cigarettes exhibit more β2* nAChR upregulation in several brain regions compared with nonmenthol smokers (Brody et al., 2013). It is unclear whether this additional upregulation occurs because menthol enhances nicotine's pharmacology, alters the metabolism of nicotine, or acts independently. Of note, smokers of menthol cigarettes have more difficulty quitting when compared with smokers of nonmenthol cigarettes (Gandhi et al., 2009).
Menthol is a negative allosteric modulator of α4β2 nAChRs and a noncompetitive antagonist of α7 and α3β4 nAChRs, with IC50 values of ∼111, ∼35, and ∼70 μm respectively (Hans et al., 2012; Ashoor et al., 2013; Ton et al., 2015). These concentrations probably exceed those found in vivo (Benowitz et al., 2004) and may not explain the observations of Brody et al. (2013). It is also suggested that menthol decreases airway irritation, and allows smokers to inhale more nicotine; or menthol may slow the metabolism of nicotine, allowing elevated concentrations of nicotine in the plasma (Benowitz et al., 2004; Alsharari et al., 2015). Nevertheless, some still suggest that menthol's ability to alter nicotine metabolism may not be sufficient to increase plasma levels of nicotine (Ashley et al., 2012).
If menthol does increase nicotine in the plasma, this may be an insufficient explanation for enhanced upregulation observed in human menthol cigarette smokers (Brody et al., 2013). Human studies using 2-[18F]fluoro-A-85380 (binds β2* nAChRs) indicate that β2* nAChR upregulation in humans is saturated with only a few cigarettes smoked daily (Staley et al., 2006), probably because β2* nAChRs are saturated even at nicotine concentrations found in low-nicotine cigarettes (Brody et al., 2009). Menthol may increase nicotine concentration in human blood by ∼20% (Ahijevych and Garrett, 2004; Benowitz et al., 2004), and this may be insufficient to explain the menthol effect noted in humans (Brody et al., 2013). Thus, actions in the airways or in the liver may not fully explain the role of menthol in nicotine dependence. This led us to hypothesize that prolonged exposure to smoking-relevant levels of menthol may act independently of nicotine to change nAChRs. To address this hypothesis, we used a combination of in vivo and in vitro methods to characterize menthol's effect on nicotine reward-related behavior and on nAChR number, function, and stoichiometry.
Materials and Methods
Reagents.
(±)-Menthol (product #63670), CI-976 (product #C3743), acetylcholine chloride (product #A6625), and (−)-nicotine hydrogen tartrate (product #1463304) were obtained from Sigma-Aldrich. All nicotine concentrations listed (both molarity and milligrams per kilogram) use the “free base” convention (Matta et al., 2007), with a molecular weight for nicotine of 162.23 g/mol. Models 1002 and 1004 osmotic minipumps were obtained from Alzet.
Animals.
All experiments were conducted in accordance with the guidelines for care and use of animals provided by the National Institutes of Health, and protocols were approved by the Institutional Animal Care and Use Committee at the California Institute of Technology. The α6-eGFP bacterial artificial chromosome (BAC) transgenic strain used in this study possessed six copies of the α6-eGFP BAC and has been described previously (Mackey et al., 2012; Henderson et al., 2014). The α4-mCherry knock-in mouse strain and the β3-GFP nAChR knock-in mouse strain have been described previously (Shih et al., 2014; Srinivasan et al., 2016). The Gene Expression Nervous System Atlas (GENSAT) TH-eGFP strain (Gong et al., 2003) was purchased from the Mutant Mouse Regional Resource Center. All experiments using mice included both sexes in approximately equal numbers. In total, 71 mice were used to accumulate the data in this manuscript. Of those, 33 were male and 38 were female. No sex differences were noted.
Menthol dose selection.
We studied effects of (±)-menthol (referred to as menthol from this point). We estimated the pharmacologically relevant dose of menthol by analogy with nicotine doses used in mouse studies. The typical menthol cigarette contains 1–20 mg of menthol (Ai et al., 2015) and ∼1 mg of nicotine (Rodgman and Perfetti, 2009). Therefore, menthol per cigarette is 1–5 times that of nicotine for the typical menthol cigarette, but up to 20 times greater for some menthol cigarette brands. Steady-state and peak concentrations of nicotine, as found in human smokers, are replicated in mice using 0.4 and 2.0 mg/kg/h doses of nicotine, respectively (Matta et al., 2007). Conditioned place preference (CPP) assays use 0.5 mg/kg nicotine (Tapper et al., 2004). Assays for in vivo upregulation use 2.0 mg/kg/h nicotine (Nashmi et al., 2007; Henderson et al., 2014). We selected 1 mg/kg and 2 mg/kg/h menthol for CPP and in vivo upregulation assays, respectively (Figs. 1, 2). Both dose selections fall within the 1:5 menthol/nicotine ratio of a typical “menthol cigarette.”
Figure 1.

Chronic menthol alone upregulates α4* and α6* nAChRs in vivo. A, Montage of 60× images from a vehicle-treated α4-mCherry mouse. Scale bar, 100 μm. B, 60× images of α4-mCherry direct fluorescence in the VTA, SNc, and SNr of mice treated with vehicle or menthol (2 mg/kg/h, 10 d). Scale bars, 10 μm. D, Montage of 60× images from a vehicle-treated α6-GFP mouse. Scale bar, 100 μm. E, 60× images of α6-GFP direct fluorescence in the VTA and SNc of mice treated with vehicle or menthol. Scale bars, 10 μm. B, E, Display lookup table “fire” was used in ImageJ. C, F, Quantification of α4-mCherry and α6-GFP intensities; transparent circles are mean intensities of individual mice. G, Representative TIRFM images of Neuro-2a cells transfected with α4-SEP and β2 nAChR subunits or with α6-SEP, β2, and β3 nAChR subunits. Menthol (500 nm) was added 24 h before imaging. Scale bars, 10 μm. In each panel, the left image is at pH 7.4 and the right image is the same cell at pH 5.4. H, PMID was quantified for SEP nAChRs. I, RNA transcript levels of α4 and β2 nAChR subunits in Neuro-2a cells, detected by RNA sequencing, are unaffected by 500 nm menthol treatment (24 h). For all panels, data are mean ± SEM; *p < 0.05; **p < 0.01; ***p < 0.005 (unpaired t test). For all panels, number in parenthesis indicates n value.
Figure 2.

In midbrain DA neurons, chronic menthol does not significantly alter ACh-induced response amplitudes but accelerates their decay and decreases the baseline firing rate. A–D, Midbrain slices; VTA DA neurons, bregma ∼−3.1 mm. B1, B2, Fluorescent neurons in slices from α6-GFP mice. C1, Currents induced by ACh puffs. C2, Baseline current-clamp traces for no drug-treated and for menthol-treated mice. E–H, Midbrain cultures; putative DA neurons. E1–E4, Fluorescent neurons from β3-GFP or TH-GFP mice. F1, Currents induced by ACh puffs. F2, Baseline current-clamp traces for no drug-treated and for chronic menthol-treated neurons. D1, D2, G1, G2, Summary of ACh response amplitude and decay time constant. D3, Histograms for DA neuron firing frequency for vehicle-treated and menthol-treated mouse brain slices (n = 16 and 11 for vehicle treated and menthol treated, respectively). G3, Histograms for DA neuron firing frequency for no drug-treated and menthol-treated cultured neurons (n = 23 and 31 for no drug treated and menthol treated, respectively). E–H, Chronic menthol treatment with cultured midbrain DA neurons was 500 nm, 7–10 d. H, Chronic menthol alters action potential properties of midbrain DA neurons. H1, Mean action potential spike waveform for a no drug-treated and menthol-treated midbrain DA neuron. H2, H3, Quantification of antipeak amplitude and action potential half-width. H2, n = 12 for both no drug-treated and menthol-treated cultured DA neurons. H3, n = 19 and 26 for no drug-treated and menthol-treated cultured DA neurons, respectively. In all cases (C1, F1), arrows indicate 100 ms puff of 300 μm ACh. D1, D2, G1, G2, For all bar graphs, number in parenthesis indicates n value. Data are mean ± SEM; *p < 0.05; **p < 0.005 (unpaired t test).
In vitro experiments with mouse neuroblastoma 2a (Neuro-2a) cells and cultured midbrain neurons were performed with 500 nm menthol (24 h and 7–10 d, respectively). We chose this concentration from preliminary menthol concentration–response studies. At 50 nm menthol, upregulation was less than at 500 nm. At 5 μm menthol, the data were too variable for systematic study. Therefore, 500 nm menthol provided the most consistent and robust upregulation response with nAChRs transiently expressed in Neuro-2a cells.
We attempted to determine the concentration of menthol in the mouse brain following our long-term exposure paradigm (see Figs. 1–4). An experimental assay that could detect menthol at concentrations ≥2.5 μm in brain tissue was developed. When the assay was used on brains from mice implanted with osmotic minipumps, no measurable amount of menthol was detected. Therefore the menthol concentration in the brains was <2.5 μm, and possibly much less.
Figure 3.

Long-term menthol treatment prevents nAChR-induced acceleration of DA neuron firing frequency. A, Current-clamp recordings from cultured midbrain DA neurons treated with no drug (A1) or treated with 500 nm menthol over 10 d (A2) before and after an ACh puff. B1, Time course of firing frequency before and after a 100 ms ACh puff. Dashed lines are mean firing frequency for no drug-treated and menthol-treated neurons before the ACh puff. B2, Quantification of firing frequency in no drug-treated and menthol-treated conditions before and after ACh puff. Mean for pre-ACh (−) and post-ACh (+) puff are the 3 s before and after the ACh puff. Data are mean ± SEM; *p < 0.05; **p < 0.001 (unpaired t test). In all cases, arrows indicate 100 ms puff of 300 μm ACh. C, Current-clamp recordings from cultured midbrain DA neurons treated with 500 nm menthol (10 d) without sulpiride (C1) or with acute exposure to 1 μm sulpiride (C2). In all traces, arrows show a 100 ms puff of 300 μm ACh. Number in parenthesis indicates n value.
Figure 4.

Chronic menthol before nicotine exposure abolishes nicotine reward-related behavior. A, Schematic of CPP assay. Mice were implanted with osmotic minipumps for 20 d to continuously deliver 2 mg/kg/h menthol or vehicle 10 d before and during CPP training. B, Saline control mice did not exhibit a change in baseline preference. Mice implanted with osmotic minipumps delivering vehicle exhibited reward-related behavior to 0.5 mg/kg nicotine, but those receiving menthol exhibited no reward-related behavior to 0.5 mg/kg nicotine. Data are mean ± SEM; one-way ANOVA, F = 8.28; *p < 0.05, post hoc Tukey's test. Number in parenthesis indicates n value. C, Mice exhibited no significant preference for the white or black chambers during the initial test phase.
Chronic drug administration with osmotic minipumps.
Chronic menthol or vehicle was administered to mice using osmotic minipumps (model 1002, Alzet) following a procedure that has been reported previously (Henderson et al., 2014). Vehicle was 60% ethanol and 40% saline. Given the infusion rate of the osmotic minipumps (0.25 μl/h), ∼4.2 mg/kg ethanol was delivered to mice each hour for a total of 0.1 g/kg in a 24 h period. This is 45–100-fold below the dose that has been used to study the reinforcing effects of ethanol with C57BL/6 mice (Grahame and Cunningham, 1997). Because the control solutions in the osmotic minipumps also contained 60% ethanol, we have confidence that effects attributed to menthol in this study do not include effects of ethanol.
CPP assays.
