

Abstract
Calorie restriction (CR) is neuroprotective in Parkinson's disease (PD) although the mechanisms are unknown. In this study we hypothesized that elevated ghrelin, a gut hormone with neuroprotective properties, during CR prevents neurodegeneration in an 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) model of PD. CR attenuated the MPTP-induced loss of substantia nigra (SN) dopamine neurons and striatal dopamine turnover in ghrelin WT but not KO mice, demonstrating that ghrelin mediates CR's neuroprotective effect. CR elevated phosphorylated AMPK and ACC levels in the striatum of WT but not KO mice suggesting that AMPK is a target for ghrelin-induced neuroprotection. Indeed, exogenous ghrelin significantly increased pAMPK in the SN. Genetic deletion of AMPKβ1 and 2 subunits only in dopamine neurons prevented ghrelin-induced AMPK phosphorylation and neuroprotection. Hence, ghrelin signaling through AMPK in SN dopamine neurons mediates CR's neuroprotective effects. We consider targeting AMPK in dopamine neurons may recapitulate neuroprotective effects of CR without requiring dietary intervention.
SIGNIFICANCE STATEMENT The neuroprotective mechanisms of calorie restriction (CR) in Parkinson's disease are unknown. Indeed, the difficulty to adhere to CR necessitates an alternative method to recapitulate the neuroprotective benefits of CR while bypassing dietary constraints. Here we show that CR increases plasma ghrelin, which targets substantia nigra dopamine to maintain neuronal survival. Selective deletion on AMPK beta1 and beta2 subunits only in DAT cre-expressing neurons shows that the ghrelin-induced neuroprotection requires activation of AMPK in substantia nigra dopamine neurons. We have discovered ghrelin as a key metabolic signal, and AMPK in dopamine neurons as its target, which links calorie restriction with neuroprotection in Parkinson's disease. Thus, targeting AMPK in dopamine neurons may provide novel neuroprotective benefits in Parkinson's disease.
Keywords: AMPK, calorie restriction, ghrelin, stereology, substantia nigra
Introduction
Parkinson's disease (PD) is the second most common neurodegenerative disease affecting ∼160 per 100,000 people with an estimated incidence number of new cases each year of 16–19 per 100,00 according to the World Health Organization, creating a substantial medical, social, and financial burden. The motor symptoms of PD include rigidity and tremor of the extremities, postural instability, and bradykinesia.
The body mass index of an individual affects PD progression, as obesity causes dopamine neuronal cell loss in the substantia nigra (SN) in a mouse model of PD (Choi et al., 2005), and midlife obesity and type-2 diabetes is associated with a greater incidence of PD in humans (Chen et al., 2014b). In contrast to obesity, calorie restriction (CR) attenuates 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced neurotoxicity in both mice (Duan and Mattson, 1999) and nonhuman primates (Maswood et al., 2004). Indeed, CR in monkeys may delay the aging process (Colman et al., 2009) and CR in humans has the potential to slow PD disease progression (Chan et al., 1997), yet the beneficial effects of CR are dependent on the adherence to strict dietary constraints that are not always practical and achievable in society. Therefore, it is paramount to identify the key molecular mechanisms linking CR and neuroprotection to circumvent the need to adhere to CR.
Ghrelin is synthesized in the stomach where pro-ghrelin is acylated in the endoplasmic reticulum by the enzyme ghrelin O-acyltransferase (GOAT). Acyl ghrelin is then released into the bloodstream where it crosses the blood–brain barrier and binds to the ghrelin receptor [growth hormone secretagogue receptor 1a (GHSR)] in the brain. In addition to its well known metabolic effects, ghrelin is neuroprotective in PD as ghrelin and GHSR KO mice exhibited significantly greater loss of SN dopaminergic neurons compared with WT controls in an MPTP model of PD (Andrews et al., 2009). The neuroprotective mechanisms include reducing apoptosis and suppressing microglial activation and local inflammatory responses in the SN (Dong et al., 2009; Moon et al., 2009). Moreover, postprandial plasma ghrelin concentrations are lower in human PD patients (Unger et al., 2011), suggesting clinical relevance.
Plasma ghrelin is elevated during periods of negative energy balance, including CR and previous studies showed that the anxiolytic and antidepressant effects of CR require GHSR signaling (Lutter et al., 2008). Ghrelin also prevented an excessive decline in blood glucose levels during CR (Zhao et al., 2010). These studies provide biological precedents that the ghrelin system mediates some of the beneficial effects of CR. Because ghrelin protects against SN dopaminergic cell loss (Jiang et al., 2008; Andrews et al., 2009; Moon et al., 2009), we reasoned that elevated plasma ghrelin during CR contributes to the neuroprotective effects of CR in PD. Indeed, cells treated with serum from CR rats show greater survivability, increased mitochondrial function, and mitochondrial biogenesis (López-Lluch et al., 2006), arguing that a hormonal signal mediates the effects of CR on mitochondrial function and cell survivability. These findings above led us to hypothesize that increased plasma ghrelin during CR acts on SN dopamine neurons to restrict SN dopamine neuronal degeneration in a mouse model of PD.
Materials and Methods
Animals.
All experiments herein were conducted in accordance with Monash University Animal Ethics Committee guidelines. Mice were maintained under standard laboratory conditions with ad libitum access to food and water at 21°C with a 12 h light/dark cycle unless otherwise stated.
Experimental protocol.
For the first set of experiments, ghrelin WT/KO mice were individually housed. Male ghrelin WT/KO mice (∼8–10 weeks old) on a C57BL/6J background were obtained from Regeneron Pharmaceuticals and bred in the Monash Animal Services facilities. Mice in ad libitum groups had ad libitum access to food, whereas the remaining mice were CR to 70% of their baseline food intake. Baseline food intake was calculated by measuring average food intake over 1 week before the initiation of the restriction period. CR mice had daily blood glucose and body weight measurements taken and then given access to a previously calculated and weighed food pellet ∼1 h before the initiation of the dark cycle (18:00 h) in an attempt to maintain normal physiological feeding times for the duration of the experiment (27 d).
In the second set of experiments to test the effect of ghrelin administration on neuronal function in the midbrain, we used group housed male C57BL/6J mice (8–10 weeks old; Monash Animal Services, Victoria, Australia) that had ad libitum access to food and water. C57BL/6J mice were randomly allocated to receive saline, a low dose of ghrelin (5 mg/kg) or a high dose of ghrelin (15 mg/kg). The mice were injected intraperitoneally and the food removed from the cage, they were subsequently culled 45 min later via decapitation after being deeply anesthetized, then the brains were dissected and snap frozen (−70°C) for HPLC and Western blot analysis.
To generate mice with selective deletion of AMPK β1 and β2 only in DAT-expressing dopamine neurons, we crossed Dat-Cre knock-in mice obtained from The Jackson Laboratory [stock no. 006660; B6.SJL-Slc6a3<tm1.1(cre)bkmn>/j] with Ampk β 1 subunit (β1) and β 2 subunit (β2) floxed mice (O'Neill et al., 2011). The resultant offspring (Dat-Cre;Ampk β 1fl/fl;Ampk β 2fl/fl designated AMPK KO or Ampk β 1fl/fl;Ampk β 2fl/fl designated AMPK WT) were used as experimental mice. To validate this model, AMPK WT and KO mice were also bred with cre-dependent loxSTOPlox tdTOMATO reporter mice [stock number 007908; B6;129S6-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J]. The resultant offspring Dat-Cre;tdTomato or Dat-Cre;Ampk β 1fl/fl;Ampk β 2fl/fl;tdTomato mice allow tdTomato visualization of DAT-expressing neurons that have undergone cre recombination. These mice were also used for fluorescence-activated cell sorting (FACS). The Dat-Cre;tdTomato were used as AMPK WT mice and Dat-Cre;Ampk β 1fl/fl;Ampk β 2fl/fl;tdTomato mice were used as AMPK KO mice. These mice were culled via inhalation anesthetic and the substantia nigra was collected. The cells were dissociated using papain (Worthington Kit, LK003150) following the kit instructions. After collection of ∼5000 tdTomato cells via FACS sorting using the influx v7 Sorter, the RNA was extracted and PCR was run to determine the presence/absence of AMPKβ1 and 2.
In the third set of experiments, to test the effects of ghrelin administration in mice lacking AMPK activation, we group housed AMPK WT and KO mice (8–10 weeks old) with ad libitum access to water. The mice were administered ghrelin (1 mg/kg) or saline daily at the beginning of the light cycle for 14 consecutive days. After injections, the food was subsequently removed for 6 h to prevent excess consumption of calories, after this period all mice had ad libitum access to food. Previous studies (Andrews et al., 2009) indicate that if calories are consumed after injection of acyl ghrelin there is no neuroprotective effect observed. On days 7 and 8, mice were injected with saline or MPTP (30 mg/kg). Mice were culled on day 14 and perfused for immunohistochemical analysis or fresh tissue collection for Western blot and HPLC analysis.
MPTP administration.
Experimental mice were injected with MPTP (30 mg/kg, i.p.) dissolved in saline as described previously (Andrews et al., 2005) over 2 consecutive days. Control animals received sterile saline using the same timeline. Animals were injected with MPTP or Saline and perfused 7 d later for immunohistochemical analysis or fresh tissue collection for HPLC and Western blot analysis.
Immunohistochemistry.
Free-floating sections were stained with both tyrosine hydroxylase (TH) and ionized calcium binding adaptor (IBA1) or glial fibrillary acidic protein (GFAP). All mice were deeply anesthetized and perfused with 0.05% PBS followed by 4% paraformaldehyde (PFA) to fix the tissue. Brains were stored in PFA overnight then transferred to a 30% sucrose solution. Coronal sections (30 μm thick) of the entire SN were collected with systematic sampling of every fifth section.
