

Abstract
An abnormal chromosome number, a condition known as aneuploidy, is a ubiquitous feature of cancer cells. A number of studies have shown that aneuploidy impairs cellular fitness. However, there is also evidence that aneuploidy can arise in response to specific challenges and can confer a selective advantage under certain environmental stresses. Cancer cells are likely exposed to a number of challenging conditions arising within the tumor microenvironment. To investigate whether aneuploidy may confer a selective advantage to cancer cells, we employed a controlled experimental system. We used the diploid, colorectal cancer cell line DLD1 and two DLD1-derived cell lines carrying single-chromosome aneuploidies to assess a number of cancer cell properties. Such properties, which included rates of proliferation and apoptosis, anchorage-independent growth, and invasiveness, were assessed both under standard culture conditions and under conditions of stress (i.e., serum starvation, drug treatment, hypoxia). Similar experiments were performed in diploid vs. aneuploid non-transformed human primary cells. Overall, our data show that aneuploidy can confer selective advantage to human cells cultured under non-standard conditions. These findings indicate that aneuploidy can increase the adaptability of cells, even those, such as cancer cells, that are already characterized by increased proliferative capacity and aggressive tumorigenic phenotypes.
Fundamental to the survival of any organism is the balance between cell proliferation and cell death, which is required to ensure organismal development and to maintain healthy tissues and organs. The death and proliferation of normal, healthy cells is ensured by their ability to respond to and modulate growth and death signals. As opposed to healthy cells, cancer cells are characterized by the ability to escape such signals, thus becoming capable of evading apoptosis and proliferating independent of growth signals1. Several other features, typically referred to as “hallmarks of cancer”1, are shared by many cancer cells independent of their origin. One such feature, ubiquitous in cancer cells, is aneuploidy2,3,4. Inspired by his studies in sea urchin embryos, Theodor Boveri proposed, over a century ago, that the abnormal chromosome numbers (aneuploidy) found in cancer cells were responsible for cancer cells’ abnormal behavior5,6. Nevertheless, the effect of aneuploidy on cancer cell behavior is still unclear and abnormal chromosome numbers are generally acknowledged to negatively affect cell function7. Indeed, aneuploidy is the leading cause of miscarriage in humans8 and mosaic aneuploidy is typically associated with inherited disorders9. Moreover, recent studies aimed at investigating the effect of aneuploidy on cell physiology have revealed that aneuploidy negatively affects cellular fitness7 in a number of experimental systems, including mouse embryonic fibroblasts10 and budding yeast11. Nevertheless, there is also evidence that aneuploidy can confer a selective advantage in certain contexts. For instance, aneuploidy was shown to be an acquired trait in strains of Candida albicans that developed resistance to antifungal drugs12,13. Similarly, acquisition of aneuploid karyotypes was shown to allow budding yeast to adapt to a number of genotypic defects, including the lack of a key molecular motor14, telomerase insufficiency15, or lack of thiol peroxidase genes16. Moreover, aneuploid budding yeast strains were shown to display a growth advantage under a number of environmental stresses, despite their reduced fitness when grown under optimal conditions17. Finally, aneuploidy was proposed to contribute to the adaptation of liver cells in response to hepatic injury18,19 and is required for normal development of the Drosophila rectum20,21. These findings suggest that aneuploidy may confer a similar selective advantage to cancer cells. Moreover, the observation that certain aneuploidies can be either recurrent in cancers of different origin or specifically recurring in cancers from individual anatomical sites22 suggests that, as observed in fungi12,13,17 or in mouse hepatocytes18, specific aneuploidies may confer selective advantage in a given environment, but not in others.
Addressing the question of whether aneuploidy may confer a selective advantage to cancer cells can be very challenging, given that cancer cell karyotypes are very complex2,22,23 and typically characterized by high degrees of aneuploidy, as well as numerous chromosome rearrangements. Moreover, many cancer cells also display chromosome numerical instability (CIN), which generates chromosome numerical heterogeneity within cancer cell populations3,24,25. To avoid such complexity, we chose to address the effect of aneuploidy on cancer cells in a simplified experimental system. Specifically, we performed a series of assays in the diploid, chromosomally stable (non-CIN), colorectal cancer cell (CRC) line DLD124 and two DLD1-derived cell lines that were previously generated via microcell-mediated chromosome transfer26 and carry an extra copy of either chromosome 7 (DLD1 + 7) or chromosome 13 (DLD1 + 13). Finally, we extended our investigation to primary human cells by performing cell proliferation experiments in diploid amniocytes (AF) and amniocytes with trisomy 13 (AF + 13). The trisomic cell lines used here (DLD1 + 7, DLD1 + 13, and AF + 13) were recently shown to display higher rates of whole-chromosome mis-segregation and to rapidly accumulate chromosome number heterogeneity compared to their diploid counterparts27.
Results
To explore whether aneuploidy confers a selective advantage to cancer cells, we made use of two trisomic cell lines derived from the diploid (2N = 46), chromosomally stable CRC cell line DLD124. The DLD1-derived trisomic cell lines used in this study carried an extra copy of either chromosome 7 (DLD1 + 7) or chromosome 13 (DLD1 + 13)26,27. This experimental set-up is advantageous for several reasons. First, it allows investigation of the specific effects of individual aneuploidies on the properties of cells that already display a transformed phenotype, thus providing insight on how aneuploidy may contribute to tumorigenesis. Second, it includes trisomies that may play different roles in cancer. Indeed, gain of chromosome 7 and chromosome 13 are both frequently found in colorectal cancer28,29. However, whereas gain of chromosome 7 is also seen in many other types of cancer22, gain of chromosome 13 appears to be specific to colorectal cancer22,28. We assessed a number of properties, including proliferative capacity, apoptosis evasion, anchorage-independent growth, and invasiveness, which are known to correlate with tumorigenic capacity1,30. Such properties were assessed under both standard culture conditions and under a number of culture conditions that are representative of possible conditions occurring in the tumor microenvironment31 concomitantly with the aneuploidies under study. We reasoned that this strategy would allow us to identify a potential selective advantage conferred by the extra chromosome. The selective conditions included nutrient starvation (serum-free culture medium), exposure to 10 μM of the chemotherapeutic drug 5-fluorouracil (5-FU), or hypoxia. The performance of the trisomic cells under such selective conditions was compared to that of the diploid parental CRC cell line.