An unbiased protocol was used, where nicotine (0.5 mg/kg) was given in the white chamber on “drug” days and saline was given in the black chamber. A standard three-chamber rectangular cage was used where one compartment was black with a stainless steel grid floor and the other was white with a stainless steel mesh floor (Med Associates). The floor of the middle compartment was lined with smooth polyvinyl chloride to make it a neutral zone. Lighting was equilibrated so that there was minimal bias for any of the three chambers. The test consisted of three stages over a 10 d period (Tapper et al., 2004). Each conditioning period was 20 min. The interval between drug injections and conditioning was <1 min as mice were injected and placed directly in the conditioning chamber for their 20 min training period. In the first stage, a mouse was placed in the central compartment and allowed free access to all chambers for 20 min. During stage 2, drug injections were given in the black (saline) or white (nicotine, dissolved in saline) chambers. The mice received intraperitoneal drug injections in the drug-associated conditioning chamber on days 2, 4, 6, and 8, and received intraperitoneal saline injections in the saline-associated chamber on days 3, 5, 7, and 9. Drug-naive mice that exhibited a severe bias toward one of the two conditioning chambers (defined as >65% time spent in one chamber) during stage 1 were excluded from the study. Following exclusions, no initial bias was observed in mice (see Fig. 3). Adult male and female C57BL/6 mice (3–6 months old) were used in CPP assays. In total, 14 males and 17 females were used (menthol osmotic minipump group, 0.5 mg/kg nicotine injection: four males, six females; vehicle osmotic mini-pump group, 0.5 mg/kg nicotine injection: four males, four females; saline only group: six males, seven females). No sex differences were observed.
Spectral confocal imaging.
Imaging mouse brain slices and cultured Neuro-2a cells containing fluorescent nAChRs followed procedures and instrumentation previously reported (Srinivasan et al., 2012; Henderson et al., 2014). An Eclipse C1si laser-scanning confocal microscope equipped with a 60× 1.2 numerical aperture VC Plan Apo water objective and 32 photomultiplier tubes was used for confocal imaging. Before an imaging session using cultured Neuro-2a cells, cell culture medium was replaced with phenol red-free CO2-independent Leibovitz (L-15) medium (Invitrogen). eGFP and mCherry were excited at 488 and 561 nm respectively. eGFP and mCherry fluorescence were obtained. eGFP and mCherry fluorescence emission spectra were captured and images were unmixed using standard spectra acquired from cells expressing eGFP or mCherry only.
For in vivo nAChR upregulation assays, male and female α6-GFP and α4-mCherry mice (∼3 months old) were used. No sex differences were observed.
Donor recovery after acceptor photobleaching.
We examined Förster resonance energy transfer (FRET) between nAChR subunits using the acceptor photobleaching method (Nashmi et al., 2003; Drenan et al., 2008a). FRET was detected by recording GFP dequenching during incremental photodestruction of mCherry. Images were acquired of mCherry bleaching at eight time points, at 10 s intervals, and at a laser excitation of 561 nm. The bleaching protocol was optimized to achieve ∼80% photodestruction of mCherry while still recording increases in GFP emission at each time point. Spectral images were unmixed into their GFP and mCherry components. GFP and mCherry components were saved in Microsoft Excel format and fluorescence intensities were normalized to the prebleach time point (100%). FRET efficiency (E) was calculated as E = 1 − (IDA/ID), where IDA represents the normalized fluorescence intensity of GFP (100%) in the presence of both donor (GFP) and acceptor (mCherry), and ID represents the normalized fluorescence intensity of GFP in the presence of donor only (complete photodestruction of mCherry). The ID value was extrapolated from a scatter plot of the fractional increase of GFP versus the fractional decrease of mCherry. Data are reported as mean ± SEM. In figures representing FRET assays, only a single imaging session is represented; however, each result has been repeated in ≥3 sessions on separate days with similar or identical results.
Brain slice preparation (electrophysiology).
Recordings were performed using brain slices prepared from 7–11-week-old C57BL/6 [wild type (WT)] mice or α6-GFP nAChR mice, using the protocol described with some modifications (Ye et al., 2006; Xiao et al., 2009). Male and female mice were used; but no sex differences were observed. Mice were killed with CO2, and were subject to cardiac perfusion with ice-cold modified glycerol-based artificial cerebral spinal fluid (GACSF) saturated with 95% O2/5% CO2 (carbogen) containing (mm) the following: 250 glycerol, 2.5 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 26 NaHCO3 and 11 glucose. The brain was subsequently removed and sliced with a microslicer (DTK-1000, Ted Pella), while immersed in GACSF. Brain slices were allowed to recover at 32°C in a holding chamber filled with carbogenated ACSF containing (mm) the following: 125 NaCl, 2.5 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 26 NaHCO3, and 11 glucose. One hour later, the holding chamber with slices was placed at room temperature. One of the slices was transferred into the recording chamber and superfused (1.5–2.0 ml/min) with carbogen-saturated ACSF at 32 ± 0.5°C.
Patch-clamp electrophysiology.
Neurons and Neuro-2a cells were visualized with an upright microscope (BX50WI, Olympus) and near-infrared or blue illumination (the latter for visualizing fluorescent proteins). Whole-cell patch-clamp techniques were used to record electrophysiological signals with a MultiClamp 700B amplifier (Molecular Devices), Digidata 1322 analog-to-digital converters (Molecular Devices), and pClamp 9.2 software (Molecular Devices). Data were sampled at 10 kHz and filtered at 2 kHz. A patch electrode had a resistance of 4–6 MΩ when filled with intracellular solutions. Those solutions were as follows: solution 1 (in mm, for most experiments): 135 K-gluconate, 5 KCl, 5 EGTA, 0.5 CaCl2, 10 HEPES, 2 Mg-ATP, and 0.1 GTP; solution 2 (in mm, for particular experiments as stated): 70 K-gluconate, 65 KCl, 5 EGTA, 0.5 CaCl2, 10 HEPES, 2 Mg-ATP, 0.1 GTP, and 2 QX-314. The pH of these solutions was adjusted to 7.2 with Tris-base, and their osmolarity was adjusted to 300 mOsm with sucrose. The junction potential between patch pipette and bath solutions was nulled just before gigaseal formation. The bath was continually perfused with ACSF. All recordings were done at a temperature of 32 ± 1°C.
For cultured cells, 50,000 Neuro-2a cells were plated onto sterilized 12-mm-diameter glass coverslips (Deckgläser), placed in 35 mm culture dishes, and cultured in a humidified incubator (37°C; 95% air, 5% CO2). Cells were transfected as described below.
Acetylcholine (ACh) was dissolved in extracellular solution containing (in mm) the following: 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose (320 mOsm, pH set to 7.3 with Tris-base). This solution with dissolved ACh was puffed (0.1 or 0.3 s; 20 psi) onto voltage-clamped cells or neurons (holding potential, −65 mV). To avoid receptor desensitization by repetitive ACh application, we applied ACh at ∼3 min intervals and continually perfused the recording chamber with extracellular solution.
For concentration–response curves (see Fig. 5), drug applications were performed using an Octaflow II perfusion system (0.5 s, 8 psi; ALA Scientific). Curve fitting was completed with KaleidaGraph 4.5 and plotted in Origin 10.
Figure 5.
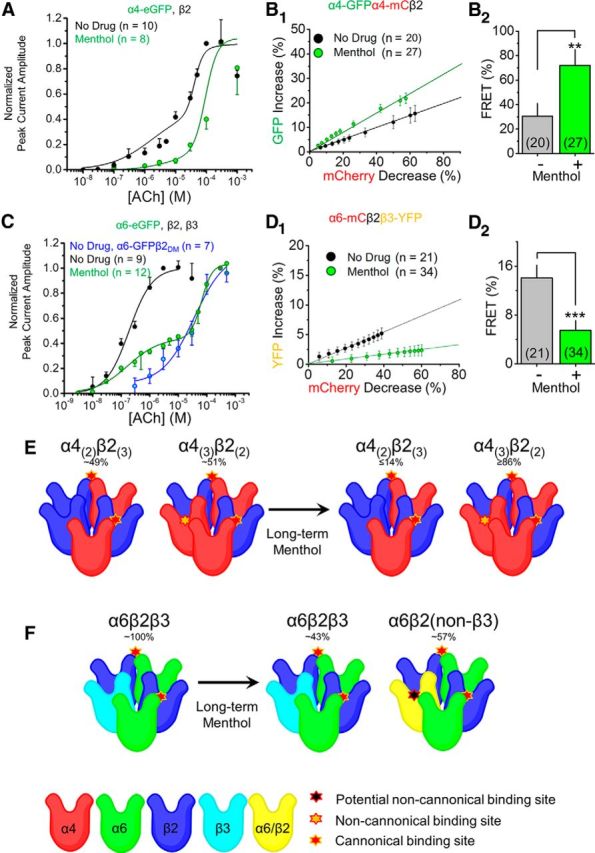
Chronic menthol alters nAChR stoichiometry and favors low-sensitivity nAChRs. A, Concentration–response curves of Neuro-2a cells transfected with α4-eGFPβ2 nAChRs treated with no drug (black) or with menthol (red; 24 h, 500 nm). C, Concentration–response curves of Neuro-2a cells transfected with α6-eGFP, β2, and β3 nAChR subunits treated with no drug (black), with menthol (red), or with α6-eGFPβ2DM nAChRs with no drug (blue). B1, D1, Linear plot of donor dequenching versus acceptor (mCherry) photodestruction for no drug and for menthol (24 h, 500 nm) treatments. B2, D2, Mean FRET efficiency. FRET efficiency was calculated from the linear plot shown in B1 and D1 as described previously (Drenan et al., 2008a; Nichols et al., 2014). B2, D2, Data are mean ± SEM; **p < 0.01; ***p < 0.001 (unpaired t test). E, F, Summary of menthol-induced changes in nAChR stoichiometry. E, α4β2 nAChRs assemble in two stoichiometries: (α4)2(β2)3 and (α4)3(β2)2. We found that under no drug treatment the α4β2 nAChR pool is mixed. Following 24 h menthol treatment, α4β2 nAChRs assemble almost entirely in the low-sensitivity (α4)3(β2)2 stoichiometry. F, α6β2β3 nAChRs assemble efficiently under no drug treatment, but following 24 h menthol treatment the receptor pool is changed to a mixture of α6β2β3 and α6β2 nAChRs. Number in parenthesis indicates n value.
Total internal reflection fluorescence microscopy.
Cultured Neuro-2a cells were imaged live at 37°C in a stage-mounted culture dish incubator (Warner Instruments) using methods and instrumentation reported previously (Richards et al., 2011; Henderson et al., 2014). Total internal reflection fluorescence microscopy (TIRFM) enables the visualization of fluorescently labeled intracellular molecules within ∼250 nm of the cell–coverslip interface. Just before imaging, growth medium was exchanged for extracellular solution (150 mm NaCl, 4 mm KCl, 10 mm HEPES, 2 mm MgCl2, 2 mm CaCl2, and 10 mm glucose) adjusted to the appropriate pH (5.4 or 7.4). Superecliptic pHluorin (SEP) was excited at 488 nm with an air-cooled argon laser (IMA101040ALS, Melles Griot). We acidified the imaging dish by perfusing the bath, normally at pH 7.4, with an otherwise identical solution adjusted to pH 5.4. The plasma membrane integrated density (PMID) was determined by taking an initial TIRFM image of each cell at pH 7.4 followed by acidification of the solution and a subsequent low-pH image (pH 5.4). Low-pH images were used to demarcate endoplasmic reticulum-localized nAChRs that were subtracted from the total footprint to determine the PMID of nAChRs.
Neuro-2a cell culture and transient transfections.