The sections was washed thoroughly in 0.1 m PB and then endogenous peroxidase activity was blocked using 1% H2O2 in 0.1 m PB for 15 min and washed again. The tissue was then transferred to 4% normal horse serum and 0.3% Triton X-100 in 0.1 m PB for 1 h, followed by a secondary mouse blocking step using AffiniPure Goat Anti-Mouse IgG (H+L; 1:200, Jackson ImmunoResearch) to prevent nonspecific binding of mouse antibodies in mouse tissue. The tissue was then incubated with the primary antibodies, in this case either anti-TH (mouse, 1:5000; Millipore) and anti-IBA1(rabbit, 1:1000; Wako) or anti-GFAP (rabbit, 1:1000; DAKO) for 24 h at 4°C. Following the primary antibody incubation the tissue was washed thoroughly and incubated in the secondary antibody goat anti-mouse IgG (H+L) AlexaFluor 488 (1:400; Invitrogen) and goat anti-rabbit IgG (H+L) AlexaFluor 594 (1:400; Invitrogen) for fluorescent staining for 90 min at room temperature. The tissue was then thoroughly washed and mounted directly onto slides and coverslipped with anti-fade mounting media.
Stereological investigation of cell number and volume.
To quantify the number of TH neurons, microglia (IBA1 stain), and astrocytes (GFAP stain) in the SN we used design-based stereology. Using the StereoInvestigator software (MicroBrightField) we analyzed both cell number (using the optical fractionator probe) and cell volume (using the nucleator probe). To visualize the cells we used a Zeiss microscope with a motorized stage and a MicroFibre digital camera connected to a computer.
Analysis of blood chemistry.
Trunk blood was collected via decapitation from deeply anesthetized mice and collected into EDTA tubes pretreated with pefabloc (SC Roche Applied Science) to achieve a concentration of 1 mg/ml. The blood was then briefly centrifuged and the plasma was collected and acidified with HCl (final concentration 0.05N). Plasma ghrelin levels were determined using Active Ghrelin or Des-acyl Ghrelin Enzyme-Linked Immunoassay Kits (Mitsubishi Chemical Medicine). Active and des-acyl ghrelin were measured according to kit instructions. Plasma insulin concentration was determined through an in-house ELISA assay.
High performance liquid chromatography.
We used high-performance liquid chromatography (HPLC) to identify, separate, and quantify dopamine (DA) and DOPAC concentrations within samples of striatal tissue. Striatal (both sides) tissue was rapidly dissected and snap frozen (∼−70°C). The samples were then sonicated in 0.4 ml cold 0.1 m perchloric acid containing internal standard. Following centrifugation, DA DOPAC and internal standard in the supernatant were extracted on alumina at pH 8.4, eluted in 0.1 m perchloric acid, separated by reverse-phase HPLC and detected using electrochemical detection. Both dopamine and DOPAC concentrations in the striatum were calculated by reference to the internal standard and external standards. The protein content of each sample was determined from the centrifuged pellet by the Lowry method. The concentrations of DA and DOPAC are expressed as nanograms per milligram of protein present (mean ± SEM).
Western blot.
Whole tissue samples of the SN and striatum or SN4741 cells were processed for Western blot analysis. Briefly, tissue was sonicated in RIPA buffer (50 mm Tris.HCl, 150 mm NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X-100) containing a protease inhibitor (Sigma-Aldrich), then centrifuged [10,000 rotations per minute (rpm), 10 min, 4°C] to remove cell debris, and the supernatant was collected. For cell culture studies SN4741 cells were maintained at 37°C in a 5% CO2 humidified environment in DMEM (41965, Life Technologies) supplemented with 10% fetal bovine serum, 2 mm glutamine, 100 U/ml penicillin, and 0.1 mg/ml streptomycin, 0.6% glucose. Once cells had reached ∼90–100% confluency, the cells were subcultured.
SN4741 cells were treated with vehicle (compound diluent), 1 μm acyl ghrelin (Tocris Bioscience), 6 nm JMV2894 (ghrelin receptor agonist, Aeterna Zentaris) or 0.5 μm oligomycin (Sigma-Aldrich) for 5 min. Cells were washed three times with ice-cold PBS and lysed in ice-cold RIPA lysis buffer (50 mm Tris HCl, pH 7.5 containing 1% NP40, 0.1% SDS, 0.5% sodium deoxycholate, and 150 mm NaCl) with 1% mammalian protease (Sigma-Aldrich, P8340) and phosphatase inhibitors (Sigma-Aldrich, P0044; Ho et al., 2013). Cell lysates were incubated at 4°C for 15 min and then centrifuged at 22,000 × g for 10 min at 4°C. The supernatant was collected and 1 volume of 2× SDS-PAGE sample loading buffer (Sigma-Aldrich, S3401) was added and left at room temperature for 1 h.
An aliquot was then used to identify the amount of protein present in each sample using a BCA kit (Pierce) according to kit instructions. The samples concentrations were then standardized and the supernatants were mixed with Laemmli buffer, and boiled for 5 min. Samples (20 μl) were loaded onto 10% acrylamide gels and separated by SDS-PAGE. The separated proteins were then transferred from the gel to the PVDF membrane (Biorad). The blots were then blocked for 1 h in Tris-buffered saline solution containing 0.1% Tween 20 (TBST) and 5% bovine serum albumin (BSA). The membranes were subsequently incubated overnight at 4°C in TBST with 5% BSA with either of the following antibodies: TH (1:1000; Milipore), Parkin (1:1000; Santa Cruz Biotechnology), PINK (1:1000; Santa Cruz Biotechnology), LC3B (1:1000; Cell Signaling Technology), pACC (1:1000; Cell Signaling Technology) or pAMPK (1:1000; Cell Signaling Technology), where AMPKα (1:1000; Cell Signaling Technology) antibodies, ACC (1:1000; Cell Signaling Technology) and anti-β actin (1:1000; Abcam) were used as controls. Blots were visualized using the chemiluminescence method (ECL; GE Healthcare) and levels were detected using ImageLab Software v4.1, Biorad.
RNA extraction and PCR.
After FACS, cells were stored in Qiazol for RNA extraction. Briefly, chloroform was added, samples were centrifuged (12,000 × g, 15 min, 4°C) and supernatant was collected. Isopropanol and glycogen was added and the samples centrifuged (12,000 × g, 10 min, 4°C). The pellet formed was washed with ethanol (75%) and vortexed. cDNA was synthesized using the iScript cDNA synthesis kit (no. 170-8890, Bio-Rad Laboratories). The cDNA collected was combined with Mastermix and primers (either AMPKβ1, AMPKβ2, or GHSR) and exposed to a heat block in the Mastercycler. We used TaqMan Gene Expression Mastermix (Applied Biosystems) and GHSR primers (GHSR forward: GCTGCTCACCGTGATGGTAT, reverse: GCTGCTCACCGTGATGGTAT) as our control. A PCR was required to amplify the AMPKβ1 and AMPKβ2 transcripts from the cDNA. We used nested PCR to enhance accuracy using two PCRs involving outer and inner primers (AMPKβ1: outer forward: CCACTCCGAAGAGATCAAGG, reverse: GTGCTGGGTCACAAGAGATG; AMPKβ1: inner forward: CACGACCTGGAAGCGAAT, reverse: CATGTAAGGCTCCTGGTGGT; AMPKβ2: outer forward: GTTATCCGCTGGTCTGAAGG, reverse: CAGCAGCGTGGTGACATACT; AMPKβ2: inner forward: GAGCACCAAGATCCCTCTGA, reverse: GGAAGTAAGGCTGGGTCACA). This process was repeated with inner primers and then visualized in a gel mounting media (agarose gel) and exposed to electrical current (120 V) for 25 min. The results were viewed using gene snap technology. The specificity of the primers was confirmed using a blast search. Positive control was hypothalamic tissue from C57BL/6 mice and negative control contained no cDNA.
Rotarod.
Mice were trained before testing by being placed on a rotating rod (Ugo Basile Rota-Rod 47600), spinning at 4 rpm for 5 min. Lane width = 5 cm. On training day, mice were subjected to incrementally increasing speed over 300 s going from 4 to 40 rpm. Each animal underwent four trials. The length of time that the mice remained on the rod was recorded and analyzed.
Statistical analysis.
All data are represented as mean ± SEM. Two-way ANOVA with a Bonferroni post hoc test was used to determine statistical significance between treatment and genotype and one-way ANOVA with a Tukey post hoc test was used to determine statistical significance between injection groups. Cell lysate analysis used a two-tailed Student's t test. p < 0.05 was considered statistically significant.
Results
Effect of CR on metabolic parameters in ghrelin WT/KO mice
CR significantly elevated acylated (Fig. 1A) and des-acyl plasma ghrelin in WT, with no detectable levels in KO mice (data not shown), confirming reports that CR increases plasma ghrelin. There were no genotypic differences in plasma insulin from ad libitum or CR mice, although CR significantly reduced plasma insulin levels compared with ad libitum mice (significant main effect ad libitum vs CR; Fig. 1B,C). Both body weight and blood glucose measurements exhibited a significant overall reduction in response to CR (data not shown).
Figure 1.