Aneuploidy suppresses cancer cell proliferation under standard culture conditions, but can confer a proliferative advantage under selective conditions
We first examined the proliferative capacity of the three CRC cell lines grown under various conditions by determining growth curves over a period of 6 days (Fig. 1). We found that under standard conditions, the DLD1 cells displayed faster growth rates compared to each aneuploid variant (Fig. 1A). Under both serum-free and 5-FU conditions (Fig. 1B,C), the proliferation rates were lower than those observed under standard culture conditions for all cell lines. However, the trisomic cell lines displayed higher proliferation rates than the parental diploid cell line, with the DLD1 + 13 cells proliferating better than both DLD1 + 7 and DLD1 cells (Fig. 1B,C). Moreover, the DLD1 + 13 cells displayed faster proliferation rates than either of the other two CRC cell lines, whereas the DLD1 + 7 cells did not display a proliferation advantage over the diploid cells under hypoxic conditions (Fig. 1D). However, it is important to note that the hypoxic conditions did not induce the overall reduction in proliferation rates observed under the other two selective conditions (compare Figs 1A–D). This may be explained by an adaptation of colorectal cells to survive in a hypoxic environment, such us the gut lumen32.
Figure 1. Aneuploidy suppresses cell proliferation under standard culture conditions, but favors proliferation under selective conditions in CRC cells.
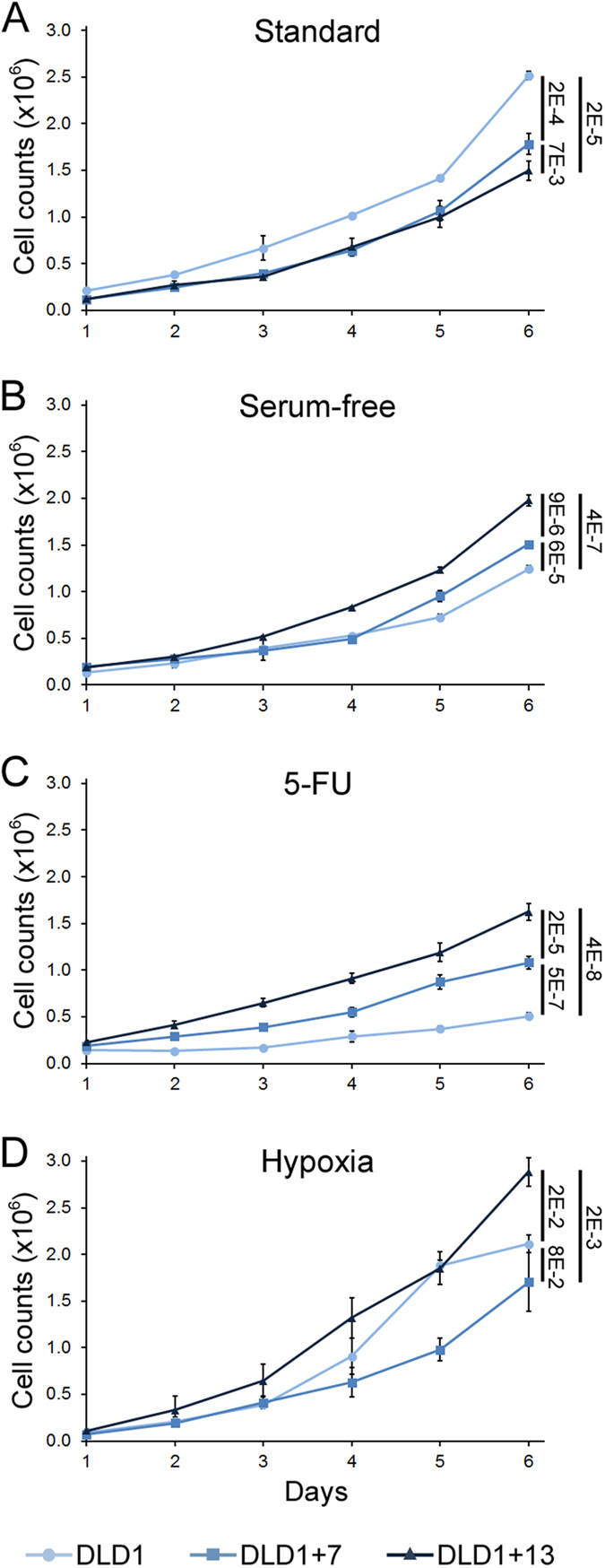
The graphs show growth curves for DLD1, DLD1 + 7, and DLD1 + 13 cells cultured for six days under different conditions. Data for cells cultured in standard media (A), serum-free media (B), media containing 10 μM 5-FU (C), or incubated under hypoxic conditions (D) are reported as mean and standard deviations from three biological replicates. Similar results were found by automated cell counting (data not shown). For statistical analysis, the growth curves in logarithmic scale were fit to linear functions and analyzed using R software package. The numbers to the right of the graphs indicate the p values for comparison of end-point data. Statistical analysis was also performed on the trends in cell growth using the geepack R package for longitudinal data analysis60 and showed statistically significant difference for all pairwise comparisons (p < 4.5E-7), except for DLD1 + 7 vs. DLD1 + 13 under standard culture conditions.