Neuro-2a cells were cultured using standard techniques (Srinivasan et al., 2011). Cells were plated by adding 90,000 cells to poly-d-lysine-coated 35 mm glass-bottom imaging dishes (MatTek) and cultured in a humidified incubator (37°C; 95% air, 5% CO2). Cells were transfected with 500 ng of each nAChR subunit cDNA. In assays using two fluorescent variants of the same nAChR subunit (α4-GFP and α4-mCherry; see Fig. 5B1,B2), 250 ng of each subunit were used so that the total amount per subunit was 500 ng. Plasmids were mixed with 250 μl of Opti-MEM. Lipofectamine 2000 was separately added to 250 μl of Opti-MEM. After 5 min at 24°C, DNA and lipofectamine solutions were mixed together and incubated for 25 min at 24°C. The solutions were then added to preplated Neuro-2a cells and incubated for 24 h. After 24 h, the Opti-MEM was removed and replaced with growth medium. Fifty nanomolars filter-sterilized nicotine was added after replacing the Opti-MEM with standard culture medium (α6β2β3 nAChRs). For α4β2 nAChRs, 100 nm nicotine was present for 48 h (nicotine was added at the time of transfection and then replenished when the media was changed). CI-976 was added (20 μm) with nicotine 24 h before imaging. Cells were imaged 48 h after transfection.
We used different concentrations of nicotine (50 or 100 nm) for α6β2β3 and α4β2 nAChRs because we have found that robust and consistent upregulation of nAChR number in TIRFM assays occurs at these concentrations using Neuro-2a cells (Richards et al., 2011; Henderson et al., 2014).
Cultured midbrain neurons.
To culture primary midbrain neurons from mouse embryos, ventral midbrain tissue from embryonic day 14 embryos was dissected out and cultured using previously published procedures (Srinivasan et al., 2016). Following digestion with papain (15 min at 37°C), cells were separated by DNase treatment and trituration, and plated at a density of 100,000 cells per dish in poly-l-ornithine and laminin-coated imaging dishes containing Neurobasal medium supplemented with B27, Glutamax, 1% Hyclone equine serum, and 100 μm ascorbate. Either control medium or 200 nm nicotine was added to the culture for 2 weeks from day 7 to day 21. Media were changed every 3 d. Recordings (peak currents, hyperpolarization-activated current, action potential duration, and baseline firing rates) did not differ among putative DA neurons from WT, β3-eGFP, or TH-eGFP cultures (See Table 1).
Table 1.
Comparison of electrophysiological characteristics among cultured midbrain neurons derived from WT, β3-eGFP, and TH-eGFP mice
| Peak current amplitude | Firing frequency | |
|---|---|---|
| WT | 54.4 ± 12.3 pA | 3.9 ± 1.9 Hz |
| β3-eGFP | 46.9 ± 8.6 pA | 3.2 ± 0.7 Hz |
| TH-eGFP | 53.8 ± 17.7 pA | 4.1 ± 0.8 Hz |
Values are represented as mean ± SEM; n = 9–13 neurons for each genotype.
Single-cell harvesting and RNA extraction using cultured midbrain DA neurons.
Cells were harvested on day 21 in culture, following previously reported procedures (Henley et al., 2013; Srinivasan et al., 2016). Glass capillary tubing (Kimax-51; 1.5–1.8 mm outer diameter) was cleaned by sonication in ethanol and then baked overnight at 200°C to inactivate RNase. Large-bore glass micropipettes (10–30 μm tip diameter) were pulled from the tubing using a Sutter P-50 microelectrode puller. Before harvesting, we removed the media from the culture dishes, rinsed them with 2 ml of Dulbecco's PBS, and replaced 1 ml of Dulbecco's PBS remaining in the dish for harvesting. Individual cultured neurons were identified using a microscope equipped with a 40× phase objective. The pipette was positioned over the cell soma using a motorized micromanipulator (Sutter Instrument); the cell was aspirated into the glass micropipette using gentle suction. The micropipette containing the cell was immediately removed from the bathing solution, placed inside a plastic, RNase-free microcentrifuge tube containing 1.5 μl of RNase-free PBS and 0.5 μl of an RNase inhibitor solution, and its tip was broken against the side of the tube near the bottom. We applied outward pressure to the broken micropipette using a large-gauge syringe needle to expel the remaining fluid at the tip of the micropipette into the tube. The tube was then centrifuged briefly using a desktop centrifuge and then frozen on dry ice. Typically, 16 cells were harvested per dish over a 1 h period at ambient temperature. The resultant mRNAs were reverse transcribed and amplified to construct single-neuron high-yield cDNA sequencing libraries.
Laser-capture microdissection of adult mouse SNc neurons.
As previously described in detail (Henley et al., 2013), whole brains from 4-month-old male mice were collected (postmortem interval of <5 min), fresh frozen over dry ice, and stored at −80°C. Midbrain cryostat sections (20 mm) were mounted on UV-treated Zeiss membrane slides (1.0 polyethylene naphthalate nuclease-free), air dried for 5 min, and stained with cresyl violet for 1 min. The sections were rinsed, dried, and then visualized under bright-field illumination at 400× magnification on a Zeiss Palm laser capture microdissection microscope. Twenty putative DA+ cell bodies from the SNc were dissected using multiple low laser energy pulses, and were catapulted into Zeiss 200 μl adhesive caps. Cell lysis solution (Illumina) containing 30 SMART reverse transcription primers and quantitation controls (“spikes”) were then added into the pool of cells before freezing.
Harvesting of RNA from Neuro-2a cells.
We used a mirVana miRNA Isolation Kit (Life Technologies) to prepare RNA samples. Cells were processed fresh (i.e., not frozen). Media in dishes containing neuro-2a cells transiently transfected with α4-SEP and β2 nAChR subunits were aspirated, rinsed in 1 ml PBS, and then the PBS wash was aspirated. Neuro-2a cells were lysed directly in the culture plate using 600 μl of lysis/binding solution. The lysate was collected with a rubber spatula and pipetted into a tube where it was pipetted to obtain a homogenous lysate. miRNA homogenate additive (one-tenth volume of lysate) was added to the tube containing the lysate, vortexed, and left on ice for 10 min, and then acid-phenol–chloroform was added at a volume equal to lysate. Lysate was vortexed for 60 s and then centrifuged for 5 min at 10,000 × g at room temperature. The aqueous phase was then removed and transferred to a fresh tube. One hundred percent ethanol was added (1.25 times the volume of lysate) to the aqueous phase. The ethanol/lysate mixture was pipetted onto a filter cartridge and centrifuged for 15 s, repeatedly, until all of the ethanol/lysate mixture passed through the filter. Seven hundred microliters of miRNA Wash Solution 1 was added to the filter and the filter was centrifuged for 10 s. Five hundred microliters of Wash Solution 2/3 was added to the filter and then centrifuged for 10 s (twice). One hundred microliters of preheated (95°C) elution solution was used to recover the RNA. The eluate, containing the RNA, was stored at −80°C with 2.5 μl RNase inhibitor. A Turbo DNA-free Kit (Life Technologies) was used to remove contaminating DNA from RNA preparations.
RNA sequencing.
We prepared amplified cDNA from RNA using Clontech's SMARTerTM Ultra Low RNA system for Illumina Sequencing (Clontech). We used Clontech's Advantage 2 PCR system for amplification of cDNA templates by long-distance PCR. Illumina's Nextera DNA Sample Preparation kit with Tn5-mediated tagmentation technology (Epicenter) was used as an in vitro transposition method to simultaneously fragment and tag the cDNA libraries with Illumina-compatible sequencing primers. After quality control measures of yield and fragment length distribution were taken using the Qubit fluorometer (Invitrogen) and the Agilent Bioanalyzer, 100 bp sequencing reads were generated on the Illumina HiSeq instrument. Each sequencing library generated >20 million unique mapping reads.
RNA sequencing computational analysis.
One hundred base pair sequence tags were mapped to the mouse genome using TopHat 1.3.2. We quantified transcript abundance [fragments per kilobase per million mapped reads (FPKM)] using Cufflinks. We annotated the transcripts with genome annotations provided by Ensembl. We conducted pairwise comparisons and calculated statistical values using Cuffdiff, part of the Cufflinks suite, to identify differentially expressed genes. We assessed ontology with DAVID (Database for Annotation, Visualization and Integrated Discovery; Huang da et al., 2009). Cuffdiff data with FPKM >1, a corrected p value of <0.05 and a fold difference of >1.5 were given as input to Ingenuity Pathway Analysis (Qiagen) for pathway analysis.
Results
Menthol alone upregulates α4* and α6* nAChR subunits in vivo
How does menthol enhance nicotine-induced upregulation? One possibility is that menthol upregulates nAChRs, independent of nicotine. To test this idea, we used mice containing mCherry-labeled α4* nAChRs or GFP-labeled α6* nAChRs and a “chronic” menthol concentration of 2 mg/kg/h for 10 d. Previous studies show that these fluorescent nAChR subunits assemble appropriately with nonlabeled subunits, traffic properly, and function in a manner comparable to WT nAChRs (Nashmi et al., 2007; Mackey et al., 2012; Henderson et al., 2014; Shih et al., 2014). In midbrain, both DA neurons and GABAergic neurons express α4* nAChRs (Nashmi et al., 2007; Mackey et al., 2012). In GABAergic neurons of the SNr, the fluorescence intensity of α4-mCherry nAChRs did not change following menthol treatment (Fig. 1A–C). In contrast, the fluorescence intensities of α4-mCherry* nAChRs were 58.5 and 54.2% greater in VTA and SNc neurons, respectively, when compared with those of vehicle-treated mice (Fig. 1A–C; p < 0.05).
In midbrain, >90% of TH-positive neurons express α6* and β3* nAChRs; GABAergic neurons do not (Drenan et al., 2008b; Mackey et al., 2012; Srinivasan et al., 2016). The fluorescence intensities of α6-GFP neurons in the VTA and SNc were 75.0% and 46.5% greater in menthol-treated mice when compared with vehicle-treated mice (Fig. 1D–F; p < 0.05). These data show that menthol alone upregulates α4* and α6* nAChR subunits in midbrain DA neurons.
Given that no α4* upregulation was noted in GABAergic neurons, menthol seems to exert some cell specificity. We have found that nicotine also exerts a cell-specific effect as it upregulates α4* nAChRs in somata of GABA neurons but does not upregulate α4* nAChRs in somata of DA neurons of the VTA or SNc (Nashmi et al., 2007). Despite observing cell-specific upregulation, we see that menthol and nicotine exhibit different cell-specific targets: menthol upregulates α4* nAChRs in somata of the VTA and SNc (Fig. 1) but nicotine upregulates α4* nAChRs only in somata of GABA neurons (Nashmi et al., 2007).
Menthol upregulates nAChRs on the PM
Using mice containing fluorescent nAChRs is an appropriate method to study nAChR upregulation in vivo, but is not readily amenable to study changes in nAChR number on the PM. To study nAChR subunit upregulation on the PM, we used nAChRs tagged to SEP, a pH-sensitive GFP analog, and TIRFM (Fig. 1G,H; Richards et al., 2011; Henderson et al., 2014; Nichols et al., 2014). We used transient heterologous expression in the neuron-like Neuro-2a cell line. As previously documented (Moss et al., 2009), Neuro-2a cells express transfected proteins at lower levels than various HEK293-derived cell lines. This allows one to control nAChR stoichiometry by varying the cDNA levels of each plasmid, and various trafficking processes remain unsaturated, so that individual steps can be studied in early and late exocytotic pathways (Son et al., 2009; Srinivasan et al., 2011, 2012; Xiao et al., 2011; Henderson et al., 2014). Neuro-2a cells also robustly express α6* nAChRs (Henderson et al., 2014).