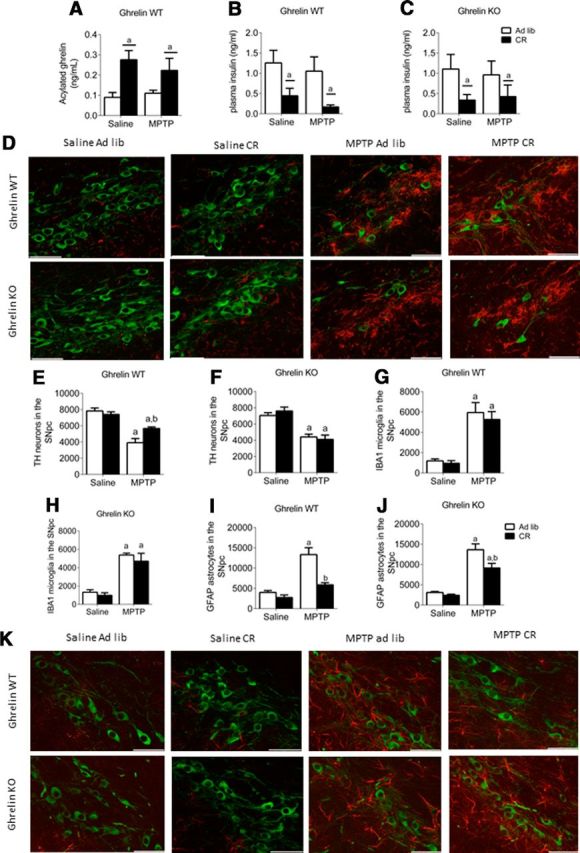
Deletion of ghrelin negates the protective effect of CR. A, CR significantly elevates plasma acylated ghrelin. B, C, Overall reduction in plasma insulin levels in response to CR in both genotypes. D, Representative images showing MPTP-induced TH cell loss in the SN and microglial (IBA) activation. E, F, Stereological quantification of TH neurons in the SN showing CR has no significant effect in MPTP-treated ghrelin KO mice (F) but is protective in ghrelin WT mice (E). G, H, Stereological quantification of IBA1 microglia in the SN shows elevated cell number following MPTP treatment but no effect of genotype. I, J, Stereological quantification of SN GFAP shows that MPTP administration increased GFAP cell number to a lesser extent in ghrelin WT compared with ghrelin KO mice. K, Representative images showing MPTP-induced astrocyte (GFAP) activation in the SN (TH, green; GFAP, red). Data are represented as mean ± SEM (n = 6–10, two-way ANOVA, p < 0.05). A, Significant compared with saline ad libitum controls. b, Significant compared with MPTP ad libitum controls. Scale bar, 50 μm.
Ghrelin restricts MPTP-induced nigrostriatal damage during CR
TH neurons
We used the stereological optical fractionator probe to estimate total TH-positive (i.e., dopamine) neurons in the SN. MPTP administration significantly reduced the number of SN TH neurons in ad libitum and CR ghrelin WT and KO mice (Fig. 1D). CR partially attenuated SN TH neuronal loss in ghrelin WT (Fig. 1E), however, this protective effect was lost in ghrelin KO mice (Fig. 1F).
Gliosis
MPTP treatment exhibited a significant elevation in microglia (IBA1+ cells) present in the SN of both genotypes, although CR did not prevent the MPTP-induced increase of IBA1 cell number in either ghrelin WT or ghrelin KO mice (Fig. 1G,H). Astrocytes, as represented by GFAP staining, are the most abundant cell type found throughout the CNS and play a critical role during cellular damage to minimize overall cell loss (Hailer et al., 2001). Elevated GFAP+ cells in any specified region indicates greater cellular damage in that area. MPTP treatment initiated a significant increase in GFAP cells in both ghrelin WT and KO ad libitum mice compared with saline controls (Fig. 1I,J). CR reduced GFAP expression in the SN of both MPTP-treated ghrelin WT and KO mice relative to MPTP ad libitum mice (Fig. 1I,J), This result indicates that CR restricts GFAP cell expression in the SN, although this does not appear to be directly mediated by ghrelin.
TH cell volume
To accurately measure cell volume we used the nucleator stereological probe. There was a significant main effect for MPTP to reduce average cell volume in ghrelin WT but not ghrelin KO (Fig. 2A,B). We performed a cell-volume distribution analysis to determine whether diet or treatment preferentially affected neuronal number within a certain volume range. MPTP treatment to CR ghrelin WT mice prevented the loss of TH neurons with volumes between 1000 and 2000 μm3 compared with ad libitum MPTP-treated ghrelin WT mice (Fig. 2C). Remarkably, no beneficial effects of CR on TH neuronal cell volume between were observed in ghrelin KO mice (Fig. 2D). Thus, ghrelin influences both TH cell number and cell volume distribution during CR.
Figure 2.
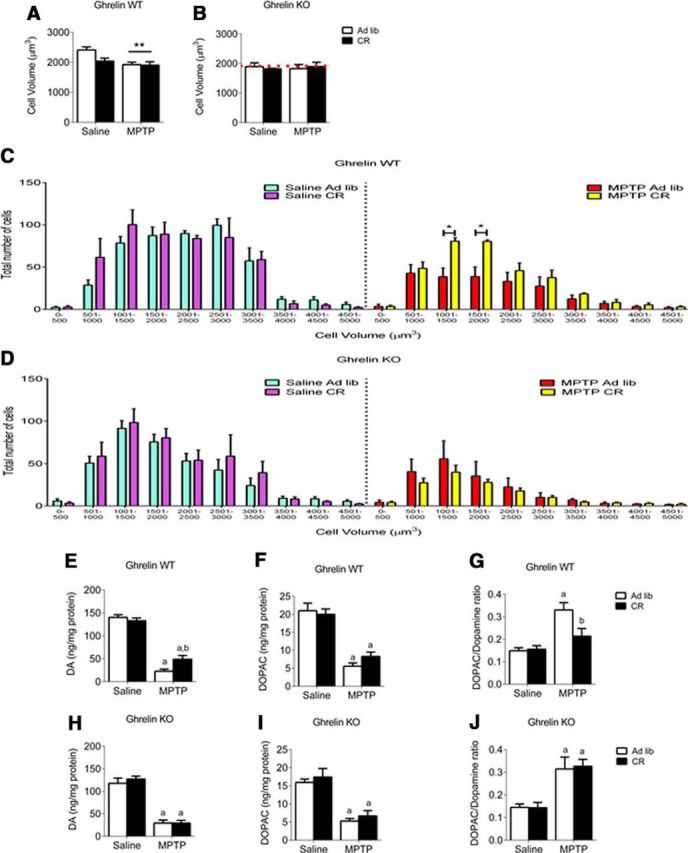
CR reduces small volume TH cell loss and enhances dopamine turnover in ghrelin WT but not KO mice. A, Overall cell volume for ghrelin WT mice showed a significant (p < 0.05) effect of MPTP administration but no effect of genotype or diet. B, Ghrelin KO mice showed no overall effect of diet, treatment, or genotype. The red dotted line represents the average cell volume of ghrelin WT MPTP-treated mice. When the cells were separated based on number and volume distribution as shown in C and D, the effect of CR is apparent. C, Ghrelin WT have a significant (p < 0.05) effect between ad libitum and CR cell volume in smaller (1000–2000 μm3) cells. There was no significant difference in the ghrelin KO mice (D). E, H, CR attenuates striatal DA loss in ghrelin WT but not ghrelin KO mice after MPTP administration. F, I, MPTP reduced DOPAC with no effect of genotype. G, J, CR reduced the elevation of the DOPAC/DA ratio in MPTP-treated mice compared with ad libitum, in ghrelin WT but not ghrelin KO mice. a, Significant compared with saline ad libitum controls. b, Significant compared with MPTP ad libitum controls.*p < 0.05, **p < 0.01. Data are represented as mean ± SEM (n = 6–10, two-way ANOVA, p < 0.05).
HPLC analysis
HPLC analysis of DA in the striatum revealed a significant overall (p < 0.05) reduction with MPTP administration and CR significantly attenuated the loss of dopamine in WT but not KO mice (Fig. 2E,H). MPTP also significantly reduced DOPAC in the striatum, however, there was no effect of diet on DOPAC levels regardless of genotype (Fig. 2F,I). CR also prevented the increase in the DOPAC/DA ratio observed after MPTP in ghrelin WT ad libitum but not ghrelin KO ad libitum mice (Fig. 2G,J).
TH protein expression
Reduced DA levels in the striatum indicate impaired dopamine synthesis, which is controlled by the rate-limiting enzyme TH. In the SN of ghrelin WT and KO mice, MPTP administration significantly reduced TH protein expression in both ad libitum fed and CR mice with no protective effect of CR in either genotype (Figs. 3A,C,D). In the striatum, however, CR significantly attenuated the reduction in TH protein levels in MPTP-treated ghrelin WT but not ghrelin KO mice (Fig. 3B,E,F). These results highlight that CR has site-specific effects acting to increase TH in the striatum but not the SN. Together, these results corroborate with the TH neuronal counts, cell volume analysis, and HPLC DA content results indicating that CR has a protective effect only in ghrelin WT mice. Overall, these results imply that ghrelin is responsible for the protective effects of CR in a mouse model of PD.
Figure 3.

The protective effect of CR is concomitant with striatal dopamine and elevated pAMPK, an effect not observed in ghrelin KO mice. A, B, Representative Western blot images of MPTP-induced reduction in TH levels in the SN and striatum. C, D, Quantification of TH levels in ghrelin WT and KO mice showed that MPTP significantly (p < 0.05) reduced TH expression in the SN. E, F, Quantification of TH levels in the striatum revealed that MPTP significantly (p < 0.05) reduced TH expression, this effect was rescued in CR ghrelin WT mice but not in KO mice. G, H, Representative Western blot images of pAMPK, AMPK, pACC, and ACC levels in the SN and striatum after either ad libitum or CR paradigms followed by MPTP or saline treatment. I, M, Quantification of pAMPK/AMPK and pACC/ACC levels in the SN reveals no effect in ghrelin WT mice, however, in KO mice there was a significant (p < 0.05) reduction between MPTP ad libitum and MPTP CR groups (J, N), showing that CR KO mice could not adapt appropriately to MPTP-induced cell degeneration. K, L, MPTP and CR individually increased striatal pAMPK/AMPK in ghrelin WT mice but not in ghrelin KO mice, as no change from baseline with either MPTP or CR was observed. O, P, MPTP-induced an increase in striatal pACC/ACC in ghrelin WT but not ghrelin KO mice, mimicking the effects seen with pAMPK/AMPK. Q, R, Representative Western blots for LC3 I and LC3 II in the SN and striatum of ghrelin WT and KO mice. S, LC3 II in the SN is significantly reduced in ghrelin WT mice after MPTP treatment; however, this was not observed in ghrelin KO mice (T). U, V, There was no effect of CR on LC3 II in the striatum from ghrelin WT and KO mice. However, there was a significant main effect of MPTP to increase LC3 II in WT but not KO mice. a, Significant compared with saline controls. b, Significant compared with a low dose of ghrelin. *p < 0.05, **p < 0.01. Data are represented as mean ± SEM (n = 5–7, one-way ANOVA, p < 0.05).