Cell division and suppression of apoptosis in CRC cells cultured under standard and non-standard conditions
To assess whether the higher proliferation rates were linked to increased cell division rates, we quantified the mitotic index for the three CRC cell lines under all culture conditions (Fig. 2A–D). We found that the diploid cells displayed a higher mitotic index compared to the aneuploid CRC cells under standard culture conditions (Fig. 2B), whereas both trisomic CRC cell lines displayed slightly higher (but not statistically significant) mitotic indices compared to DLD1 cells grown under serum-free or 5-FU conditions (Fig. 2B). Under hypoxic conditions, the DLD1 + 7 cells displayed the lowest mitotic index (although the difference was not statistically significant), whereas DLD1 and DLD1 + 13 cells displayed similar mitotic indices (Fig. 2B). However, it is worth noting that the mitotic indices displayed by all cell lines under hypoxic conditions were higher (statistically significant for both DLD1 and DLD1 + 13) than those observed under either of the other two selective conditions and similar to those observed under standard culture conditions (Fig. 2C,D). More strikingly, the mitotic index for DLD1 + 13 cells subjected to hypoxia was higher than the mitotic index measured for DLD1 + 13 cells under any other culture condition (Fig. 2C,D), consistent with the high proliferation rate of these cells in hypoxia (Fig. 1D) and suggesting a karyotype-specific effect of trisomy 13 in adaptation to hypoxia in colorectal cancer.
Figure 2. Mitotic index and apoptosis in diploid vs. aneuploid CRC cells.

(A) Representative images of DLD1, DLD1 + 7, and DLD1 + 13 cells grown under standard culture conditions and stained with DAPI. Arrows point to mitotic cells. Images are maximum intensity projections of Z-stacks acquired at 0.6 μm steps. Scale bar, 10 μm. (B,C) Mitotic indices for the three cell lines cultured under different conditions and reported with data grouped either by culture conditions (B) or by cell line (C). The data are reported as mean and standard errors from three biological replicates. Statistical analysis was performed using the χ2 test and only p values that were significant or close to significance are reported in the figure. (D) The same data reported in (C) are shown here as normalized value over the mitotic index of the given cell line cultured under standard conditions. (E) Representative TUNEL assay image of DLD1 + 13 cells cultured under standard conditions. Arrow points to TUNEL-positive cell. Images are maximum intensity projections of Z-stacks acquired at 0.6 μm steps. Scale bar, 10 μm. (F,G) Quantification of TUNEL-positive cells in the three cell lines cultured under different conditions and reported with data grouped either by culture conditions (F) or by cell line (G). The data are reported as mean and standard errors from three biological replicates. Statistical analysis was performed using the χ2 test and only p values that were significant or close to significance are reported in the figure. (H) The same data reported in (G) are shown here as normalized value over the TUNEL-positive values of the given cell line cultured under standard conditions.
Because overall proliferation rates are determined by the combined rates of cell division and cell death, and because cancer cells are known to evade apoptosis1, we also quantified the rates of apoptosis by TUNEL assay in the three CRC cell lines under all culture conditions (Fig. 2E–H). Under standard culture conditions, DLD1 cells showed the lowest incidence of apoptosis, whereas their rate of apoptosis was the highest among the three cell lines both under serum-free and 5-FU conditions (Fig. 2F). Under hypoxic conditions, the DLD1 + 7 cells displayed the highest rate of apoptosis among the three CRC cell lines (Fig. 2F), which is consistent with the observation that DLD1 + 7 cells displayed the lowest proliferation rates among the three cell lines under hypoxic conditions (Fig. 1D). The TUNEL assay data also showed that the rates of apoptosis did not vary significantly for DLD1 + 13 cells grown under different conditions (Fig. 2G,H), whereas the DLD1 cells showed higher rates of apoptosis under all selective conditions compared to standard culture conditions (Fig. 2G,H), and DLD1 + 7 cells displayed lower rates of apoptosis under standard and serum-free conditions compared to their rates of apoptosis in 5-FU and hypoxia (Fig. 2G,H).
To determine whether the observed mitotic indices and rates of apoptosis could explain the growth trends of the three CRC cell lines cultured under different conditions, we used a deterministic mathematical model (supplementary information) that took into account the experimentally observed mitotic indices and rates of apoptosis (Fig. 2) as well as mitotic timing (Table S1) for the three CRC cell lines. Simulation data obtained using this model showed that the experimentally observed growth curves indicated slower proliferation than what could be accounted for based on mitotic indices and rates of apoptotic death, suggesting that cell death may occur in our CRC cell lines at rates higher than those detected by TUNEL assay (see supplementary information and data therein for further information and discussion).
Aneuploidy can increase anchorage-independent growth of CRC cells
We next performed a soft agar assay to examine the ability of the three CRC cell lines to grow independent of anchorage in vitro, a phenotype that correlates with tumorigenicity30. As a negative control we used the non-transformed, immortalized, hTERT-RPE1 cell line. As expected, these cells were unable to form colonies in soft agar (Fig. 3A). On the other hand, all three DLD1 cell lines formed colonies under all culture conditions (Fig. 3B–H), consistent with anchorage-independent growth being the phenotype most consistently associated with tumorigenicity30. The three CRC cell lines formed similar numbers of colonies under most culture conditions (Fig. 3E–H), with the exception of hypoxia, in which DLD1 + 7 CRC cells formed more colonies compared to the diploid DLD1 cells (Fig. 3H). Moreover, in most cases the colonies formed by the aneuploid CRC cells were significantly larger than those formed by their diploid counterparts (Fig. 3I–L). Notably, the aneuploid CRC cell lines also formed larger colonies compared to diploid cells when cultured under standard conditions (Fig. 3I).
Figure 3. Aneuploidy can increase anchorage-independent growth in CRC cells.