Cells expressing α4-SEPβ2 nAChRs were treated (24 h) with no drug or with 500 nm menthol (Fig. 1G,H). α4-SEPβ2 nAChRs treated with menthol exhibited a significant (p < 0.05) twofold increase in PMID (Fig. 1H). Cells transfected with α6-SEP, β2, and β3 nAChR subunits, treated with menthol, exhibited a threefold increase in PMID (Fig. 1H), which is similar to previous observations with 24 h nicotine treatment (Henderson et al., 2014). We assessed gene expression on α4 and β2 nAChR subunits in transfected Neuro-2a cells treated with no drug or with 500 nm menthol for 24 h (Fig. 1I). We detected no change in α4 or β2 nAChR subunit transcripts in menthol-treated versus no-drug-treated Neuro-2a cells. From these data, we conclude that menthol alone robustly upregulates α4* and α6* nAChRs on the PM, via post-translational mechanism(s).
Chronic menthol exposure alters native nAChR function
In addition to increasing nAChR number in midbrain neurons, nicotine also alters the function of midbrain neurons of the DA reward pathway (Nashmi et al., 2007; Xiao et al., 2009; Faure et al., 2014). Thus, using electrophysiology, we investigated how long-term menthol treatment affects the function of midbrain VTA neurons (Fig. 2). As mentioned, α6* nAChRs in the midbrain are selectively expressed in DA neurons of the VTA and SNc (Drenan and Lester, 2012; Mackey et al., 2012). We prepared brain slices from α6-GFP mice to facilitate identification of VTA DA neurons (Fig. 2A,B1,B2). Interestingly, treatment with menthol (2.0 mg/kg, 10 d) did not significantly change peak current amplitude (Fig. 2C1,D1), but did accelerate the decay phase of nAChR currents stimulated by 300 μm ACh (100 ms puff; Fig. 2C1,D2). This functional modification of nAChRs following chronic menthol treatment was not accompanied by a significant change in baseline firing rates of VTA DA neurons (Fig. 2C2,D3; 4.25 ± 0.61 and 3.85 ± 0.88 Hz for vehicle and menthol-treated mice, respectively).
We also examined effects of long-term, submicromolar menthol (500 nm, 7–10 d) on native nAChRs using cultured midbrain neurons (Fig. 2E,H). To facilitate the identification of DA neurons, we used primary cultures from β3-eGFP knock-in mice (like α6*, selectively expressed on DA neurons of the VTA and SNc; Shih et al., 2014) or from TH-eGFP mice (Fig. 2E1–E4; Srinivasan et al., 2016). Recordings (peak currents, hyperpolarization-activated current, action potential duration, and baseline firing rates) did not differ among putative DA neurons from WT, β3-eGFP, or TH-eGFP cultures (Table 1). We chose not to use cultures from α6-GFP embryos because these arise from heterozygote × WT crosses (Mackey et al., 2012; Srinivasan et al., 2016) and produce fewer fluorescent neurons in culture than the available embryos from homozygote × homozygote crosses (β3-eGFP). We found that the TH-eGFP cultures gave the most robust fluorescent signals and allowed for easiest identification of putative DA neurons. Thus, the majority of electrophysiological data were acquired using cultures from TH-eGFP mice.
In agreement with recordings from mouse brain slices, long-term menthol significantly accelerated nAChR decay rates elicited by 300 μm ACh (100 ms puff; Fig. 2F1,G2) and did not alter peak current amplitude (Fig. 2F1,G1). Menthol treatment significantly decreased baseline firing rates of midbrain DA neurons (Fig. 2F2,G3). Additionally, we observed that menthol treatment increased the half-width of DA neuron action potentials (p < 0.05; Fig. 2H1–H3). With mouse brain slices and cultured midbrain neurons, we observed no effects of menthol when applied acutely (500 nm; data not shown).
Classical data show that nAChR agonists acutely increase firing of midbrain DA neurons (Brodie, 1991; Liu et al., 2012). Previous data also show that this effect is blunted by chronic nicotine treatment (Nashmi et al., 2007). Since menthol decreases baseline firing (Fig. 2F2,G3) similar to nicotine (Nashmi et al., 2007; Xiao et al., 2009), we studied whether menthol also alters the agonist-induced transient increase in DA neuron firing frequency. ACh application (300 μm, 100 ms) transiently increased cultured DA neuron firing frequency (Fig. 3A1,B1,B2). Following long-term treatment with menthol (500 nm, 7–10 d), cultured DA neurons displayed a transient decrease in firing frequency following ACh application (Fig. 3A2,B1,B2). Here we see that long-term menthol alone exerts an effect opposite to that of long-term nicotine. Nicotine treatment permits a transient increase in DA neuron firing frequency following nAChR agonist application (Nashmi et al., 2007). Menthol treatment, in contrast, causes a transient decrease in DA neuron firing frequency following nAChR agonist application. In 80% of the recordings, cultured DA neurons were hyperpolarized following an ACh puff (Fig. 3A2). In previous reports, midbrain DA neurons were similarly hyperpolarized by acute DA application (Ingram et al., 2002). To determine whether the hyperpolarization we observed in menthol-treated DA neurons is mediated through dopamine D2-like receptors [D2Rs, but possibly including D3 receptors (Collo et al., 2013)], we used the D2R antagonist sulpiride. Cultured midbrain DA neurons treated with 500 nm menthol (7–10 d) were recorded in current-clamp mode (as in Fig. 3A1,A2,B1,B2) and exposed to ACh (100 ms, 300 μm). DA neurons were then bath perfused with extracellular solution containing 1 μm sulpiride to block D2Rs, and then re-exposed to ACh (100 ms, 300 μm; Fig. 3C1,C2). DA neurons that were hyperpolarized following an ACh puff (Fig. 3C1) lacked this response during sulpiride exposure (Fig. 3C2). This suggests that menthol may also play a role in altering DA neuron excitability via somatic DA release that results from activation of D2Rs or activation of G-protein-activated inward rectifier channels.
Nicotine's ability to reduce DA neuron baseline firing frequency is believed to be one contributing feature of addiction as this may enhance the fraction of DA release during burst firing (Sulzer, 2011). Long-term menthol's ability to reduce baseline firing frequency of midbrain DA neurons may cause an analogous alteration in DA release. However, nAChR activation decreases DA neuron firing frequency transiently following long-term menthol treatment, which is opposite to what happens following acute nAChR activation, which has the classical effect of transiently increasing the firing frequency of DA neurons. Therefore long-term menthol treatment may alter nicotine reward-related behavior. To address this, we studied effects of long-term exposure to menthol on nicotine reward-related behavior.
Chronic menthol abolishes nicotine reward-like behavior
CPP is appropriate for studying nicotine reward-related behavior (Tapper et al., 2004). We tested how long-term (10 d) menthol treatment prior to nicotine exposure alters CPP. Similar to treatment before microscopy and electrophysiology measurements, mice were treated with vehicle or 2.0 mg/kg/h menthol for 10 d using osmotic minipumps; we then began the conventional 10 d training and testing regimen for CPP (Fig. 4A). On day 11 (after 10 d of vehicle or menthol exposure), mice were tested for initial preference, and training began on day 12 for CPP assays using nicotine (0.5 mg/kg). Final testing was conducted on day 20. During the training and testing for CPP (days 11–20), mice continued to receive vehicle or menthol (Fig. 4A). Mice receiving saline only exhibited no reward-related behavior (Fig. 4B). Mice treated long-term with vehicle and receiving 0.5 mg/kg nicotine in the drug-associated chamber during training exhibited a change in preference toward the drug-associated chamber (Fig. 4B). In contrast, mice treated long-term with menthol prior to receiving 0.5 mg/kg nicotine showed a slight change in preference away from the drug-associated chamber (Fig. 4B). Thus, long-term treatment with menthol displayed no measureable nicotine reward-related behavior (Fig. 4B; p < 0.05). In initial testing, none of the mice showed a severe bias toward the black or white chambers (Fig. 4C).
We observed this menthol-dependent decrease or abolition of nicotine reward-related behavior, despite recent data that simultaneous exposure to menthol and nicotine enhances nicotine reward and reinforcement (Gandhi et al., 2009; Ahijevych and Garrett, 2010; Wang et al., 2014). These contrasting observations point to the fact that menthol may act differently alone than when combined with nicotine. To identify the mechanism of how this may occur, we examined menthol's ability to alter nAChR assembly and stoichiometry.
Menthol alters α4β2 and α6β2* nAChR assembly and stabilizes lower-sensitivity nAChRs
Previous studies show that α4β2 nAChRs assemble in two stoichiometries: the higher-sensitivity and lower-sensitivity forms are (α4)2(β2)3 and (α4)3(β2)2, respectively (Nelson et al., 2003; Kuryatov et al., 2005). Similarly, α6β2* nAChRs assemble in at least two forms, depending on the inclusion of a β3 subunit: the (α6)2(β2)2β3 form has a higher agonist sensitivity than the α6β2 form (the stoichiometry of the latter is unknown; Tumkosit et al., 2006; Xiao et al., 2011; Henderson et al., 2014). Eponymously, these higher-sensitivity and lower-sensitivity stoichiometries are manifested by concentration–response curves (Nelson et al., 2003; Kuryatov et al., 2005). We therefore studied menthol's effect on ACh sensitivity with α4β2 and α6β2* nAChRs.
Neuro-2a cells were transiently transfected with α4-eGFPβ2 or α6-eGFPβ2β3 nAChRs and were treated with no drug or with menthol (500 nm, 24 h). As expected (Nelson et al., 2003), we observed a biphasic concentration–response curve with α4-GFPβ2 nAChRs (Fig. 5A; Table 2). Following menthol exposure, we observed a >85% monophasic concentration–response relationship with an EC50 value of 95.2 μm (Fig. 5A; Table 2). This suggests that menthol shifts the stoichiometry of α4β2 nAChRs toward a lower-sensitivity assembly [(α4)3(β2)2]. It is important to note that the (α4)3(β2)2 stoichiometry also has an inherent robust response to low agonist concentrations (Harpsøe et al., 2011; Wang et al., 2015), for instance, those produced by nicotine during smoking.
Table 2.
Concentration–response analysis reveals that chronic menthol alters α4β2 nAChR stoichiometry
| Treatment condition |
||
|---|---|---|
| α4-GFPβ2 no drug | α4-GFPβ2 + menthol | |
| EC50 value (higher sensitivity) | 1.8 ± 3.1 μm | 7.8 ± 4.9 μm |
| nh (higher) | 0.645 | 0.57 |
| EC50 value (lower sensitivity) | 41.9 ± 4.7 μma | 95.2 ± 14.0 μma,b |
| nh (lower) | 2.2 | 2.2 |
| Higher fraction | 0.49 | 0.14 |
Values are represented as mean ± SEM.
aEC50 values are statistically different from HS α4-GFPβ2 nAChRs (no drug). p < 0.005, unpaired t test.
bEC50 values are statistically different from lower-sensitivity α4-GFPβ2 nAChRs (no drug). p < 0.005, unpaired t test.