Ghrelin influences AMPK activation in the striatum
Ghrelin enhances AMPK activity in the hypothalamus (Andrews et al., 2008) and AMPK also increases mitochondrial biogenesis and function in the periphery (Bergeron et al., 2001; Horvath et al., 2011). Thus, we reasoned that the neuroprotective actions of CR induce a ghrelin-dependent increase in AMPK function in SN TH neurons. We found that both metabolic (CR) and chemical (MPTP) stress increased AMPK phosphorylation (pAMPK/AMPK ratio) and subsequent acetyl CoA carboxylase (ACC) in the striatum, but not the SN as seen for TH expression, in ghrelin WT but not ghrelin KO mice (Fig. 3G–P). CR in ghrelin KO MPTP-treated mice significantly reduced AMPK and ACC phosphorylation in the SN compared with ghrelin KO MPTP ad libitum mice (Fig. 3J,N). The maintenance of autophagy is one downstream effect of AMPK activation (Mihaylova and Shaw, 2011), therefore we examined LC3 II, the membrane-bound form of autophagosomes (Kimura et al., 2007). We observed significantly reduced LC3 II in the SN of CR ghrelin WT mice compared with ad libitum controls (Fig. 3S), with a significant overall elevation in response to MPTP in striatum (Fig. 3U). No effect was observed in the SN (Fig. 3T) or striatum (Fig. 3V) of ghrelin KO mice. The LC3 II results in the SN are inversely related to SN TH cell counts suggesting there is less autophagosome formation required in cells with less MPTP-induced degeneration.
PINK1 and Parkin regulate mitophagy and mutations in PINK and Parkin cause early onset PD, therefore, we also measured the expression of these two proteins in the SN and striatum from CR and ad libitum ghrelin WT and KO mice. PINK1 and Parkin expression showed a significant reduction in protein expression post-MPTP administration in the striatum with no significant effect of CR or genotype (data not shown). There was no change in protein levels in response to metabolic state, MPTP, or genotype in the SN (data not shown).
Exogenous ghrelin influences the phosphorylation of AMPK and ACC
To support the notion that increased endogenous ghrelin is the critical to CR-induced neuroprotection, we examined the effects of exogenous acyl ghrelin on AMPK and ACC phosphorylation both in vivo and in vitro. The addition of either acyl ghrelin or the ghrelin agonist JMV2894 increased AMPK activation in cultured dopaminergic cell line SN4741 (Fig. 4A–D).
Figure 4.
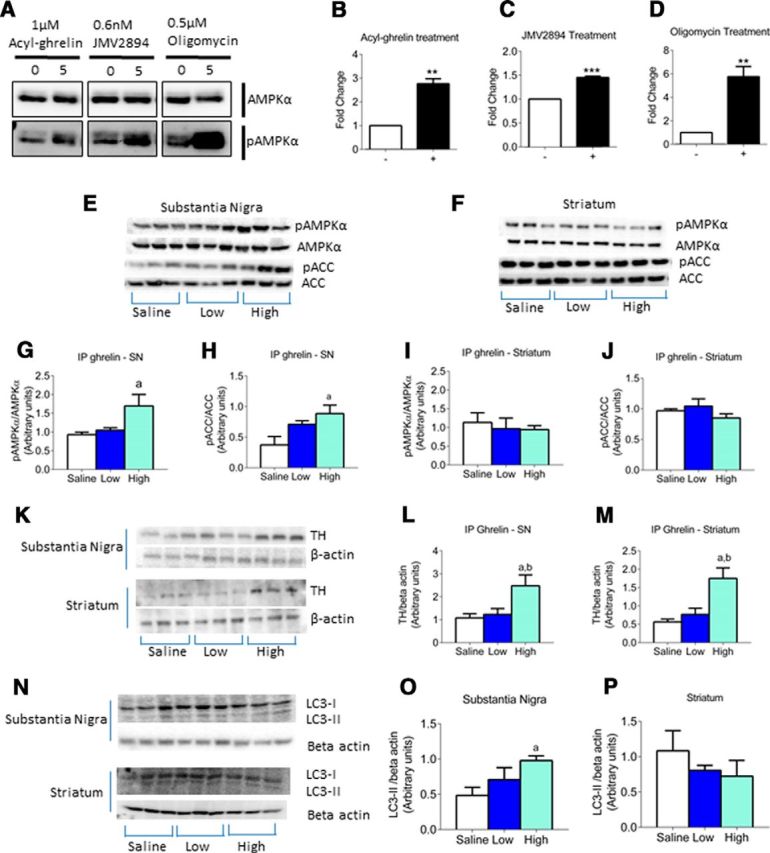
Exogenous ghrelin elevates TH and AMPK activation. A, Representative Western blot images of cultured dopaminergic neurons shows an increase in pAMPK levels in response to acyl ghrelin, JMV2894 (ghrelin agonist) or oligomycin treatment. Quantification of pAMPK/AMPK levels reveals a significant increase in response to acyl ghrelin (B), JMV2894 (C), and oligomycin (D) treatment. E, F, Representative Western blot images of pAMPK, AMPK, pACC, and ACC levels in the SN and striatum. G, H, Quantification of the pAMPK/AMPK and pACC/ACC in the SN (G) in response to a high dose of ghrelin reveals a significant elevation in response to the high dose of ghrelin. I, J, Quantification of pAMPK/AMPK and pACC/ACC in the striatum reveals no change between saline and ghrelin doses. K, Representative Western blot images of TH levels in the SN and striatum. Quantification of TH levels in the SN (L) and striatum (M) show that intraperitoneal ghrelin significantly increases TH expression in response to a high dose of ghrelin. Representative Western blot images of LC3 II expression in the SN and striatum (N). Quantification of LC3 II revealed high dose caused a significant increase in the SN (O) but not the striatum (P). a, Significant compared with saline/saline controls. b, Significant compared with saline/MPTP controls. Data are represented as mean ± SEM (n = 6–8, two-way ANOVA, p < 0.05).
For in vivo studies, we injected acyl ghrelin intraperitoneally at two different doses (low, 5 mg/kg; high, 15 mg/kg). The high dose of acyl ghrelin significantly increased AMPK and ACC phosphorylation in the SN (Fig. 4E,G,H). However, there was no significant difference in the striatum in response to either a low or high dose of acyl ghrelin (Fig. 4F,I,J). This is in contrast to the effect of CR on AMPK activation, as we observed a significant difference in the striatum but not the SN.
Injection of intraperitoneal acyl ghrelin at a high dose significantly increased TH expression in both the SN and striatum (Fig. 4K–M). Moreover, intraperitoneal acyl ghrelin increased LC3 II in the SN but not the striatum (Fig. 4N–P). There was no change in PINK1 expression in either the SN or the striatum (data not shown). Parkin expression remained unchanged in the SN; however, in the striatum there was a significant increase with a high dose of acyl ghrelin (data not shown). These results indicate that peripheral acyl ghrelin injection affects AMPK and ACC phosphorylation, as well as TH, Parkin, and LC3 II protein expression in the nigrostriatal system.
Exogenous ghrelin requires AMPK in dopamine neurons to elicit neuroprotection
To prove that ghrelin-induced neuroprotection requires AMPK activation in SN dopamine neurons, we generated a novel mouse line in which AMPK activation was disabled in dopaminergic neurons. These mice were generated by cross breeding Dat-Cre mice with Ampk β 1fl/fl; Ampk β 2fl/fl mice to generate AMPK WT and AMPK KO mice. AMPK β1 and β2 are regulatory subunits required for AMPK activity (O'Neill et al., 2011). To determine the specificity of the knock-out, we bred AMPK WT and KO with Rosa26loxSTOPlox tdTomato reporter mice to generate Dat-Cre;tdTomato and Dat-Cre;Ampk β 1fl/fl;Ampk β 2fl/fl;tdTomato mice. TH and tdTomato coexpression in the SN was >90% (Fig. 5A–C), indicating cre recombination had occurred in >90% of SN TH neurons. Deletion of AMPKβ1 and AMPKβ2 in dopamine neurons was confirmed by FACS of tdTomato-labeled neurons from midbrain dissections from Dat-Cre;tdTomato and Dat-Cre;Ampk β 1fl/fl;Ampk β 2fl/fl;tdTomato mice, and nested PCR for AMPKβ1 and AMPKβ2 (Fig. 5D). Positive bands for both AMPKβ1 and AMPKβ2 were observed in AMPK WT but not AMPK KO mice (Fig. 5D). As a positive control, nested PCR for GHSR was performed to confirm the presence of the ghrelin receptor in both AMPK WT and KO mice (Fig. 5D).
Figure 5.