Anchorage-independent growth was assessed by testing the ability of DLD1, DLD1 + 7, and DLD1 + 13 cells to form colonies on soft agar when cultured under different conditions. (A) Representative image of hTERT-RPE1 cells on soft agar. These non-transformed cells were used as a negative control and as a baseline for colony size measurements. Individual dot-like structures (yellow arrow), corresponding to individual non-proliferating cells, were observed at low density. The average size of these structures was 15 μm, and this was considered as the lower size limit for colony size measurements in DLD1 cell lines, although most of the cancer cell line colonies were well above 15 μm in size. (B–D) Representative colonies formed by DLD1, DLD1 + 7, and DLD1 + 13 on soft agar under standard culture conditions. Scale bar, 50 μm. (E–H) Total number of colonies from ten randomly selected fields of view of soft agar plates with the various cell lines under different culture conditions. (I–L) Average size of colonies formed by the various cell lines under different culture conditions. For (E–L), the data are reported as mean and standard errors from three biological replicates. Statistical analysis was performed using the t-test and only p values that were significant or close to significance are reported in the figure.
Aneuploidy increases the invasive capacity of CRC cells
To assess the invasive ability of aneuploid CRC cells compared to the diploid parental cell line, we performed a matrigel invasion assay (Fig. 4). We found that under all culture conditions, the number of cells migrating through the matrigel was higher for the aneuploid CRC cell lines compared to the diploid parental cell line (t-test, p < 10−4 for each aneuploid cell line compared to diploid cells under all culture conditions; Fig. 4E–H).
Figure 4. Aneuploidy increases invasiveness of CRC cells.

The invasive capacity of the three different cell lines was assessed using a matrigel invasion assay. (A–D) Examples of Giemsa-stained invasive DLD1, DLD1 + 7, and DLD1 + 13 cells cultured under different conditions. Scale bar, 100 μm. (E–H) Quantification of invasive DLD1, DLD1 + 7, and DLD1 + 13 cells cultured under different conditions. The data are reported as mean and s.e.m. from three biological replicates. Statistical analysis showed that significantly larger numbers of aneuploid compared to diploid cells migrated through the matrigel layer (t-test, p < 10−4 for each aneuploid CRC cell line compared to diploid cells under all culture conditions).
Trisomy 13 confers a proliferative advantage to primary human cells cultured under selective conditions
We next assessed the ability of aneuploidy to confer a selective advantage to non-transformed human cells by using diploid amniocytes (AF) and amniocytes with trisomy 13 (AF + 13). The proliferative capacity of these cells was assessed under standard culture conditions, in serum-free media, or in the presence of 5 μM 5-FU. We found that under standard culture conditions the diploid cells proliferated slightly better than the AF + 13 cells (Fig. 5A). However, we found that under both selective conditions, proliferation of the diploid cells was completely abrogated, whereas the aneuploid amniocytes were still capable of proliferating (Fig. 5B,C). Although the rates of proliferation of AF + 13 cells was dramatically reduced under selective conditions compared to their proliferation under standard culture conditions, the rates of proliferation of AF + 13 cells were significantly higher than their diploid counterparts cultured under the same selective conditions (Fig. 5B,C).
Figure 5. Aneuploidy favors proliferation of non-transformed amniotic fibroblasts grown under non-standard conditions.
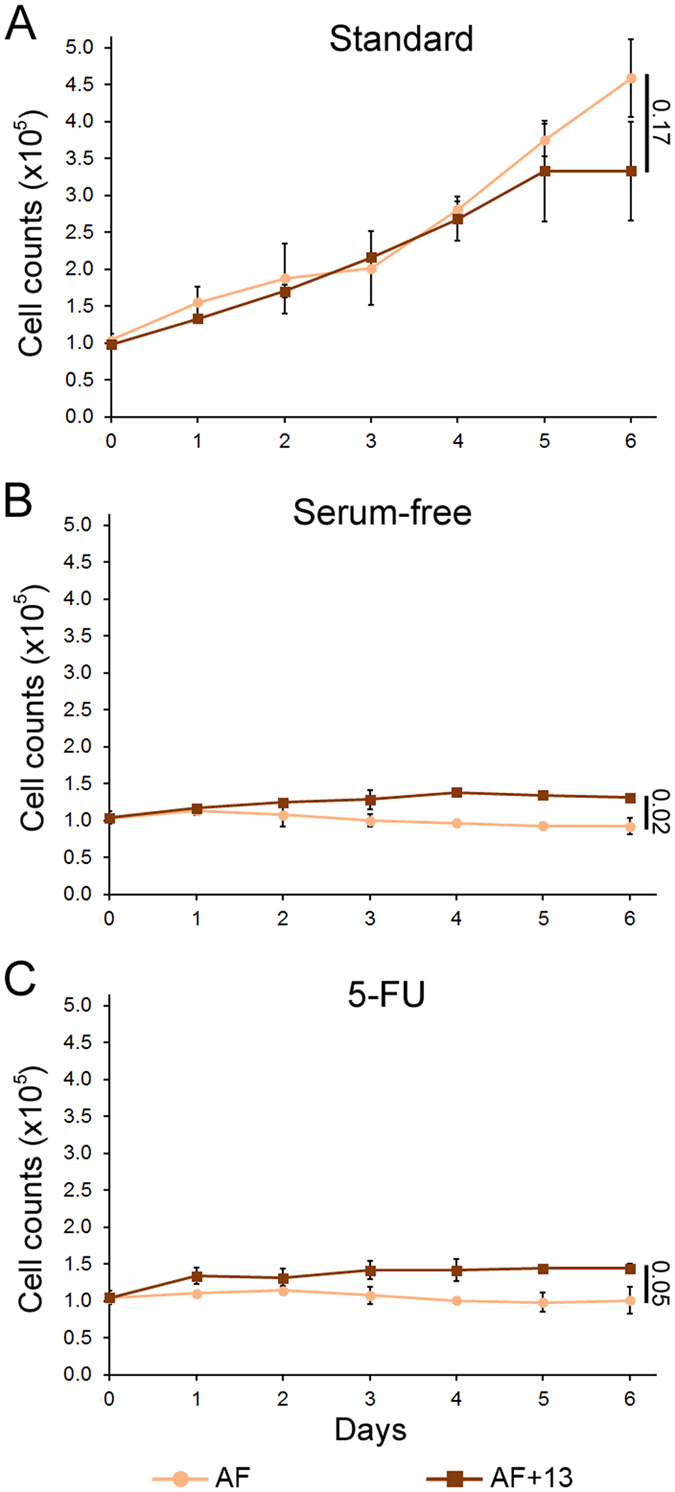
The graphs show growth curves for AF and AF + 13 cells cultured for six days under different conditions (treatment starting at day 0). Data for cells cultured in standard media (A), serum-free media (B), or media containing 5 μM 5-FU (C) are reported as mean and standard deviations from 2–3 biological replicates. For statistical analysis, the growth curves in logarithmic scale were fit to linear functions and analyzed using R software package. The numbers to the right of the graphs indicate the p values for comparison of end-point data. Statistical analysis was also performed on the trends in cell growth using the geepack R package for longitudinal data analysis60 and showed statistically significant difference for all pairwise comparisons (Standard, p = 0.02; Serum-free and 5-FU, p < 1E-6).