In cells transiently transfected with α6-GFP, β2, and β3 nAChR subunits, ACh produced a monophasic concentration–response curve with an EC50 value of 0.2 μm (Fig. 5C; Table 3), which is similar to previous observations (Henderson et al., 2014). Following menthol treatment, we observed a biphasic concentration–response relationship with EC50 values of 0.17 and 65.0 μm (Fig. 5C; Table 3). As noted above, multiple stoichiometries of α6β2* nAChRs are not well characterized, but it is likely that the two stoichiometries are α6β2β3 and α6β2(non-β3) nAChRs. To compare this biphasic curve to a “pure” population of α6β2(non-β3) nAChRs, we used a mutant β2 nAChR subunit (β2DM) that enhances exit from the endoplasmic reticulum and therefore enhances PM nAChRs levels (Srinivasan et al., 2011; Xiao et al., 2011; Henderson et al., 2014). ACh produced a monophasic concentration–response relationship curve on α6β2DM nAChRs with an EC50 value of 35.9 μm, resembling the less-sensitive phase of the menthol condition's responses (Fig. 5C; Table 3). Thus, the biphasic dose–response relationship produced by menthol likely represents a mixed population of α6β2β3 and α6β2 nAChRs.
Table 3.
Concentration–response analysis reveals that chronic menthol alters α6β2* nAChR stoichiometry
| Treatment condition |
|||
|---|---|---|---|
| α6-GFP, β2, β3 no drug | α6-GFPβ2DM no drug | α6-GFP, β2, β3 + menthol | |
| EC50 value (higher sensitivity) | 0.20 ± 0.07 μm | — | 0.17 ± 0.04 μm |
| nh (higher) | 1.01 | — | 0.78 |
| EC50 value (lower sensitivity) | — | 35.9 ± 20.7 μma | 65.0 ± 4.7 μma |
| nh (lower) | — | 0.79 | 2.3 |
| Higher fraction | — | — | 0.43 |
Values are represented as mean ± SEM.
aEC50 values are statistically different from α6-GFPβ2β3 nAChRs (no drug). p < 0.005, unpaired t test.
In addition to using concentration–response assays, we also used FRET to study menthol's effect on nAChR assembly and stoichiometry (Fig. 5B1,B2,D1,D2). Donor recovery after acceptor photobleaching was used with Neuro-2a cells transiently expressing α4-GFPα4-mCherryβ2 nAChRs, treated with no drug or with menthol (500 nm, 24 h). We observed an increase in FRET between α4-GFP and α4-mCherry nAChR subunits following 24 h treatment with menthol (Fig. 5B1,B2). This suggests that chronic menthol treatment increases the number of lower-sensitivity (α4)3(β2)2 nAChRs and supports the results from the concentration–response studies with α4β2 nAChRs (Fig. 5A).
To continue studying menthol-induced changes in α6β2* nAChRs, Neuro-2a cells transiently expressing α6-mCherryβ2β3-YFP nAChRs were treated with no drug or with menthol (500 nm, 24 h; Fig. 5D1,D2). The FRET intensity between α6-mCherry and β3-YFP subunits decreased following 24 h menthol treatment (Fig. 5D1,D2). This suggests that α6β2* nAChRs shift to the lower-sensitivity α6β2(non-β3) nAChR assembly after chronic treatment with menthol and support our observations in the concentration–response studies with α6β2β3 nAChRs (Fig. 5C).
Previous studies show that nicotine stabilizes higher-sensitivity α4β2* nAChRs (Kuryatov et al., 2005; Vallejo et al., 2005; Srinivasan et al., 2011), and this is likely an important mechanism involved in nicotine reward, sensitization, and tolerance (Tapper et al., 2004; Nashmi et al., 2007; Govind et al., 2009). Given that menthol instead stabilizes lower-sensitivity nAChRs, this may explain how long-term menthol exposure, before nicotine, may prevent or abolish nicotine reward-related behavior. It is possible that the menthol-induced stabilization of lower-sensitivity nAChRs (Fig. 5E,F) prevents the necessary stabilization and activation of higher-sensitivity nAChRs.
Long-term menthol treatment alters nAChR desensitization
To understand further how long-term menthol alone may abrogate nicotine reward-related behavior, we began to study additional functional characteristics of nAChRs and how they are altered by exposure to menthol. We used Neuro-2a cells transiently transfected with specific nAChRs to study α4β2 and α6β2β3 nAChRs. Neuro-2a cells transiently transfected with α4-eGFPβ2 or α6-eGFPβ2β3 nAChRs were treated with no drug or with menthol (500 nm, 24 h). We first verified that acute administration of menthol (≤4 μm) did not affect peak current amplitude or decay kinetics of α6β2β3 nAChRs (data not shown). Hans et al. (2012) established that acute menthol does not significantly alter α4β2 nAChR function at ≤4 μm. Menthol treatment (24 h, 500 nm) had no effect on the peak current amplitude of α4-eGFPβ2 nAChRs when stimulated by 300 μm ACh (Fig. 6A1,A2,B1). We also assessed whether acute exposure to menthol altered the deactivation kinetics of α4β2 nAChRs using 10 s applications of 300 μm ACh (Fig. 6B1,B2). Menthol did not alter the shape of α4-GFPβ2 nAChR waveforms (Fig. 6B1), including the decay time constant (Fig. 6B2).
Figure 6.

Long-term menthol decreases α6β2β3 nAChR peak current amplitude and alters α4* and α6* nAChR desensitization. A1, B1, C1, D1, Mean waveforms recorded from Neuro-2a cells expressing α6-eGFPβ2β3 or α4-eGFPβ2 nAChRs in the absence or presence of menthol (24 h, 500 nm). A1, C1, 300 ms puff of ACh. B1, D1, 10 s puff of ACh. B1, 10 s puffs reveal a sustained rebound current that is likely caused by recovery from agonist-mediated channel block. A2, C2, Mean of peak amplitude of 300 ms puffs. B2, D2, Mean decay time constants for 10 s puffs. Numbers in parenthesis indicate n value. E, F, α4-GFPβ2 and α6-GFPβ2β3 nAChRs treated with no drug (black) or with menthol (green). nAChRs were desensitized with 1 μm nicotine (10 min) and then allowed to recover. In all panels, data are mean ± SEM; *p < 0.05; ***p < 0.005 (unpaired t test). In all cases, menthol treatment was 24 h, 500 nm. In all cases, menthol-containing media was removed at the start of assays. Number in parenthesis indicates n value.
Next, we assessed menthol's effect on the functional properties of α6β2β3 nAChRs. Twenty-four hour exposure to submicromolar menthol led to a significant decrease (p < 0.005) in the peak current amplitude of α6-eGFPβ2β3 nAChRs (Fig. 6C1,C2,D1). Although the peak amplitude decreased following chronic treatment with menthol, the α6-eGFPβ2β3 nAChRs retained a phase that desensitized partially, with a time constant of ∼8 s (Fig. 6D1,D2). α6β2(Non-β3) nAChRs are not preferentially assembled when β3* nAChR subunits are available (Tumkosit et al., 2006), but they exhibit smaller peak current amplitudes (Xiao et al., 2011; Henderson et al., 2014) that resemble our observations following menthol treatment (Fig. 6C1,C2). The decrease in peak current amplitude (Fig. 6C1,C2) is not due to acute antagonism, but rather is likely due to the change in α6* nAChR stoichiometry (Fig. 5).
Interestingly, menthol (500 nm, 24 h) did increase α6β2 nAChR peak current amplitude by twofold (Fig. 7A,B1,B2). TIRFM revealed that menthol treatment resulted in a >2-fold increase in α6-SEPβ2 nAChR PMID (Fig. 7C). Here, menthol differs from nicotine, as nicotine is unable to upregulate α6β2(non-β3) nAChR number or function (Henderson et al., 2014). These results support our previous observations (Fig. 5) and suggest that menthol may selectively upregulate α6β2 nAChRs, causing a shift from primarily α6β2β3 nAChRs to a mixture of α6β2 and α6β2β3 nAChRs.
Figure 7.

Menthol upregulates α6β2(non-β3) nAChRs. Neuro-2a cells were transfected with α6-eGFP and β2 nAChR subunits. Where indicated, menthol (500 nm) was present for 24 h before imaging. A, Representative traces showing 300 μm ACh-induced currents for cells treated with no drug or with menthol (24 h, 500 nm). B1, B2, Summary of peak current and percentage of cells exhibiting function. C1, Representative TIRF images. Scale bars, 10 μm. In each panel, left image is at pH 7.4 and right is the same cell imaged at pH 5.4. C2, Mean PMID (mean ± SEM). B1, C2, *p < 0.05, unpaired t test. Number in parenthesis indicates n value.
Next we began to study how menthol may alter nAChR desensitization (Fig. 6E,F). We desensitized α4β2 and α6β2β3 nAChRs transiently transfected in Neuro-2a cells using bath perfusion of 1 μm nicotine. After 10 min, perfusion with nicotine was stopped and the nAChRs were allowed to recover for 20 min. The flow rates were kept constant for all experiments to ensure that any differences would not be altered by washout rates of nicotine. The α4β2 nAChRs desensitized completely during nicotine exposure and recovered only 20% of peak current amplitudes (Fig. 6E). Menthol-treated (24 h, 500 nm) α4β2 nAChRs exposed to nicotine desensitized, but not completely, and recovered ∼80% of their function (Fig. 6E).
The α6β2β3 nAChRs desensitized completely during nicotine application, then recovered to 40% of peak function (Fig. 6F). Menthol-treated α6β2β3 nAChRs desensitized rapidly and recovered 60% of function (Fig. 6F). The rate of recovery, following chronic menthol treatment, was faster when compared with no drug treatment (p < 0.05), but maximal recovery did not differ significantly between chronic menthol and no drug treatments. These data suggest that chronic exposure to menthol may protect nAChRs from complete desensitization and provide additional insight into how menthol alters nicotine reward-like behavior.
Menthol action on nAChRs begins in early exocytotic pathways
Recent data show that upregulation of nAChRs by nicotine requires the intracellular cycling of receptors between the Golgi and the endoplasmic reticulum (Henderson et al., 2014). To determine whether upregulation by menthol also requires Golgi–endoplasmic reticulum cycling, we used a previously characterized drug, CI-976, which inhibits coatomer protein complex I (COPI)-mediated retrograde trafficking (Yang et al., 2011; Henderson et al., 2014). Neuro-2a cells expressing α4-SEPβ2 and α6-SEPβ2β3 nAChRs were treated with no drug, with 500 nm menthol, with 20 μm CI-976, or with both drugs (Fig. 8A,B). Both α4-SEPβ2 and α6-SEPβ2β3 nAChRs displayed an increase in PMID following treatment with 500 nm menthol (Fig. 8A2,B2). Treatment with CI-976 alone did not change the basal PMID of either α4-SEPβ2 or α6-SEPβ2β3 nAChRs (Fig. 8A2,B2). Following treatment with both menthol and CI-976, we observed no increase in the PMID of α4-SEPβ2 or α6-SEPβ2β3 nAChRs (Fig. 8A2,B2). This suggests that, like effects of nicotine, the increase of nAChR number produced by menthol requires the cycling of nAChRs between the Golgi and endoplasmic reticulum.
Figure 8.

Long-term menthol upregulates nAChRs via endoplasmic reticulum–Golgi trafficking. A1, A2, Representative TIRFM images of Neuro-2a cells transfected with α4-SEP plus β2 or with α6-SEP plus β2 plus β3 nAChR subunits. Nicotine (50 or 100 nm), menthol (500 nm), or CI-976 (20 μm) were present 24 h before imaging. Scale bars, 10 μm. In each panel, the left image is at pH 7.4 and right image is the same cell at pH 5.4. B1, B2, PMID was quantified for SEP nAChRs. C1–C4, Representative confocal images of a Neuro-2a cell transfected with α6-mCherry, β2, and β3 subunits plus Sec24D-eGFP. Scale bars, 5 μm. C1, Sec24D-eGFP fluorescence. C2, α6 Fluorescence. C3, Merged C1 and C2 image. C4, ERES demarcated for quantification. D, Quantification of the α4-mCherry or α6-mCherry fluorescence in ERES. Data are mean ± SEM. *p < 0.05; **p < 0.005 (ANOVA, post hoc Tukey's test). Number in parenthesis indicates n value.