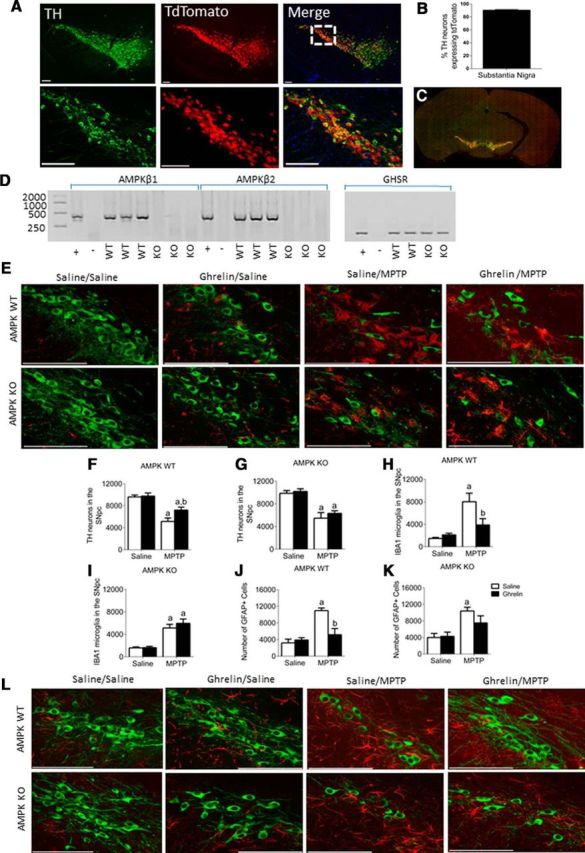
Ghrelin activates AMPK to elicit neuroprotection in an AMPK-dependent manner. A, DAT CRE mice crossed with the tdTomato line shows a >90% colocalization (B) between TH (green) and tdTomato (red) neurons. Scale bar, 100 μm. C, Representative tiled image showing TH (green) and tdTomato (red) where each tile represents a 20× image. D, tdTomato labeled TH neurons were sorted via FACs to show the selective deletion of AMPKβ1 and AMPKβ2 in AMPK WT but not AMPK KO mice. Product size for AMPKβ1 = 386 bp, AMPKβ2 = 395 bp. The ghrelin receptor (GHSR) is unaffected by deletion of AMPKβ1 and AMPKβ2 in SN TH neurons. E, Representative images showing TH neurons from AMPK WT and KO mice after chronic ghrelin treatment. F, G, Stereological quantification of TH neurons from AMPK WT (F) and KO (G) mice shows a protective effect of ghrelin treatment in WT but not KO mice. H, Stereological quantification of IBA1 microglia in the SN shows that ghrelin suppresses IBA1 cells relative to saline controls following MPTP treatment; however, this is not observed in AMPK KO mice (I). J, K, Stereological quantification of GFAP in the SN shows that ghrelin attenuates the MPTP-induced increase in GFAP cell numbers in AMPK WT (J) but not AMPK KO (K) mice. L, Representative images showing MPTP-induced astrocyte (GFAP) activation in the SN (TH, green; GFAP, red). a, Significant compared with saline/saline controls. b, Significant compared with saline/MPTP controls. Data are represented as mean ± SEM (n = 6–8, two-way ANOVA, p < 0.05). Scale bar, 100 μm.
To show that ghrelin elicits neuroprotection in a mouse model of PD, we chronically administered acyl ghrelin to AMPK WT and KO mice. In AMPK WT mice, acyl ghrelin administration significantly attenuated TH cell loss in MPTP-treated mice (Fig. 5E,F). This effect was abolished in the AMPK KO mice (Fig. 5G). Acyl-ghrelin reduced IBA1+ cell number in AMPK WT MPTP-treated mice (Fig. 5H), however, no significant effect was observed in AMPK KO mice (Fig. 5I). A similar pattern was observed with GFAP cells, in which acyl ghrelin reduced GFAP cell number in AMPK WT but not AMPK KO mice (Fig. 5J,K).
Despite the attenuated TH cell loss in the acyl ghrelin-treated AMPK WT mice there was no overall change in cell volume or distribution (Fig. 6A–D). HPLC analysis of dopamine and DOPAC in the striatum revealed that acyl ghrelin attenuated the MPTP-induced loss of dopamine and prevented the MPTP-induced rise in the DOPAC/dopamine ratio in AMPK WT but not AMPK KO mice (Fig. 6E–J). Changes in motor behavior were determined using an accelerating Rotarod by measuring latency to fall. There was no overall change between saline or ghrelin-treated AMPK WT and KO mice without MPTP treatment (Fig. 6K,L). When pretreated with saline and given MPTP there was no effect of genotype (Fig. 6M), however, there was a protective effect of ghrelin administration before MPTP in AMPK WT but not KO mice (Fig. 6N). Collectively, these experiments highlight that ghrelin activates AMPK in SN dopamine neurons, restricts dopaminergic cell loss, maintains striatal dopamine concentrations and promotes locomotor behavior after MPTP treatment to provide a neuroprotective effect.
Figure 6.
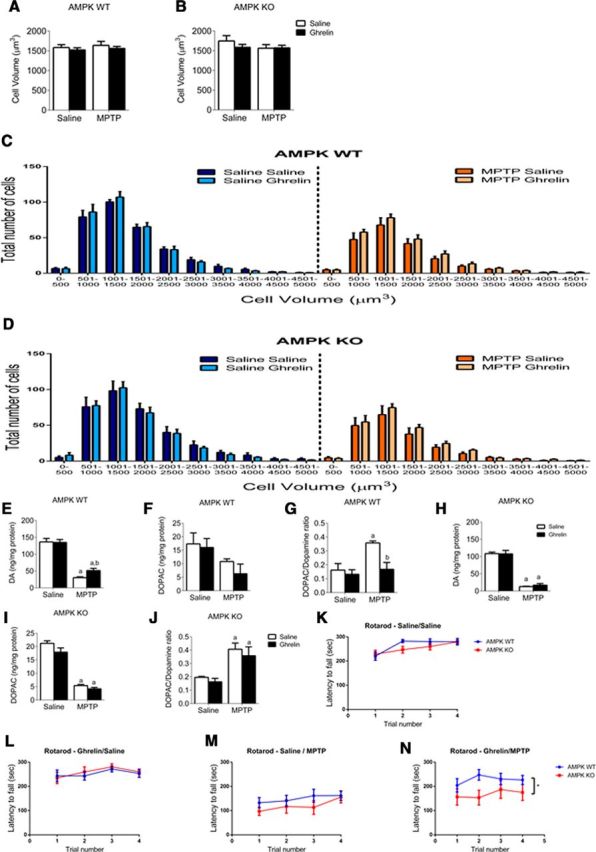
Chronic ghrelin injection enhances dopamine turnover and behavioral outcomes in AMPK WT but not KO mice. A, B, Overall cell volume showing no reduction in response to genotype or treatment. When cells were separated based on number and volume distribution as shown in C and D, there was no overall effect of genotype or treatment. HPLC data show that ghrelin significantly attenuates the MPTP-induced decrease in striatal dopamine concentration in AMPK WT but not AMPK KO mice (E, H). MPTP reduced DOPAC with no effect of genotype (F, I). Ghrelin treatment significantly attenuates the MPTP-induced increase in the DOPAC/dopamine ration in AMPK WT but not AMPK KO mice (G, J). K–N, Behavioral analysis showing latency to fall on an accelerating rotarod. K, L, No difference in latency to fall in mice not exposed to MPTP. In mice given MPTP, latency to fall is not effected by genotype in mice pretreated with saline (M). However, in mice pretreated with ghrelin there is a significant protective effect in AMPK WT but not KO as evidence by increased latency to fall (N). a, Significant compared with saline/saline controls. b, Significant compared with saline/MPTP controls. Data are represented as mean ± SEM (n = 6–12, two-way ANOVA, p < 0.05).
We previously showed that intraperitoneal ghrelin can elicit an increase in the pAMPK/AMPK and pACC/ACC ratio in SN (Fig. 4G,H) and that chronic ghrelin treatment to AMPK WT, but not AMPK KO is neuroprotective in a mouse model of PD. In further support of this neuroprotection, ghrelin treatment attenuated the MPTP-induced loss of TH in both the SN and striatum of AMPK WT but not AMPK KO mice (Fig. 7A–F). To determine whether chronic ghrelin differentially affected AMPK and subsequent ACC phosphorylation, we measured the pAMPK/AMPK and pACC/ACC ratio in AMPK WT and KO mice in response chronic daily ghrelin injections. There was no effect of chronic ghrelin treatment on the pAMPK/AMPK (Fig. 7G, I,J) or the pACC/ACC ratio (Fig. 7M,N) in the SN of either AMPK WT or AMPK KO mice. However, MPTP treatment elicited an increase in the pAMPK/AMPK (Fig. 7H,K,L) and pACC/ACC ratios (Fig. 7O,P) in the striatum of AMPK WT mice, but not AMPK KO mice. The mitophagy proteins PINK1 and Parkin (data not shown) and the autophagosome marker LC3 II were not significantly different between genotypes and treatment (Fig. 7Q–V).
Figure 7.

Chronic ghrelin injections increase nigrostriatal TH expression and AMPK activation in an AMPK-dependent manner. A, B, representative Western blot images of the SN (A) and striatum (B) showing TH levels. In both the SN and the striatum there is a significant protective effect of ghrelin administration on TH levels in AMPK WT mice (C, E) that is absent in AMPK KO mice (D, F). G, H, Representative Western blot images showing pAMPK, AMPK, pACC, and ACC levels in the SN (G) and striatum (H). There was no significant change in the pAMPK/AMPK (I, J) or pACC/ACC (M, N) ratio in the SN of AMPK WT or AMPK KO mice in response to MPTP or ghrelin. MPTP-induced an increase in the pAMPK/AMPK and pACC/ACC ratio in AMPK WT mice (K, O) but not AMPK KO mice (L, P). In the striatum, MPTP induced an increase in the pAMPK/AMPK and pACC/ACC ratio AMPK WT mice (K, O) but not AMPK KO mice (L, P). Q, R, Representative Western blot images of LC3 II expression in the SN (Q) and striatum (R). There was no significant effect of MPTP or ghrelin administration on LC3-II levels in the SN (S, T) or striatum (U, V). a, Significant compared with saline/saline controls. b, Significant compared with saline/MPTP controls. Data are represented as mean ± SEM (n = 6–8, two-way ANOVA, p < 0.05).