Discussion
Taken together, our data show that aneuploidy can confer a selective advantage to human cells under conditions of environmental stress, although in some of our assays and under certain culture conditions, there was no difference in behavior between diploid and aneuploid CRC cells. For both the CRC cells and the amniocytes, the diploid cells proliferated better under standard culture conditions. A similar proliferative advantage of euploid vs. aneuploid cells under standard culture conditions was previously shown in both yeast and mouse embryonic fibroblasts10,11,17,33. On the other hand, we found that the aneuploid cells displayed a proliferative advantage under selective culture conditions, indicating that aneuploidy can confer a selective advantage to cancer cells, as previously shown in yeast14,17,33. Our results are also consistent with the previous finding that trisomy 7 was associated with an acquired resistance of colon epithelial cells to growth in serum-free conditions34. However, our findings are even more striking in that they show that aneuploidy increases the tolerance of CRC cells to environmental stresses beyond that seen in cells that are already transformed and tumorigenic, and presumably already adapted to proliferate under certain environmental stresses.
Aneuploidy has been shown to cause transcriptomic changes that, for the most part, correlate with the specific aneuploid chromosome(s)11,17,26,35,36,37. However, when examining the proteomic changes linked to aneuploidy, the situation is more complex. Whereas some studies have reported proteomic changes to scale up with changes in chromosome copy number in yeast17, other studies in yeast and human cells reported that protein abundance does not vary significantly as a result of aneuploidy and/or that the proteomic changes do not necessarily correlate with the specific aneuploidy11,36,37,38. Nonetheless, a general link between aneuploidy and phenotypic traits is strongly supported by experimental data17,39,40,41. Moreover, aneuploidy-specific phenotypes, resulting from the altered levels of proteins encoded by genes on the aneuploid chromosomes, have been reported in a number of different organisms and contexts. For instance, the chromosome 5 aneuploidy emerging in Candida albicans exposed to fluconazole12,13 was shown to confer drug resistance by inducing overexpression of two genes encoding, respectively, for the drug target and for a transcriptional regulator of efflux pumps42. Similarly, the acquisition of trisomy 7 in colon epithelial cells cultured in serum-free conditions was associated with overexpression of the epidermal growth factor receptor, encoded on chromosome 739. Finally, a specific cytokinesis failure phenotype was recently shown to be associated with trisomy 13 and to be caused by overexpression of Spartin, a protein encoded by a gene (SPG20) on chromosome 1327. The selective advantage we observed in our study seems unlikely to depend on overexpression of specific genes on the aneuploid chromosomes. If that were the case, we would expect to find consistently significant differences between trisomy 7 and trisomy 13. In other words, we would expect trisomy 7 to confer selective advantage under a given culture condition and trisomy 13 to confer selective advantage under a different culture condition, which was not the case in our experiments. Therefore, although the specificity of chromosome 13 gain in colorectal cancer22 may suggest a specific advantage conferred by this chromosome to CRC cells, such advantage may be linked to conditions other than those tested in our study. Instead, for the selective conditions assessed here, we found a general selective advantage conferred by aneuploidy. This observation suggests that the advantage conferred by these two chromosomes may depend on a common phenotypic effect observed for both trisomies. Such an effect may be the increased rate of chromosome mis-segregation recently reported for DLD1 + 7, DLD1 + 13, and AF + 13 cells compared to diploid DLD1 and AF cells27. In DLD1 + 7, DLD1 + 13, and AF + 13 cells, high rates of chromosome mis-segregation are associated with high rates of karyotypic heterogeneity, which become apparent in the population even when cells are examined at relatively low passages27; this is consistent with findings from other studies showing a link between aneuploidy and chromosome instability (CIN) in both yeast and human cells39,41,43,44,45,46. Cell populations with high rates of karyotypic heterogeneity are expected to display high degrees of phenotypic heterogeneity33,47 and this karyotypic/phenotypic heterogeneity will allow aneuploid cells to be more “adaptable”33,47. Indeed, high karyotypic/phenotypic heterogeneity increases the chance that some cells within the population may possess the phenotype required for increased fitness in a certain environment.