Nicotine upregulates nAChRs by binding to and chaperoning nAChRs located in the endoplasmic reticulum (Kuryatov et al., 2005; Sallette et al., 2005; Henderson and Lester, 2015). The later exocytotic machinery then moves these nAChRs to the PM (Whiteaker et al., 1998; Sallette et al., 2005; Srinivasan et al., 2011). The formation of endoplasmic reticulum exit sites (ERES) with nicotine is an indicator of an event closely following the nAChR chaperoning process. To determine whether menthol causes a similar increase in ERES, we quantified the formation of condensed ERES following chronic treatment with menthol (Fig. 8C,D). Neuro-2a cells were cotransfected with α4-mCherryβ2 or α6-mCherryβ2β3 nAChRs and with Sec24D-eGFP (a marker for active ERES) and the density of α4-mCherry* and α6-mCherry* nAChRs inside ERES was calculated (Fig. 8C,D). Menthol elicited a twofold increase of α4β2 nAChRs in ERES (p < 0.05) and a threefold increase in α6β2β3 nAChRs in ERES (p < 0.005; Fig. 8D). Therefore menthol, like nicotine, increases ERES in addition to the increase in nAChR number.
TRP channels do not mediate chronic menthol effects on nAChRs
Menthol is most known as a potent ligand for TRP channels. While we believe that the actions we observed in mouse brain slices, cultured neurons, and heterologous systems arise, directly or indirectly, from effects on nAChRs, we devised experiments to determine whether TRP channels also played a role (Table 4).
Table 4.
mRNA profile of TRP family genes
| Gene | Whole brain | Laser-captured single DA neurona | Single cultured DA neuron | Neuro-2a cells |
|---|---|---|---|---|
| TRPM8b | − | − | − | − |
| TRPA1b | − | − | − | − |
| TRPV1 | − | − | − | − |
| TRPV2 | + | + | + | + |
| TRPM2 | + | + | + | + |
| TRPM7 | + | + | + | + |
| TRPC2 | + | + | + | + |
| TRPMl1 | + | + | + | + |
aFrom SNc of adult mouse brain.
bA TRP family gene encoding a menthol-activated channel.
+, >1.0 FPKM.
We used RNA sequencing to assess the presence of mRNAs for TRP channels in adult mice, cultured midbrain neurons, and Neuro-2a cells. In laser-capture microdissected DA neurons from the SNc of adult mice, we detected mRNA from >10,000 genes at levels >0.1 FPKM (Henley et al., 2013). We also identified DA neurons in cultures from embryonic mouse midbrains, using either TH-eGFP mice from GENSAT (Gong et al., 2003) or nAChR β3-GFP mice (Shih et al., 2014; as in Fig. 3). In these cultured DA neurons, we detected mRNA for >8000 genes at levels >0.1 FPKM (Henley et al., 2013; Srinivasan et al., 2016). In DA neurons from both adult mouse and midbrain cultures, TRPA1, TRPV1, and TRPM8 transcript levels were <0.1 FPKM. In Neuro-2a cells, TRPA1, TRPV1, and TRPM8 were not detected (Table 4). Because mRNAs for the major TRP channels sensitive to menthol are undetectable, we conclude that the observed effects with menthol in mouse brain (Figs. 1, 2), cultured DA neurons (Figs. 2, 3), or Neuro-2a cells (Figs. 1, 5–8) were not mediated by TRP channels.
We also experimented with icilin, a well characterized TRPM8 and TRPA1 ligand. In Neuro-2a cells expressing α4SEP-β2 nAChRs, we tested five icilin concentrations ranging from 50 to 20 μm, applied for 24 h. Icilin had no significant effect on nAChR number (Fig. 9). Therefore, we believe that the effects of chronic exposure to 500 nm menthol (Figs. 1G,H, 5–8) are not mediated by TRP channels.
Figure 9.

TrpM8 agonist icilin does not alter PM nAChR number. A1, B1, Representative TIRFM images of Neuro-2a cells transfected with α4-SEPβ2 and α6-SEPβ2β3 nAChRs. Icilin, at listed concentrations, was present 24 h before imaging. Scale bars, 10 μm. In each panel, the left image is at pH 7.4 and the right image is the same cell at pH 5.4. A2, B2, PMID was quantified for SEP nAChRs. Number in parenthesis indicates n value.
Discussion
Does menthol abolish nicotine reward-related behavior?
Interestingly, menthol applied long-term before nicotine exposure inhibits nicotine reward-related behavior. Long-term nicotine exposure results in several well characterized neurobiological and pharmacological consequences: (1) nAChR numbers are increased (Richards et al., 2011; Srinivasan et al., 2011; Henderson et al., 2014); (2) the “upregulated” nAChRs exhibit a higher sensitivity to nicotine (Kuryatov et al., 2005; Govind et al., 2012; Henderson and Lester, 2015); and (3) the cell-specific upregulation leads to changes in reward-related DA release (Nashmi et al., 2007; Sulzer, 2011; Faure et al., 2014). Additionally, activation of these higher-sensitivity nAChRs is likely both necessary and sufficient to initiate nicotine reward-related behavior (Tapper et al., 2004; Nashmi et al., 2007; Faure et al., 2014).
We observed that menthol, in the absence of nicotine, exerts changes in nAChR number, assembly, and function, which differ from sequelae previously described for nicotine. Menthol's actions on nAChR assembly are opposite to the actions of nicotine. Instead of stabilizing higher-sensitivity nAChRs, menthol stabilized lower-sensitivity nAChRs, according to our observations. Menthol also exhibited cell-specific upregulation of midbrain α4* nAChRs that differs from effects of nicotine. Nicotine robustly upregulates α4* nAChRs on SNr and VTA GABA neurons (Nashmi et al., 2007); in contrast, menthol upregulates α4* nAChRs on SNc and VTA DA neurons (Fig. 10). The nicotine-induced, cell-specific upregulation of α4* nAChRs on GABAergic neurons provides an increased inhibitory tone on midbrain DA neurons of the VTA and SNc (Nashmi et al., 2007; Xiao et al., 2009). This increased inhibition results in decreased activity of the target DA neurons and a decrease in tonic DA release in the nucleus accumbens. Acute nicotine exposure causes a rebound disinhibition that results in increased phasic DA release, driving reward and reinforcement (Faure et al., 2014). Since menthol does not alter α4* nAChR number on GABAergic neurons of the SNr but upregulates nAChRs on DA neurons of the VTA and SNc, it is possible that nicotine's circuit-based effect on midbrain GABA and DA neurons is partially negated.
Figure 10.

Chronic menthol and chronic nicotine differentially alter midbrain DA neurons. Schematic of nAChRs residing in midbrain GABA (SNr) and DA (VTA and SNc) neurons. Nicotine robustly increases higher-sensitivity α4* nAChR number in SNr GABAergic neurons, but does not change α4* nAChR number in SNc or VTA DA neurons (Nashmi et al., 2007; Xiao et al., 2009). Long-term nicotine exposure decreases firing frequency of DA neurons, but this is transiently accelerated upon exposure to nAChR agonists (i.e., nicotine; Nashmi et al., 2007; Xiao et al., 2009). Long-term menthol exposure increases lower-sensitivity α4* and α6* nAChR number in SNC and VTA DA neurons, but does not alter SNr GABAergic α4* nAChR number. DA neuron firing frequency, which decreases following long-term menthol exposure, does not accelerate following exposure to nAChR agonists. The opposing effects of nicotine and menthol on midbrain neurons may explain how long-term menthol treatment before nicotine exposure abrogates nicotine reward-related behavior.
The different pharmacology of high-sensitivity and low-sensitivity nAChRs presumably plays a role. Higher-sensitivity nAChRs (stabilized by nicotine) are robustly activated by low concentrations of nicotine (<1 μm; Nelson et al., 2003; Kuryatov and Lindstrom, 2011). Lower-sensitivity α4* and α6* nAChRs (stabilized by menthol) are robustly activated by concentrations of nicotine ≥30 μm (Fig. 5). However, circulating concentrations of nicotine in human smokers (and mice following our administration protocol) are 100–500 nm. This suggests that long-term menthol alone may reduce the efficacy of circulating nicotine and its acute effects.
We note that menthol altered midbrain DA neurons so that transient nAChR activation results, via D2R, in a decrease in DA neuron firing frequency (Fig. 3). This appears to oppose the actions of nicotine (Fig. 10). In summary, long-term menthol treatment alone may reduce or prevent the “burst-like” firing due to nAChR activation by nicotine, important for reward and reinforcement. Reduced burst-like firing may contribute to our findings that menthol alone prevents nicotine reward-related behavior.
These observations occurred with chronic exposure to menthol alone, or in the case of our behavioral studies, menthol alone followed by nicotine during continued maintained exposure to menthol. In other studies, when previously menthol-naive animals are repeatedly coexposed to nicotine plus menthol, there is an enhancement of nicotine reinforcement (Wang et al., 2014) and nicotine withdrawal (Alsharari et al., 2015). Experiments are necessary, and underway, to understand additional cellular and behavioral effects of coexposure to nicotine plus menthol.
Menthol may act directly on midbrain DA neurons
Previous data show that menthol acts in the lungs (Ahijevych and Garrett, 2010) and alters nicotine metabolism in humans (Benowitz et al., 2004) and mice (Alsharari et al., 2015). Because we observed marked effects by chronic menthol on nAChR function with mice and with cultured DA neurons in the absence of nicotine, we conclude that menthol exerts a neural effect(s) by itself, in addition to modifications that may occur when nicotine is present. In using osmotic minipumps to deliver menthol to mice, we largely avoid the pharmacokinetics of exposure through the lungs. We infer that menthol enters the mouse brain and is present at <2.5 μm after 10 d. Due to our present detection limits, we cannot provide a true concentration of menthol in mouse brains. This was one motivation to employ cultured midbrain neurons to study the function of native nAChRs on DA neurons with a known concentration of menthol; we observed local actions of menthol at 500 nm. Therefore, if menthol alters nicotine metabolism or levels of nicotine, this augments the local actions of menthol alone on midbrain neurons.
Menthol as a candidate chemical chaperone
Our data show that chronic exposure to low-dose menthol acts on processes also influenced by chronic exposure to low-dose nicotine. These actions occur at the subcellular level, the cellular level, and the behavioral level; but actions differ between nicotine and menthol (Fig. 10). Many mechanistic facts are available about so-called “inside-out” actions of chronic nicotine beginning when nicotine acts as a selective pharmacological chaperone by binding to highly nicotine-sensitive nAChRs within the endoplasmic reticulum and cis-Golgi (Henderson and Lester 2015). Several thermodynamic, pharmacokinetic, and pharmacodynamic properties allow nicotine to function in this way at concentrations orders of magnitude lower than the EC50 value for transiently activating nAChRs on the PM. Now we report that exposure to menthol also appears to act within the early exocytotic pathway, at maintained concentrations orders of magnitude lower than the effective concentrations for transient activation of TRPM8 or for allosteric modulation of Cys-loop receptors (Hans et al., 2012; Ashoor et al., 2013; Ton et al., 2015).