Discussion
CR protects against a number of pathological conditions including diabetes, cancer, heart disease, and neurodegeneration. In PD, an alternate-day feeding schedule where rats consumed 30–40% less calories than ad libitum controls was neuroprotective post-MPTP exposure (Duan and Mattson, 1999). Mice also elicited a neuroprotective response when alternate day feeding begun after exposure to MPTP (Holmer et al., 2005). Primates with a chronic overall 30% reduction in food intake were also resistant to MPTP-induced neurotoxicity (Maswood et al., 2004). These studies prove that CR is beneficial in PD; however, the difficulty to adhere to CR necessitates an alternative method to recapitulate the neuroprotective benefits of CR while bypassing dietary constraints. Evidence from cells treated with serum from CR rats suggests a hormonal factor improves mitochondrial function and cell viability (López-Lluch et al., 2006). We hypothesized that ghrelin may be this hormonal factor, because CR increases plasma acyl ghrelin (Lutter et al., 2008) and ghrelin restricts degeneration in PD (Andrews et al., 2009). In this study, we show for the first time that ghrelin mediates the neuroprotective effect of CR in a mouse model of PD by attenuating MPTP-induced loss of TH neurons, TH neuronal volume and dopamine content in the striatum. Further, we show that AMPK in SN dopamine neurons is a molecular target for ghrelin's neuroprotective effects, as deletion of AMPK β1 and β2 subunits only in dopamine neurons prevented ghrelin-induced neuroprotection. These results suggest that ghrelin, and its downstream target AMPK, has a potential therapeutic application in the treatment of PD to mimic the neuroprotective effect of CR without the need for strict dietary constraints.
Although this is the first study to show that ghrelin mediates the neuroprotective effects of CR in a mouse model of PD, it supports an increasing number of observations that ghrelin restricts the negative consequences of CR or negative energy balance. For example, ghrelin prevents the excessive decline in blood glucose during severe CR (Zhao et al., 2010) and the anxiolytic effects of CR require GHSR signaling (Lutter et al., 2008). A recent study by McFarlane et al. (2014) shows that adult-ablation of ghrelin secreting cells has no effect on food intake, body weight, and fed blood glucose. Only under CR did these mice show deficits in blood glucose. Moreover, CR reduces hippocampal cell death in GHSR WT but not GHSR KO mice (Walker et al., 2015) and CR induces neurogenesis in a GHSR-dependent manner (Hornsby et al., 2016). Collectively, these studies show that the major function of ghrelin is to act as a feedback signal of CR (negative energy balance) and maintain physiological and neurological function during this time.
Our data show that AMPK in SN dopamine neurons is a molecular target of ghrelin during CR to maintain neuronal function. First, metabolic stress (CR) and/or toxic stress (MPTP) promoted AMPK activity in striatal dopamine nerve terminals in ghrelin WT but not ghrelin KO. The ability of MPTP to increase AMPK activity is supported by previous studies in mice and cells (Choi et al., 2010). AMPK enhances mitochondrial function and biogenesis (Reznick and Shulman, 2006), as such, we suggest CR-induced AMPK phosphorylation at the nerve terminal promotes neuronal energy metabolism and supports ongoing dopaminergic neuronal activity, which is supported by the reduced striatal DOPAC/dopamine ratio of both CR ghrelin and AMPK WT mice but not their respective KO mice. Moreover, AMPK activity diminishes with age (Reznick et al., 2007) consistent with the age-related neurodegeneration that contributes to the onset of PD. Thus, the ability of CR to maintain AMPK activity in a ghrelin-dependent manner may restrict age-related decline in the nigrostriatal system. This possibility is further strengthened by data showing that plasma ghrelin and ghrelin's function diminishes with age, an effect that can be reversed with CR (Englander et al., 2004; Smith et al., 2007; Sun et al., 2007; Yang et al., 2007; Takeda et al., 2010). Further, PD patients have reduced postprandial plasma ghrelin levels (Unger et al., 2011).
In cultured dopaminergic neurons both acyl ghrelin and a ghrelin agonist elicited a robust increase in AMPK activation. Acute acyl ghrelin injection in vivo increased both AMPK and ACC phosphorylation in the SN but not the striatum. This is the first in vivo study that shows ghrelin activates AMPK activity in the midbrain, similar to numerous reports showing ghrelin activates AMPK activity in the hypothalamus (Andersson et al., 2004; Kola et al., 2005; Andrews et al., 2008). As noted above, CR drives ghrelin-induced AMPK phosphorylation in the striatum, but not the SN, yet acute ghrelin injection in vivo increased AMPK phosphorylation in the SN but not the striatum. We consider this discrepancy may be due to chronically elevated ghrelin versus an acute ghrelin injection. Chronically high plasma ghrelin, as seen in CR ghrelin WT mice, activates SN dopamine neurons via the GHSR which then facilitates and propagates AMPK phosphorylation in areas of metabolic need, in this case striatal nerve terminals to prevent degeneration. Although acute injection of ghrelin increases the pAMPK/AMPK ratio in the SN after 45 min, this narrow time frame presumably prevents propagation of AMPK phosphorylation in the striatum. It is important to note that the ghrelin receptor, GHSR, is abundantly expressed in the SN with little or no expression in the striatum (Zigman et al., 2006).
Importantly, we conclusively demonstrate that AMPK activity in dopamine neurons is necessary for ghrelin-induced neuroprotection in a mouse model of PD. We generated a model in which AMPKβ1 and AMPKβ2 were successfully deleted in DAT-expressing neurons. Deletion of both AMPKβ1 and AMPKβ2 in muscle ablated AMPK phosphorylation and lead to impaired glucose homeostasis (O'Neill et al., 2011). Using the model, we showed that ghrelin prevents nigrostriatal degeneration in MPTP-treated AMPK WT but not AMPK KO mice, clearly establishing AMPK as a critical molecular mechanism mediating the neuroprotective effects of ghrelin on the nigrostriatal system. Our genetic model also deletes AMPKβ1 and AMPKβ2 in all DAT-cre-expressing neurons including populations not associated with PD, such as the hypothalamic and VTA dopamine neurons. However, MPTP predominantly affects SN dopamine neurons (Seniuk et al., 1990; Muthane et al., 1994), which strengthens the specific and important neuroprotective actions of ghrelin on AMPK activity in the SN. We should note that we did not detect a change in pAMPK/AMPK ratio in the SN or striatum of chronic ghrelin-treated AMPK WT or KO mice, whereas acute ghrelin injection affected the pAMPK/AMPK ratio in the SN. There are many potential reasons for this including the dosage and time of tissue collection after last injection. However, the most plausible reason is due to the tissue collection, because we measured pAMPK/AMPK in a dissected piece of tissue, of which only a small proportion represents SN dopamine neurons. Nevertheless, in response to MPTP treatment AMPK WT mice produced an increase in pAMPK/AMPK ratio in the striatum, which was not observed in AMPK KO. In fact, the significant main effect of MPTP to suppress AMPK phosphorylation, independent from ghrelin treatment, in both the SN and striatum of AMPK KO illustrates the important role of AMPKβ1 and AMPKβ2 in SN dopamine neurons to combat cellular stress caused by MPTP. Moreover, CR ghrelin KO mice also did not show a compensatory increase in MPTP-induced AMPK phosphorylation in the striatum, further supporting the idea that ghrelin targets AMPK in SN dopamine neurons during CR to prevent degeneration.
Intriguingly, we noted differential effects of CR and ghrelin treatment on gliosis. In the CR experiment, the microglial response to MPTP was not different in ghrelin WT and KO mice despite the greater TH cell loss in ghrelin KO mice. This is somewhat unexpected given microglia become activated to remove neuronal damage by phagocytosis (Neumann et al., 2009). It is possible that a threshold level of cell loss elicits the same microglial response, perhaps mediated by the release of caspase signal (Burguillos et al., 2011). Moreover, GFAP cell number was increased after MPTP and suppressed in CR mice regardless of genotype. In primates CR elicited a protective effect by limiting astrogliosis in the hippocampus (Sridharan et al., 2013). These results suggest that the effects of CR on gliosis are independent from changes in plasma ghrelin. However, chronic ghrelin-treatment to AMPK WT and AMPK KO mice showed that ghrelin reduced microglia and GFAP in AMPK WT but not AMPK KO mice treated with MPTP. This effect of ghrelin treatment is consistent with in vitro studies that indicate ghrelin directly inhibits glial activation to diminish the inflammatory response (Lee and Yune, 2014). Moreover, that lack of an effect in AMPK KO mice suggests ghrelin acts directly on AMPK in SN dopamine to restrict microglia and GFAP expression, a hypothesis supported by studies showing that AMPK activity influences gliosis (Lu et al., 2010; Yi et al., 2011; Chen et al., 2014a; Han et al., 2014; Zhou et al., 2014).
This is the first study to show the important neuroprotective in vivo actions of AMPK in dopamine neurons, although a number of studies implicate AMPK as an intracellular energy sensor promoting neuroprotection in models of PD. For example, AMPK attenuates mitochondrial and dopaminergic dysfunction in drosophila models of PD (Ng et al., 2012), and pharmacological activators of AMPK, such as Resveratrol (Jin et al., 2008) and guanidino-propionic acid (Horvath et al., 2011), were neuroprotective in vivo. Metformin treatment in cells overexpressing α synuclein, to model PD, also activated AMPK and restricted cell death (Dulovic et al., 2014). However, in vitro studies recently demonstrated that AMPK overactivation has a detrimental effect and promoted α synuclein accumulation and inhibited neurite growth (Jiang et al., 2013).