Recent studies have shown that despite a general correlation between genes on the aneuploid chromosome and gene expression/proteomic profiles10,17,35,36,48,49,50, upregulation of transcripts and/or proteins involved in stress response can occur independent of the specific aneuploidy36,49,50,51. This observation suggests that the upregulation of stress response pathways may make aneuploid cells “primed” for a response to environmental changes, thus contributing (alone or in concert with the karyotypic/phenotypic heterogeneity arising in aneuploid cell populations) to confer a selective advantage to aneuploid cells.
Our soft agar and matrigel invasion assays provided interesting observations in that aneuploid CRC cells were found to grow larger colonies and to be more invasive than diploid CRC cells not only under selective conditions, but also under standard culture conditions. One could envision anchorage-independent growth or migration through an extra-cellular matrix as challenges of their own. Therefore, the observation that aneuploid cells are more anchorage-independent and migrate better through matrigel regardless of the culture conditions, reinforces the idea that aneuploidy increases the fitness of cells, even those, such as cancer cells, that are already characterized by increased proliferative capacity and aggressive tumorigenic phenotypes.
In several of our assays, DLD1 + 13 cells performed better than DLD1 + 7 cells, despite both displaying an advantage over DLD1 cells. DLD1 + 13 cells were recently shown to fail cytokinesis and acquire tetraploid/near-tetraploid karyotypes at significantly higher rates compared to the other two CRC cell lines27. Interestingly, tetraploidy was recently shown to increase the tolerance for mitotic errors and CIN52,53. Thus, the better fitness of DLD1 + 13 over DLD1 + 7 may be explained by the ability of tetraploidy to buffer the overall effects of aneuploidy and confer tolerance to phenomena/events other than mitotic errors and CIN.
Given the advantage provided to cancer cells by aneuploidy, it is surprising that non-CIN CRC cells (such as DLD1) exist. However, this may be due to the fact that a combination of the high rates of mutation displayed by non-CIN CRC cells54 and the increase in CIN triggered by aneuploidy may not be tolerated and may cause cell death rather than a proliferative advantage. This is consistent with the previously suggested tumor-suppressing effect of very high rates of CIN55.
In conclusion, our work shows that addition of one extra chromosome is enough to confer a selective advantage to both primary and cancer cells, allowing aneuploid cells to perform better than diploid cells under a number of environmental challenges. These findings are consistent with the widespread aneuploidy observed in solid tumors4, with the association between aneuploidy/CIN and poor patient prognosis56,57, and with the link between CIN and drug resistance58,59.
Materials and Methods
CRC and hTERT-RPE1 cell lines: general information, culture conditions, and treatments
The DLD1 cell line was obtained from American Type Culture Collection (ATCC, VA, USA); the DLD1 + 7 and DLD1 + 13 cell lines were generated previously by microcell-mediated chromosome transfer26, and DLD1 + 13 cells were sub-cloned as previously described27. All the DLD1 cell lines were maintained in RPMI 1640 medium (ATCC, VA, USA) supplemented with 10% FBS (Gibco, NY, USA) and antibiotic/antimycotic mixture (Gibco, NY, USA). The hTERT-RPE1 cells (ATCC, VA, USA) were maintained in DMEM/F12 medium (Gibco, NY, USA) supplemented with 10% FBS (Gibco, NY, USA) and antibiotic/antimycotic mixture (Gibco, NY, USA). All cells were kept in a humidified incubator at 37 °C with 5% CO2.
For exposure to selective conditions, culture medium was prepared as follows: standard culture medium without FBS (Serum-free); standard culture medium with 10 μM 5-fluorouracil (5-FU) (Sigma-Aldrich, MO, USA); standard culture medium pre-conditioned in hypoxia chamber for 48 hrs (Hypoxia). For all experiments, except those in hypoxic conditions, cells were incubated with the appropriate culture media in a humidified incubator at 37 °C with 5% CO2 for the duration of the experiment. For experiments in hypoxic conditions, cells were maintained inside a modular incubator chamber (Hillups-Rothenberg, CA, USA) in which humidity was maintained by addition of a 60 mm Petri dish containing 5 ml of water. Prior to each use, the chamber was flushed with 5% CO2, 1% O2, and N2 before being sealed and stored at 37 °C in an incubator. Cells were removed from the chamber for not more than one hour to conduct experiments, after which time the chamber was again flushed with 5% CO2, 1% O2, and N2 before being sealed and stored at 37 °C. Hypoxic conditions were confirmed using a Cyto-ID Hypoxia Detection Kit (Enzo Life Sciences, NY, USA) and fluorescence microscopy detection according to the manufacturer’s instructions.
Amniotic fibroblasts: general information, culture conditions, and treatments
Passage 1–3 fibroblast cultures were established from surplus amniocentesis samples used in pre-natal diagnosis. Two cases of constitutional trisomy 13 (gestational age 16 and 22 weeks) and three diploid controls (gestational age 13, 15, and 16 weeks) were used in our study. FISH analysis of interphase nuclei stained with a chromosome 13 specific probe showed that the trisomic cell populations used in this study were highly homogenous (>95%) for the trisomic karyotype. The study acknowledged the ethics guidelines under Portugal national rules and according to the principles of the Declaration of Helsinki, and was approved by the Ethics Committee of Hospital de S. João-Porto (dispatch 14, Nov 2012). Informed consent forms with detailed information were provided to all patients. The study did not imply collection of extra material from the healthy female donors (only surplus cells/tissues were used); the study did not bring any direct benefits to the volunteers; there were no risks or costs for the volunteers; there was no access to patient clinical data (samples were obtained in anonymous form from the Hospital Genetics Department); participation was volunteer and could be interrupted at any time; there are no ethical impacts predicted; there will be no commercial interests. Amniotic fibroblasts were maintained in EMEM (Lonza, Switzerland) supplemented with 15% FBS, 2.5 mM glutamine, and 1x antibiotic-antimycotic solution (all from Gibco, NY, USA). All cells were kept in a humidified incubator at 37 °C with 5% CO2. For exposure to selective conditions, culture medium was prepared as follows: standard culture medium without FBS (serum-free); standard culture medium with 5 μM 5-fluorouracil (5-FU) (Sigma-Aldrich, MO, USA). A lower dose of 5-FU was used in AF compared to DLD1 cells due to the AF’s higher sensitivity to the drug.