We ruled out participation by TRPM8 or TRPA1, and these concentrations of menthol produce no acute effects on the two classes of nAChRs studied here (Hans et al., 2012). We do not believe that menthol is acting as a selective pharmacological chaperone for nAChRs. “Chemical chaperoning” may more appropriately describe effects of chronic menthol exposure at nAChRs. Chemical chaperones, such as butyrate, 4-pheylbutyric acid, and valproic acid, stabilize partially or improperly folded proteins. These compounds do increase the expression of nAChRs, mostly at micromolar concentrations (Sine et al., 1990; Kuryatov et al., 2013), via post-translational mechanisms. In one case, valproic acid upregulated α7 nAChRs in neuroblastoma cells at concentrations just 10-fold higher than the menthol concentrations studied here (Kuryatov et al., 2013). The present data are novel in providing information that a tobacco additive associated with nicotine dependence stabilizes certain nAChR stoichiometries, that the upregulation occurs in vivo, and that the changes have behavioral components.
From electrophysiology and FRET assays, this study shows that menthol alone stabilizes lower-sensitivity α4* and α6* nAChRs. In this effect, menthol phenomenologically resembles the nAChR partial agonist cytisine or the endogenous protein lynx1, which also stabilize lower-sensitivity (α4)3(β2)2 nAChRs (Richards et al., 2012; Srinivasan et al., 2012; Nichols et al., 2014). It is possible that menthol acts like lynx1, which binds to and stabilizes α–α nAChR interfaces and acts as a chaperone for these α–α-containing nAChRs. The FRET methods in this paper are dominated by the nAChRs residing in the endoplasmic reticulum and Golgi, with additional contribution from the PM. Therefore, we know that the receptors existing in the early secretory pathway are also lower-sensitivity (α4)3(β2)2 and α6β2(non-β3) nAChRs following menthol exposure.
However, we lack data proving that menthol exerts its chronic actions on nAChRs by binding directly to those nAChRs. Chemical chaperones have less selective actions than pharmacological chaperones. It is prudent to suppose that menthol binds directly to several proteins, perhaps including nAChRs. In particular, we do not yet know how chronic menthol exposure produces strong coupling between nAChR activation and D2R-mediated GIRK activation.
Although “vehicle” solutions contained ethanol, one asks whether the 60% ethanol in the osmotic minipumps interacted with the menthol delivered from those pumps. The calculated plasma ethanol concentration was 45–100-fold lower than that affecting reward-related pathways in other reports (see Materials and Methods). For the cell culture experiments, there was no ethanol. The extensive biochemical and pharmacological correspondences among the in vivo experiments and cultures argue against a synergistic interaction, at the behavioral level, between ethanol and menthol.
Menthol, more than a tobacco flavorant
According to the estimate that 30% of the 15 billion cigarettes consumed daily contain menthol, and the assumption that they contain on average 3 mg apiece, one calculates that smokers inhale 15 tons of menthol daily. Along with nicotine, menthol is one of the most inhaled substances on Earth. We have observed that menthol alone can upregulate nAChRs, decrease DA neuron baseline firing, alter nAChR stoichiometry, alter nAChR desensitization, increase endoplasmic reticulum exit of nAChRs, and suppress nicotine reward-related behavior. We have observed these effects at estimated pharmacologically relevant levels of menthol, acting locally on midbrain DA neurons relevant to reward-related pathways. Others have shown that menthol may enhance nicotine reinforcement (Wang et al., 2014) and nicotine withdrawal (Alsharari et al., 2015). Thus, emerging data show that menthol may not be just a harmless tobacco additive.
Footnotes
This work was supported by the National Institutes of Health (NIH; DA017279, DA019375, DA033721, DA036061, DA037161, and DA037743), the National Institute on Aging (NIH) Intramural Research Program, and the California Tobacco-Related Disease Research Program (17RT0127). We thank Michael Marks, Sharon Grady, and Ying Xie for menthol assays. We thank Heather Gold for help with bioinformatics.
The authors declare no competing financial interests.
References
- Ahijevych K, Garrett BE. Menthol pharmacology and its potential impact on cigarette smoking behavior. Nicotine Tob Res. 2004;6(Suppl 1):S17–S28. doi: 10.1080/14622200310001649469. [DOI] [PubMed] [Google Scholar]
- Ahijevych K, Garrett BE. The role of menthol in cigarettes as a reinforcer of smoking behavior. Nicotine Tob Res. 2010;12(Suppl 2):S110–S116. doi: 10.1093/ntr/ntq203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ai J, Taylor KM, Lisko JG, Tran H, Watson CH, Holman MR. Menthol content in US marketed cigarettes. Nicotine Tob Res. 2015;pii:mtv162. doi: 10.1093/ntr/ntv162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsharari SD, King JR, Nordman JC, Muldoon PP, Jackson A, Zhu AZ, Tyndale RF, Kabbani N, Damaj MI. Effects of menthol on nicotine pharmacokinetic, pharmacology and dependence in mice. PloS One. 2015;10:e0137070. doi: 10.1371/journal.pone.0137070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley M, Dixon M, Sisodiya A, Prasad K. Lack of effect of menthol level and type on smokers' estimated mouth level exposures to tar and nicotine and perceived sensory characteristics of cigarette smoke. Regul Toxicol Pharmacol. 2012;63:381–390. doi: 10.1016/j.yrtph.2012.04.010. [DOI] [PubMed] [Google Scholar]
- Ashoor A, Nordman JC, Veltri D, Yang KH, Al Kury L, Shuba Y, Mahgoub M, Howarth FC, Sadek B, Shehu A, Kabbani N, Oz M. Menthol binding and inhibition of α7-nicotinic acetylcholine receptors. PloS One. 2013;8:e67674. doi: 10.1371/journal.pone.0067674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benowitz NL, Herrera B, Jacob P., 3rd Mentholated cigarette smoking inhibits nicotine metabolism. J Pharmacol Exp Ther. 2004a;310:1208–1215. doi: 10.1124/jpet.104.066902. [DOI] [PubMed] [Google Scholar]
- Breese CR, Marks MJ, Logel J, Adams CE, Sullivan B, Collins AC, Leonard S. Effect of smoking history on [3H]nicotine binding in human postmortem brain. J Pharmacol Exp Ther. 1997;282:7–13. [PubMed] [Google Scholar]
- Brodie M. Low concentrations of nicotine increase the firing rate of neurons of the rat ventral tegmental area in vitro. In: Adlkoffer F, editor. Advances in pharmacological sciences. Basel: Birkhäuser; 1991. pp. 373–377. [Google Scholar]
- Brody AL, Mandelkern MA, Costello MR, Abrams AL, Scheibal D, Farahi J, London ED, Olmstead RE, Rose JE, Mukhin AG. Brain nicotinic acetylcholine receptor occupancy: effect of smoking a denicotinized cigarette. Int J Neuropsychopharmacol. 2009;12:305–316. doi: 10.1017/S146114570800922X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody AL, Mukhin AG, La Charite J, Ta K, Farahi J, Sugar CA, Mamoun MS, Vellios E, Archie M, Kozman M, Phuong J, Arlorio F, Mandelkern MA. Up-regulation of nicotinic acetylcholine receptors in menthol cigarette smokers. Int J Neuropsychopharmacol. 2013;16:957–966. doi: 10.1017/S1461145712001022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collo G, Bono F, Cavalleri L, Plebani L, Mitola S, Merlo Pich E, Millan MJ, Zoli M, Maskos U, Spano P, Missale C. Nicotine-induced structural plasticity in mesencephalic dopaminergic neurons is mediated by dopamine D3 receptors and Akt-mTORC1 signaling. Mol Pharmacol. 2013;83:1176–1189. doi: 10.1124/mol.113.084863. [DOI] [PubMed] [Google Scholar]
- Drenan RM, Lester HA. Insights into the neurobiology of the nicotinic cholinergic system and nicotine addiction from mice expressing nicotinic receptors harboring gain-of-function mutations. Pharmacol Rev. 2012;64:869–879. doi: 10.1124/pr.111.004671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenan RM, Nashmi R, Imoukhuede P, Just H, McKinney S, Lester HA. Subcellular trafficking, pentameric assembly and subunit stoichiometry of neuronal nicotinic ACh receptors containing fluorescently-labeled α6 and β3 subunits. Mol Pharmacol. 2008a;73:27–41. doi: 10.1124/mol.107.039180. [DOI] [PubMed] [Google Scholar]
- Drenan RM, Grady SR, Whiteaker P, McClure-Begley T, McKinney S, Miwa JM, Bupp S, Heintz N, McIntosh JM, Bencherif M, Marks MJ, Lester HA. In vivo activation of midbrain dopamine neurons via sensitized, high-affinity α6 nicotinic acetylcholine receptors. Neuron. 2008b;60:123–136. doi: 10.1016/j.neuron.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faure P, Tolu S, Valverde S, Naude J. Role of nicotinic acetylcholine receptors in regulating dopamine neuron activity. Neurosci. 2014;282:86–100. doi: 10.1016/j.neuroscience.2014.05.040. [DOI] [PubMed] [Google Scholar]
- Gandhi KK, Foulds J, Steinberg MB, Lu SE, Williams JM. Lower quit rates among African American and Latino menthol cigarette smokers at a tobacco treatment clinic. Int J Clin Pract. 2009;63:360–367. doi: 10.1111/j.1742-1241.2008.01969.x. [DOI] [PubMed] [Google Scholar]
- Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, Heintz N. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- Govind AP, Vezina P, Green WN. Nicotine-induced upregulation of nicotinic receptors: underlying mechanisms and relevance to nicotine addiction. Biochem Pharmacol. 2009;78:756–765. doi: 10.1016/j.bcp.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govind AP, Walsh H, Green WN. Nicotine-induced upregulation of native neuronal nicotinic receptors is caused by multiple mechanisms. J Neurosci. 2012;32:2227–2238. doi: 10.1523/JNEUROSCI.5438-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grahame NJ, Cunningham CL. Intravenous ethanol self-administration in C57BL/6J and DBA/2J mice. Alcohol Clin Exp. 1997;21:56–62. doi: 10.1111/j.1530-0277.1997.tb03728.x. [DOI] [PubMed] [Google Scholar]
- Hans M, Wilhelm M, Swandulla D. Menthol suppresses nicotinic acetylcholine receptor functioning in sensory neurons via allosteric modulation. Chem Senses. 2012;37:463–469. doi: 10.1093/chemse/bjr128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harpsøe K, Ahring PK, Christensen JK, Jensen ML, Peters D, Balle T. Unraveling the high- and low-sensitivity agonist responses of nicotinic acetylcholine receptors. J Neurosci. 2011;31:10759–10766. doi: 10.1523/JNEUROSCI.1509-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson BJ, Lester HA. Inside-out neuropharmacology of nicotinic drugs. Neuropharm. 2015;96(Pt B):178–193. doi: 10.1016/j.neuropharm.2015.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson BJ, Srinivasan R, Nichols WA, Dilworth CN, Gutierrez DF, Mackey ED, McKinney S, Drenan RM, Richards CI, Lester HA. Nicotine exploits a COPI-mediated process for chaperone-mediated up-regulation of its receptors. J Gen Physiol. 2014;143:51–66. doi: 10.1085/jgp.201311102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henley BM, Williams BA, Srinivasan R, Cohen BN, Xiao C, Mackey ED, Wold BJ, Lester HA. Transcriptional regulation by nicotine in dopaminergic neurons. Biochem Pharmacol. 2013;86:1074–1083. doi: 10.1016/j.bcp.2013.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Ingram SL, Prasad BM, Amara SG. Dopamine transporter-mediated conductances increase excitability of midbrain dopamine neurons. Nat Neurosci. 2002;5:971–978. doi: 10.1038/nn920. [DOI] [PubMed] [Google Scholar]