In conclusion, CR is perhaps the most robust and reproducible mechanism to enhance lifespan and promote healthy aging. The exact mechanism/s that achieve this are currently unknown, however, several theories include altered stress response pathways, altered signaling pathways involving SIRT1, FOXO, UCP2, and AMPK (Andrews, 2010) as well as alterations in metabolic hormones such as ghrelin and insulin. We consider CR induces a mild stress and encourages compensatory metabolic changes that favor improved intracellular mitochondrial health. Although CR promotes metabolic health and reduces neurodegeneration there is a poor compliance in the general population, as it requires ∼20–40% reduced calorie intake over years to achieve maximal benefits. Consequently, there is a need to recapitulate these beneficial effects without restricting calorie intake. We have discovered a novel pathway where circulating ghrelin, which is elevated during CR, has a protective role in the nigrostriatal system via enhanced AMPK activity. This ghrelin-induced neuroprotection is dependent on AMPK activity in dopamine neurons. Future research should focus on exploiting this pathway to determine the in vivo neuroprotective effects that restrict neurodegeneration without the need to adhere to strict dietary regimes.
Footnotes
This work was supported by grants and fellowships from the Australian National Health and Medical Research Council to Z.B.A. (546131, 1084344) and B.E.K.; the Australian Research Council to Z.B.A. (FT100100966); and NIH NS056181 to J.H; and supported in part by the Victorian Government's Operational Infrastructure Support Program (B.E.K.), a St David's Medical Foundation Seed-Corn Grant (J.S.D.) and a Monash University Fellowship to Z.B.A.
The authors declare no competing financial interests.
References
- Andersson U, Filipsson K, Abbott CR, Woods A, Smith K, Bloom SR, Carling D, Small CJ. AMP-activated protein kinase plays a role in the control of food intake. J Biol Chem. 2004;279:12005–12008. doi: 10.1074/jbc.C300557200. [DOI] [PubMed] [Google Scholar]
- Andrews ZB. Uncoupling protein-2 and the potential link between metabolism and longevity. Curr Aging Sci. 2010;3:102–112. doi: 10.2174/1874609811003020102. [DOI] [PubMed] [Google Scholar]
- Andrews ZB, Horvath B, Barnstable CJ, Elseworth J, Yang L, Beal MF, Roth RH, Matthews RT, Horvath TL. Uncoupling protein-2 is critical for nigral dopamine cell survival in a mouse model of Parkinson's disease. J Neurosci. 2005;25:184–191. doi: 10.1523/JNEUROSCI.4269-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews ZB, Liu ZW, Walllingford N, Erion DM, Borok E, Friedman JM, Tschöp MH, Shanabrough M, Cline G, Shulman GI, Coppola A, Gao XB, Horvath TL, Diano S. UCP2 mediates ghrelin's action on NPY/AgRP neurons by lowering free radicals. Nature. 2008;454:846–851. doi: 10.1038/nature07181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews ZB, Erion D, Beiler R, Liu ZW, Abizaid A, Zigman J, Elsworth JD, Savitt JM, DiMarchi R, Tschoep M, Roth RH, Gao XB, Horvath TL. Ghrelin promotes and protects nigrostriatal dopamine function via a UCP2-dependent mitochondrial mechanism. J Neurosci. 2009;29:14057–14065. doi: 10.1523/JNEUROSCI.3890-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron R, Ren JM, Cadman KS, Moore IK, Perret P, Pypaert M, Young LH, Semenkovich CF, Shulman GI. Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am J Physiol Endocrinol Metab. 2001;281:E1340–E1346. doi: 10.1152/ajpendo.2001.281.6.E1340. [DOI] [PubMed] [Google Scholar]
- Burguillos MA, Deierborg T, Kavanagh E, Persson A, Hajji N, Garcia-Quintanilla A, Cano J, Brundin P, Englund E, Venero JL, Joseph B. Caspase signalling controls microglia activation and neurotoxicity. Nature. 2011;472:319–324. doi: 10.1038/nature09788. [DOI] [PubMed] [Google Scholar]
- Chan YC, Suzuki M, Yamamoto S. Dietary, anthropometric, hematological and biochemical assessment of the nutritional status of centenarians and elderly people in Okinawa, Japan. J Am Coll Nutr. 1997;16:229–235. doi: 10.1080/07315724.1997.10718679. [DOI] [PubMed] [Google Scholar]
- Chen CC, Lin JT, Cheng YF, Kuo CY, Huang CF, Kao SH, Liang YJ, Cheng CY, Chen HM. Amelioration of LPS-induced inflammation response in microglia by AMPK activation. Biomed Res Int. 2014a;2014:692061. doi: 10.1155/2014/692061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Guan Z, Wang L, Song G, Ma B, Wang Y. Meta-analysis: overweight, obesity, and Parkinson's disease. Int J Endocrinol. 2014b;2014:203930. doi: 10.1155/2014/203930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JS, Park C, Jeong JW. AMP-activated protein kinase is activated in Parkinson's disease models mediated by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Biochem Biophys Res Commun. 2010;391:147–151. doi: 10.1016/j.bbrc.2009.11.022. [DOI] [PubMed] [Google Scholar]
- Choi JY, Jang EH, Park CS, Kang JH. Enhanced susceptibility to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity in high-fat diet-induced obesity. Free Radic Biol Med. 2005;38:806–816. doi: 10.1016/j.freeradbiomed.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, Allison DB, Cruzen C, Simmons HA, Kemnitz JW, Weindruch R. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325:201–204. doi: 10.1126/science.1173635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Song N, Xie J, Jiang H. Ghrelin antagonized 1-methyl-4-phenylpyridinium (MPP+)-induced apoptosis in MES23.5 cells. J Mol Neurosci. 2009;37:182–189. doi: 10.1007/s12031-008-9162-7. [DOI] [PubMed] [Google Scholar]
- Duan W, Mattson MP. Dietary restriction and 2-deoxyglucose administration improve behavioral outcome and reduce degeneration of dopaminergic neurons in models of Parkinson's disease. J Neurosci Res. 1999;57:195–206. doi: 10.1002/(SICI)1097-4547(19990715)57:2%3C195::AID-JNR5%3E3.0.CO%3B2-P. [DOI] [PubMed] [Google Scholar]
- Dulovic M, Jovanovic M, Xilouri M, Stefanis L, Harhaji-Trajkovic L, Kravic-Stevovic T, Paunovic V, Ardah MT, El-Agnaf OM, Kostic V, Markovic I, Trajkovic V. The protective role of AMP-activated protein kinase in alpha-synuclein neurotoxicity in vitro. Neurobiol Dis. 2014;63:1–11. doi: 10.1016/j.nbd.2013.11.002. [DOI] [PubMed] [Google Scholar]
- Englander EW, Gomez GA, Greeley GH., Jr Alterations in stomach ghrelin production and in ghrelin-induced growth hormone secretion in the aged rat. Mech Ageing Dev. 2004;125:871–875. doi: 10.1016/j.mad.2004.06.011. [DOI] [PubMed] [Google Scholar]
- Hailer NP, Wirjatijasa F, Roser N, Hischebeth GT, Korf HW, Dehghani F. Astrocytic factors protect neuronal integrity and reduce microglial activation in an in vitro model of N-methyl-d-aspartate-induced excitotoxic injury in organotypic hippocampal slice cultures. Eur J Neurosci. 2001;14:315–326. doi: 10.1046/j.0953-816x.2001.01649.x. [DOI] [PubMed] [Google Scholar]
- Han Y, Jiang C, Tang J, Wang C, Wu P, Zhang G, Liu W, Jamangulova N, Wu X, Song X. Resveratrol reduces morphine tolerance by inhibiting microglial activation via AMPK signalling. Eur J Pain. 2014;18:1458–1470. doi: 10.1002/ejp.511. [DOI] [PubMed] [Google Scholar]
- Ho L, Titus AS, Banerjee KK, George S, Lin W, Deota S, Saha AK, Nakamura K, Gut P, Verdin E, Kolthur-Seetharam U. SIRT4 regulates ATP homeostasis and mediates a retrograde signaling via AMPK. Aging. 2013;5:835–849. doi: 10.18632/aging.100616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmer HK, Keyghobadi M, Moore C, Menashe RA, Meshul CK. Dietary restriction affects striatal glutamate in the MPTP-induced mouse model of nigrostriatal degeneration. Synapse. 2005;57:100–112. doi: 10.1002/syn.20163. [DOI] [PubMed] [Google Scholar]
- Hornsby AK, Redhead YT, Rees DJ, Ratcliff MS, Reichenbach A, Wells T, Francis L, Amstalden K, Andrews ZB, Davies JS. Short-term calorie restriction enhances adult hippocampal neurogenesis and remote fear memory in a Ghsr-dependent manner. Psychoneuroendocrinology. 2016;63:198–207. doi: 10.1016/j.psyneuen.2015.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath TL, Erion DM, Elsworth JD, Roth RH, Shulman GI, Andrews ZB. GPA protects the nigrostriatal dopamine system by enhancing mitochondrial function. Neurobiol Dis. 2011;43:152–162. doi: 10.1016/j.nbd.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Li LJ, Wang J, Xie JX. Ghrelin antagonizes MPTP-induced neurotoxicity to the dopaminergic neurons in mouse substantia nigra. Exp Neurol. 2008;212:532–537. doi: 10.1016/j.expneurol.2008.05.006. [DOI] [PubMed] [Google Scholar]