Growth curves
To determine the proliferative capacity of CRC cells over time, we plated each DLD1 cell line in a 6-well plate (6 wells total) at a density of 1.5 × 105 cells per well. Following 24 hrs of growth in standard media, the cells were washed twice in PBS and re-incubated under selective culture conditions. For the 4-6-day samples, the media was replenished half-way through the experiment. At 24-hour intervals for six days, the media from one well of each DLD1 cell line was removed, the cells were washed twice with PBS and trypsinized. Cells were then vigorously re-suspended in media to a final total volume of 3 ml. From this, a 100 μl sample was thoroughly pipetted and mixed with an equal amount of 0.4% trypan blue solution (Gibco, NY, USA), and subjected to cell counting. Cell counts were performed by hemocytometer (Bright-Line, PA, USA) and averaged from four samples.
To determine the proliferative capacity of amniotic fibroblasts over time, we were unable to use the same method used in CRC cells due to technical difficulties. Instead, we used a different method, as described below. We plated each trisomy 13 and diploid control in 6-well plates (7 wells total) at a density of 0.8–1 × 105 cells per well. Following 24 hrs of growth in standard media, the cells were rinsed in PBS and re-incubated under selective culture conditions. One well was immediately fixed with cold methanol and stained with DAPI for nuclei quantification (day 0). The other six wells were fixed in methanol and DAPI-stained at 24-hour intervals over a six-day period (days 1–6). To quantify the number of cells, images of stained nuclei in a total of 100 neighboring fields (covering a 1 cm2 area) at two different locations in each well were acquired with a 10× objective on a Leica DMI 6000B motorized inverted epi-fluorescence microscope. The results from the two different stitched fields were averaged representing the number of nuclei per cm2. This number was then extrapolated to the total area of the well (9.6 cm2). The growth curves represent the mean ± S.D. values calculated from independent experiments using different trisomic and diploid samples.
Mitotic index analysis
1.5 × 105 CRC cells suspended in standard media were plated on 22 × 22 mm acid-washed sterile glass coverslips inside 35 mm Petri dishes. After 24 hours, cells were incubated under the appropriate culture conditions for additional 24 hrs. Cells were then washed twice with PBS, fixed with freshly prepared 4% paraformaldehyde (Fischer Scientific, NJ, USA), washed twice more with PBS, and DAPI stained. For each sample, the number of mitotic cells was counted over a total of at least 1,000 cells.
TUNEL assay
0.5 × 105 cells suspended in standard media were plated on 22 × 22 mm acid-washed sterile glass coverslips inside 35 mm Petri dishes. After 24 hours, cells were incubated under the appropriate culture conditions for additional 24 hrs. Cells were then washed twice with PBS, fixed with freshly prepared 4% paraformaldehyde (Fischer Scientific, NJ, USA), washed twice more with PBS + 0.01% sodium citrate, and finally stained using an in situ cell death detection kit (Roche, Basel, Switzerland). Cells were then washed three times with PBS before DAPI staining. For each sample, the number of TUNEL positive cells was recorded over a total of at least 1,000 cells. Positive and negative controls were used according to the manufacturer’s guidelines.
Fluorescence microscopy
For both mitotic index and TUNEL assay quantification, samples were viewed on a Nikon Eclipse TE2000-U inverted microscope (Nikon Instruments Inc., NY, USA) equipped with a swept field confocal system (Prairie Technologies, WI, USA), a 100×/1.4 NA Plan-Apochromatic objective, and an automated ProScan stage (Prior Scientific, Cambridge, UK). The confocal head was accessorized with a multiband pass filter set for illumination at 405, 488, 561, and 640 nm and illumination was obtained through an Agilent MLC400 monolithic laser combiner (Agilent Technologies, CA, USA) controlled by a four channel acousto-optic tunable filter. Digital images were acquired with a HQ2 CCD camera (Photometrics, AZ, USA). Stacks of images were acquired through the Z axis at 0.6 μm steps. Exposure time, Z-axis position, laser line power, and confocal system were all controlled by NIS Elements AR software (Nikon Instruments Inc., NY, USA) on a PC computer (Dell, TX, USA).
Soft agar assay
1.5 ml of a 1:1 mixture of 2% agar (Fischer Scientific, NJ, USA) and 2× DMEM (Gibco, NY, USA) media (prepared according to the specific selective condition for each sample) was poured evenly into a 35 mm Petri dish. After cooling, 1.5 ml of a 1:1 mixture of 1.4% agar (Fischer Scientific, NJ, USA) and 2× DMEM (prepared according to the specific culture condition for each sample) media was used to re-suspend 1 × 105 cells and then poured on the bottom solidified agar layer. After solidification of the top agar layer, 2 ml of RPMI 1640 media (prepared according to the specific selective condition for each sample) were added and the samples incubated in a humidified incubator at 37 °C with 5% CO2, or in the hypoxia chamber. The colonies were grown for 22 days and media changed every 4–5 days. Ten randomly selected fields of view were imaged on a Nikon Eclipse Ti inverted microscope (Nikon Instruments Inc., NY, USA) equipped with a 20×/0.4 NA ADL phase contrast objective, phase-contrast transillumination, transmitted light shutter, ProScan automated stage (Prior Scientific, MA, USA), and a HQ2 CCD camera (Photometrics, AZ, USA). For each field of view, Z-stacks were acquired by imaging 10 focal planes at 100 μm steps and colony number and size were measured for colonies present on different focal planes. The total number of colonies was quantified for all ten fields of view and the size of each colony was quantified by measuring the length of the longest axis. hTERT-RPE1 cells were used as a negative control and as a baseline for colony size measurements. Individual dot-like structures, corresponding to 1-few non-proliferating cells, were observed at low density in hTERT-RPE1 samples. The average size of these structures was 15 μm, and this was considered as the lower size limit for colony size measurements in DLD1 cell lines.