- Kuryatov A, Lindstrom J. Expression of functional human α6β2β3 acetylcholine receptors in Xenopus laevis oocytes achieved through subunit chimeras and concatamers. Mol Pharmacol. 2011;79:126–140. doi: 10.1124/mol.110.066159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuryatov A, Luo J, Cooper J, Lindstrom J. Nicotine acts as a pharmacological chaperone to up-regulate human α4β2 acetylcholine receptors. Mol Pharmacol. 2005;68:1839–1851. doi: 10.1124/mol.105.012419. [DOI] [PubMed] [Google Scholar]
- Kuryatov A, Mukherjee J, Lindstrom J. Chemical chaperones exceed the chaperone effects of RIC-3 in promoting assembly of functional α7 AChRs. PloS One. 2013;8:e62246. doi: 10.1371/journal.pone.0062246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester HA, Xiao C, Srinivasan R, Son CD, Miwa J, Pantoja R, Dougherty DA, Banghart MR, Goate AM, Wang JC. Nicotine is a selective pharmacological chaperone of acetylcholine receptor number and stoichiometry. Implications for drug discovery. AAPS J. 2009;11:167–177. doi: 10.1208/s12248-009-9090-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Zhao-Shea R, McIntosh JM, Gardner PD, Tapper AR. Nicotine persistently activates ventral tegmental area dopaminergic neurons via nicotinic acetylcholine receptors containing α4 and α6 subunits. Mol Pharmacol. 2012;81:541–548. doi: 10.1124/mol.111.076661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackey ED, Engle SE, Kim MR, O'Neill HC, Wageman CR, Patzlaff NE, Wang Y, Grady SR, McIntosh JM, Marks MJ, Lester HA, Drenan RM. α6* Nicotinic acetylcholine receptor expression and function in a visual salience circuit. J Neurosci. 2012;32:10226–10237. doi: 10.1523/JNEUROSCI.0007-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matta SG, Balfour DJ, Benowitz NL, Boyd RT, Buccafusco JJ, Caggiula AR, Craig CR, Collins AC, Damaj MI, Donny EC, Gardiner PS, Grady SR, Heberlein U, Leonard SS, Levin ED, Lukas RJ, Markou A, Marks MJ, McCallum SE, Parameswaran N, et al. Guidelines on nicotine dose selection for in vivo research. Psychopharmacology (Berl) 2007;190:269–319. doi: 10.1007/s00213-006-0441-0. [DOI] [PubMed] [Google Scholar]
- McCarthy WJ, Caskey NH, Jarvik ME, Gross TM, Rosenblatt MR, Carpenter C. Menthol vs nonmenthol cigarettes: effects on smoking behavior. Am J Public Health. 1995;85:67–72. doi: 10.2105/AJPH.85.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss FJ, Imoukhuede PI, Scott K, Hu J, Jankowsky JL, Quick MW, Lester HA. GABA transporter function, oligomerization state, and anchoring: correlates with subcellularly resolved FRET. J Gen Physiol. 2009;134:489–521. doi: 10.1085/jgp.200910314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhin AG, Kimes AS, Chefer SI, Matochik JA, Contoreggi CS, Horti AG, Vaupel DB, Pavlova O, Stein EA. Greater nicotinic acetylcholine receptor density in smokers than in nonsmokers: a PET study with 2-18F-FA-85380. J Nucl Med. 2008;49:1628–1635. doi: 10.2967/jnumed.108.050716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nashmi R, Dickinson ME, McKinney S, Jareb M, Labarca C, Fraser SE, Lester HA. Assembly of α4β2 nicotinic acetylcholine receptors assessed with functional fluorescently labeled subunits: effects of localization, trafficking, and nicotine-induced upregulation in clonal mammalian cells and in cultured midbrain neurons. J Neurosci. 2003;23:11554–11567. doi: 10.1523/JNEUROSCI.23-37-11554.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nashmi R, Xiao C, Deshpande P, McKinney S, Grady SR, Whiteaker P, Huang Q, McClure-Begley T, Lindstrom JM, Labarca C, Collins AC, Marks MJ, Lester HA. Chronic nicotine cell specifically upregulates functional α4* nicotinic receptors: basis for both tolerance in midbrain and enhanced long-term potentiation in perforant path. J Neurosci. 2007;27:8202–8218. doi: 10.1523/JNEUROSCI.2199-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson ME, Kuryatov A, Choi CH, Zhou Y, Lindstrom J. Alternate stoichiometries of α4β2 nicotinic acetylcholine receptors. Mol Pharmacol. 2003;63:332–341. doi: 10.1124/mol.63.2.332. [DOI] [PubMed] [Google Scholar]
- Nichols WA, Henderson BJ, Yu C, Parker RL, Richards CI, Lester HA, Miwa JM. Lynx1 shifts α4β2 nicotinic receptor subunit stoichiometry by affecting assembly in the endoplasmic reticulum. J Biol Chem. 2014;289:31423–31432. doi: 10.1074/jbc.M114.573667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards CI, Srinivasan R, Xiao C, Mackey ED, Miwa JM, Lester HA. Trafficking of α4* nicotinic receptors revealed by superecliptic phluorin: effects of a β4 amyotrophic lateral sclerosis-associated mutation and chronic exposure to nicotine. J Biol Chem. 2011;286:31241–31249. doi: 10.1074/jbc.M111.256024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards CI, Luong K, Srinivasan R, Turner SW, Dougherty DA, Korlach J, Lester HA. Live-cell imaging of single receptor composition using zero-mode waveguide nanostructures. Nano Lett. 2012;12:3690–3694. doi: 10.1021/nl301480h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgman A, Perfetti TA. The chemical components of tobacco and tobacco smoke. Boca Raton, FL: CRC; 2009. [Google Scholar]
- Sallette J, Pons S, Devillers-Thiery A, Soudant M, Prado de Carvalho L, Changeux JP, Corringer PJ. Nicotine upregulates its own receptors through enhanced intracellular maturation. Neuron. 2005;46:595–607. doi: 10.1016/j.neuron.2005.03.029. [DOI] [PubMed] [Google Scholar]
- Shih PY, Engle SE, Oh G, Deshpande P, Puskar NL, Lester HA, Drenan RM. Differential expression and function of nicotinic acetylcholine receptors in subdivisions of medial habenula. J Neurosci. 2014;34:9789–9802. doi: 10.1523/JNEUROSCI.0476-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sine SM, Claudio T, Sigworth FJ. Activation of Torpedo acetylcholine receptors expressed in mouse fibroblasts. J Gen Physiol. 1990;96:395–437. doi: 10.1085/jgp.96.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son CD, Moss FJ, Cohen BN, Lester HA. Nicotine normalizes intracellular subunit stoichiometry of nicotinic receptors carrying mutations linked to autosomal dominant nocturnal frontal lobe epilepsy. Mol Pharmacol. 2009;75:1137–1148. doi: 10.1124/mol.108.054494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan R, Pantoja R, Moss FJ, Mackey ED, Son CD, Miwa J, Lester HA. Nicotine upregulates α4β2 nicotinic receptors and ER exit sites via stoichiometry-dependent chaperoning. J Gen Physiol. 2011;137:59–79. doi: 10.1085/jgp.201010532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan R, Richards CI, Xiao C, Rhee D, Pantoja R, Dougherty DA, Miwa JM, Lester HA. Pharmacological chaperoning of nicotinic acetylcholine receptors reduces the endoplasmic reticulum stress response. Mol Pharmacol. 2012;81:759–769. doi: 10.1124/mol.112.077792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan R, Henley BM, Henderson BJ, Henley B, Indersmitten T, Cohen BN, Kim CH, McKinney S, Deshpande P, Xiao C, Lester HA. Smoking-relevant nicotine concentration attenuates the unfolded protein response in dopaminergic neurons. J Neurosci. 2016;36:65–79. doi: 10.1523/JNEUROSCI.2126-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley JK, Krishnan-Sarin S, Cosgrove KP, Krantzler E, Frohlich E, Perry E, Dubin JA, Estok K, Brenner E, Baldwin RM, Tamagnan GD, Seibyl JP, Jatlow P, Picciotto MR, London ED, O'Malley S, van Dyck CH. Human tobacco smokers in early abstinence have higher levels of β2* nicotinic acetylcholine receptors than nonsmokers. J Neurosci. 2006;26:8707–8714. doi: 10.1523/JNEUROSCI.0546-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D. How addictive drugs disrupt presynaptic dopamine neurotransmission. Neuron. 2011;69:628–649. doi: 10.1016/j.neuron.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapper AR, McKinney SL, Nashmi R, Schwarz J, Deshpande P, Labarca C, Whiteaker P, Marks MJ, Collins AC, Lester HA. Nicotine activation of α4* receptors: sufficient for reward, tolerance and sensitization. Science. 2004;306:1029–1032. doi: 10.1126/science.1099420. [DOI] [PubMed] [Google Scholar]
- Ton HT, Smart AE, Aguilar BL, Olson TT, Kellar KJ, Ahern GP. Menthol enhances the desensitization of human α3β4 nicotinic acetylcholine receptors. Mol Pharmacol. 2015;88:256–264. doi: 10.1124/mol.115.098285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumkosit P, Kuryatov A, Luo J, Lindstrom J. β3 Subunits promote expression and nicotine-induced up-regulation of human nicotinic α6* nicotinic acetylcholine receptors expressed in transfected cell lines. Mol Pharmacol. 2006;70:1358–1368. doi: 10.1124/mol.106.027326. [DOI] [PubMed] [Google Scholar]
- Vallejo YF, Buisson B, Bertrand D, Green WN. Chronic nicotine exposure upregulates nicotinic receptors by a novel mechanism. J Neurosci. 2005;25:5563–5572. doi: 10.1523/JNEUROSCI.5240-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Kuryatov A, Sriram A, Jin Z, Kamenecka TM, Kenny PJ, Lindstrom J. An accessory agonist binding site promotes activation of α4β2* nicotinic acetylcholine receptors. J Biol Chem. 2015;290:13907–13918. doi: 10.1074/jbc.M115.646786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Wang B, Chen H. Menthol facilitates the intravenous self-administration of nicotine in rats. Front Behav Neurosci. 2014;8:437. doi: 10.3389/fnbeh.2014.00437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteaker P, Sharples CG, Wonnacott S. Agonist-induced up-regulation of α4β2 nicotinic acetylcholine receptors in M10 cells: pharmacological and spatial definition. Mol Pharmacol. 1998;53:950–962. [PubMed] [Google Scholar]
- World Health Organization. The MPOWER package. Geneva: World Health Organization; 2008. Report on the global tobacco epidemic. [Google Scholar]
- Xiao C, Nashmi R, McKinney S, Cai H, McIntosh JM, Lester HA. Chronic nicotine selectively enhances α4β2* nicotinic acetylcholine receptors in the nigrostriatal dopamine pathway. J Neurosci. 2009;29:12428–12439. doi: 10.1523/JNEUROSCI.2939-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao C, Srinivasan R, Drenan RM, Mackey ED, McIntosh JM, Lester HA. Characterizing functional α6β2 nicotinic acetylcholine receptors in vitro: mutant β2 subunits improve membrane expression, and fluorescent proteins reveal responsive cells. Biochem Pharmacol. 2011;82:852–861. doi: 10.1016/j.bcp.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JS, Valente C, Polishchuk RS, Turacchio G, Layre E, Moody DB, Leslie CC, Gelb MH, Brown WJ, Corda D, Luini A, Hsu VW. COPI acts in both vesicular and tubular transport. Nat Cell Biol. 2011;13:996–1003. doi: 10.1038/ncb2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye JH, Zhang J, Xiao C, Kong JQ. Patch-clamp studies in the CNS illustrate a simple new method for obtaining viable neurons in rat brain slices: glycerol replacement of NaCl protects CNS neurons. J Neurosci Methods. 2006;158:251–259. doi: 10.1016/j.jneumeth.2006.06.006. [DOI] [PubMed] [Google Scholar]