- Jiang P, Gan M, Ebrahim AS, Castanedes-Casey M, Dickson DW, Yen SH. Adenosine monophosphate-activated protein kinase overactivation leads to accumulation of alpha-synuclein oligomers and decrease of neurites. Neurobiol Aging. 2013;34:1504–1515. doi: 10.1016/j.neurobiolaging.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin F, Wu Q, Lu YF, Gong QH, Shi JS. Neuroprotective effect of resveratrol on 6-OHDA-induced Parkinson's disease in rats. Eur J Pharmacol. 2008;600:78–82. doi: 10.1016/j.ejphar.2008.10.005. [DOI] [PubMed] [Google Scholar]
- Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452–460. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- Kola B, Hubina E, Tucci SA, Kirkham TC, Garcia EA, Mitchell SE, Williams LM, Hawley SA, Hardie DG, Grossman AB, Korbonits M. Cannabinoids and ghrelin have both central and peripheral metabolic and cardiac effects via AMP-activated protein kinase. J Biol Chem. 2005;280:25196–25201. doi: 10.1074/jbc.C500175200. [DOI] [PubMed] [Google Scholar]
- Lee JY, Yune TY. Ghrelin inhibits oligodendrocyte cell death by attenuating microglial activation. Endocrinol Metab (Seoul) 2014;29:371–378. doi: 10.3803/EnM.2014.29.3.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Lluch G, Hunt N, Jones B, Zhu M, Jamieson H, Hilmer S, Cascajo MV, Allard J, Ingram DK, Navas P, de Cabo R. Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency. Proc Natl Acad Sci U S A. 2006;103:1768–1773. doi: 10.1073/pnas.0510452103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu DY, Tang CH, Chen YH, Wei IH. Berberine suppresses neuroinflammatory responses through AMP-activated protein kinase activation in BV-2 microglia. J Cell Biochem. 2010;110:697–705. doi: 10.1002/jcb.22580. [DOI] [PubMed] [Google Scholar]
- Lutter M, Sakata I, Osborne-Lawrence S, Rovinsky SA, Anderson JG, Jung S, Birnbaum S, Yanagisawa M, Elmquist JK, Nestler EJ, Zigman JM. The orexigenic hormone ghrelin defends against depressive symptoms of chronic stress. Nat Neurosci. 2008;11:752–753. doi: 10.1038/nn.2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maswood N, Young J, Tilmont E, Zhang Z, Gash DM, Gerhardt GA, Grondin R, Roth GS, Mattison J, Lane MA, Carson RE, Cohen RM, Mouton PR, Quigley C, Mattson MP, Ingram DK. Caloric restriction increases neurotrophic factor levels and attenuates neurochemical and behavioral deficits in a primate model of Parkinson's disease. Proc Natl Acad Sci U S A. 2004;101:18171–18176. doi: 10.1073/pnas.0405831102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarlane MR, Brown MS, Goldstein JL, Zhao TJ. Induced ablation of ghrelin cells in adult mice does not decrease food intake, body weight, or response to high-fat diet. Cell Metab. 2014;20:54–60. doi: 10.1016/j.cmet.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon M, Kim HG, Hwang L, Seo JH, Kim S, Hwang S, Kim S, Lee D, Chung H, Oh MS, Lee KT, Park S. Neuroprotective effect of ghrelin in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson's disease by blocking microglial activation. Neurotox Res. 2009;15:332–347. doi: 10.1007/s12640-009-9037-x. [DOI] [PubMed] [Google Scholar]
- Muthane U, Ramsay KA, Jiang H, Jackson-Lewis V, Donaldson D, Fernando S, Ferreira M, Przedborski S. Differences in nigral neuron number and sensitivity to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in C57/bl and CD-1 mice. Exp Neurol. 1994;126:195–204. doi: 10.1006/exnr.1994.1058. [DOI] [PubMed] [Google Scholar]
- Neumann H, Kotter MR, Franklin RJ. Debris clearance by microglia: an essential link between degeneration and regeneration. Brain. 2009;132:288–295. doi: 10.1093/brain/awn109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng CH, Guan MS, Koh C, Ouyang X, Yu F, Tan EK, O'Neill SP, Zhang X, Chung J, Lim KL. AMP kinase activation mitigates dopaminergic dysfunction and mitochondrial abnormalities in Drosophila models of Parkinson's disease. J Neurosci. 2012;32:14311–14317. doi: 10.1523/JNEUROSCI.0499-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill HM, Maarbjerg SJ, Crane JD, Jeppesen J, Jørgensen SB, Schertzer JD, Shyroka O, Kiens B, van Denderen BJ, Tarnopolsky MA, Kemp BE, Richter EA, Steinberg GR. AMP-activated protein kinase (AMPK) β1β2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc Natl Acad Sci U S A. 2011;108:16092–16097. doi: 10.1073/pnas.1105062108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reznick RM, Shulman GI. The role of AMP-activated protein kinase in mitochondrial biogenesis. J Physiol. 2006;574:33–39. doi: 10.1113/jphysiol.2006.109512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reznick RM, Zong H, Li J, Morino K, Moore IK, Yu HJ, Liu ZX, Dong J, Mustard KJ, Hawley SA, Befroy D, Pypaert M, Hardie DG, Young LH, Shulman GI. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab. 2007;5:151–156. doi: 10.1016/j.cmet.2007.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seniuk NA, Tatton WG, Greenwood CE. Dose-dependent destruction of the coeruleus-cortical and nigral-striatal projections by MPTP. Brain Res. 1990;527:7–20. doi: 10.1016/0006-8993(90)91055-L. [DOI] [PubMed] [Google Scholar]
- Smith RG, Sun Y, Jiang H, Albarran-Zeckler R, Timchenko N. Ghrelin receptor (GHS-R1A) agonists show potential as interventive agents during aging. Ann N Y Acad Sci. 2007;1119:147–164. doi: 10.1196/annals.1404.023. [DOI] [PubMed] [Google Scholar]
- Sridharan A, Pehar M, Salamat MS, Pugh TD, Bendlin BB, Willette AA, Anderson RM, Kemnitz JW, Colman RJ, Weindruch RH, Puglielli L, Johnson SC. Calorie restriction attenuates astrogliosis but not amyloid plaque load in aged rhesus macaques: a preliminary quantitative imaging study. Brain Res. 2013;1508:1–8. doi: 10.1016/j.brainres.2013.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Garcia JM, Smith RG. Ghrelin and growth hormone secretagogue receptor expression in mice during aging. Endocrinology. 2007;148:1323–1329. doi: 10.1210/en.2006-0782. [DOI] [PubMed] [Google Scholar]
- Takeda H, Muto S, Hattori T, Sadakane C, Tsuchiya K, Katsurada T, Ohkawara T, Oridate N, Asaka M. Rikkunshito ameliorates the aging-associated decrease in ghrelin receptor reactivity via phosphodiesterase III inhibition. Endocrinology. 2010;151:244–252. doi: 10.1210/en.2009-0633. [DOI] [PubMed] [Google Scholar]
- Unger MM, Möller JC, Mankel K, Eggert KM, Bohne K, Bodden M, Stiasny-Kolster K, Kann PH, Mayer G, Tebbe JJ, Oertel WH. Postprandial ghrelin response is reduced in patients with Parkinson's disease and idiopathic REM sleep behaviour disorder: a peripheral biomarker for early Parkinson's disease? J Neurol. 2011;258:982–990. doi: 10.1007/s00415-010-5864-1. [DOI] [PubMed] [Google Scholar]
- Walker AK, Rivera PD, Wang Q, Chuang JC, Tran S, Osborne-Lawrence S, Estill SJ, Starwalt R, Huntington P, Morlock L, Naidoo J, Williams NS, Ready JM, Eisch AJ, Pieper AA, Zigman JM. The P7C3 class of neuroprotective compounds exerts antidepressant efficacy in mice by increasing hippocampal neurogenesis. Mol Psychiatry. 2015;20:500–508. doi: 10.1038/mp.2014.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Youm YH, Nakata C, Dixit VD. Chronic caloric restriction induces forestomach hypertrophy with enhanced ghrelin levels during aging. Peptides. 2007;28:1931–1936. doi: 10.1016/j.peptides.2007.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi CO, Jeon BT, Shin HJ, Jeong EA, Chang KC, Lee JE, Lee DH, Kim HJ, Kang SS, Cho GJ, Choi WS, Roh GS. Resveratrol activates AMPK and suppresses LPS-induced NF-κB-dependent COX-2 activation in RAW 264.7 macrophage cells. Anat Cell Biol. 2011;44:194–203. doi: 10.5115/acb.2011.44.3.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao TJ, Liang G, Li RL, Xie X, Sleeman MW, Murphy AJ, Valenzuela DM, Yancopoulos GD, Goldstein JL, Brown MS. Ghrelin O-acyltransferase (GOAT) is essential for growth hormone-mediated survival of calorie-restricted mice. Proc Natl Acad Sci U S A. 2010;107:7467–7472. doi: 10.1073/pnas.1002271107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Cao Y, Ao G, Hu L, Liu H, Wu J, Wang X, Jin M, Zheng S, Zhen X, Alkayed NJ, Jia J, Cheng J. CaMKKbeta-dependent activation of AMP-activated protein kinase is critical to suppressive effects of hydrogen sulfide on neuroinflammation. Antioxid Redox Signal. 2014;21:1741–1758. doi: 10.1089/ars.2013.5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zigman JM, Jones JE, Lee CE, Saper CB, Elmquist JK. Expression of ghrelin receptor mRNA in the rat and the mouse brain. J Comp Neurol. 2006;494:528–548. doi: 10.1002/cne.20823. [DOI] [PMC free article] [PubMed] [Google Scholar]