Matrigel invasion assay
The matrigel invasion assay was performed using an 8 μm pore transwell PET membrane (BD Biosciences Inc., MA, USA). The matrigel mixture (Corning Inc., MA, USA) was reconstituted with the appropriate culture media to a concentration of 200 μg/ml and poured evenly over the transwell PET membrane; 105 cells were re-suspended in RPMI 1640 media and plated onto the matrigel; 500 μl of media were also added to the bottom well and the chambers were incubated. After 24 hrs, both the media in the bottom well and that in the top chamber were replaced with media prepared according to the specific culture condition for each sample. After incubating for additional 24 hrs, non-invasive cells were scraped off the chamber side of the transwell membrane and invasive cells were fixed with 100% methanol, washed twice with PBS, and stained with a 1% Giemsa solution. Quantification was performed by light microscopy on a Nikon Eclipse TS100, using a 20×/0.4 NA ADL phase contrast objective and counting the total number of cells from 6 random fields of view.
Additional Information
How to cite this article: Rutledge, S. D. et al. Selective advantage of trisomic human cells cultured in non-standard conditions. Sci. Rep. 6, 22828; doi: 10.1038/srep22828 (2016).
Supplementary Material
Acknowledgments
We would like to acknowledge Thomas Ried (National Cancer Institute, NIH) for providing the aneuploid CRC cell lines. We also thank Silke Hauf and Rich Walker for providing important feedback throughout the execution of this project. Finally, we thank all the members of the Hauf and Cimini labs for helpful comments and discussions. SDR was partly supported through Graduate Teaching Assistantships provided by the Dept. of Biological Sciences (Virginia Tech). Work in the Cimini lab partly funded by NSF grants MCB-0842551 and MCB-1517506 and HFSP grant RGY0069/2010. We further acknowledge the Virginia Tech University Libraries for covering the publication fees through the Open Access Subvention Fund. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Author Contributions Conceived and designed experiments: S.D.R., T.A.D. and D.C. Performed experiments: S.D.R., T.A.D., J.M.N., C.L.K., D.W., M.B.-V., S.D.K. and E.L. Analyzed and interpreted data: S.D.R., T.A.D., J.M.N., M.V.-C., C.L.K., M.B.-V., E.L. and D.C. Contributed reagents/materials/analysis tools: M.V.-C., D.W., S.D.K., E.L. and D.C. Wrote the paper: S.D.R., T.A.D., E.L. and D.C. All authors approved the final manuscript.
References
- Hanahan D. & Weinberg R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674, 10.1016/j.cell.2011.02.013 (2011). [DOI] [PubMed] [Google Scholar]
- Cimini D. Merotelic kinetochore orientation, aneuploidy, and cancer. Biochim Biophys Acta 1786, 32–40, 10.1016/j.bbcan.2008.05.003 (2008). [DOI] [PubMed] [Google Scholar]
- Nicholson J. M. & Cimini D. How mitotic errors contribute to karyotypic diversity in cancer. Adv Cancer Res 112, 43–75, 10.1016/B978-0-12-387688-1.00003-X (2011). [DOI] [PubMed] [Google Scholar]
- Mitelman F., Johansson B. & Mertens F. Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer http://cgap.nci.nih.gov/Chromosomes/Mitelman (2014).
- Boveri T. Zur Frage der Entstehung maligner Tumoren. (Gustav Fischer Verlag, 1914). [Google Scholar]
- Boveri T. Concerning the origin of malignant tumours by Theodor Boveri. Translated and annotated by Henry Harris. J Cell Sci 121, Supplement 1 1–84 (2008). [DOI] [PubMed] [Google Scholar]
- Torres E. M., Williams B. R. & Amon A. Aneuploidy: cells losing their balance. Genetics 179, 737–746, 10.1534/genetics.108.090878 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassold T. & Hunt P. To err (meiotically) is human: the genesis of human aneuploidy. Nat Rev Genet 2, 280–291, 10.1038/35066065 (2001). [DOI] [PubMed] [Google Scholar]
- Biesecker L. G. & Spinner N. B. A genomic view of mosaicism and human disease. Nat Rev Genet 14, 307–320, 10.1038/nrg3424 (2013). [DOI] [PubMed] [Google Scholar]
- Williams B. R. et al. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science 322, 703–709, 10.1126/science.1160058 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres E. M. et al. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 317, 916–924, 10.1126/science.1142210 (2007). [DOI] [PubMed] [Google Scholar]
- Selmecki A., Forche A. & Berman J. Aneuploidy and isochromosome formation in drug-resistant Candida albicans. Science 313, 367–370, 10.1126/science.1128242 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmecki A. M., Dulmage K., Cowen L. E., Anderson J. B. & Berman J. Acquisition of aneuploidy provides increased fitness during the evolution of antifungal drug resistance. PLoS genetics 5, e1000705, 10.1371/journal.pgen.1000705 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rancati G. et al. Aneuploidy underlies rapid adaptive evolution of yeast cells deprived of a conserved cytokinesis motor. Cell 135, 879–893, 10.1016/j.cell.2008.09.039 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millet C., Ausiannikava D., Le Bihan T., Granneman S. & Makovets S. Cell populations can use aneuploidy to survive telomerase insufficiency. Nat Commun 6, 8664, 10.1038/ncomms9664 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaya A. et al. Adaptive aneuploidy protects against thiol peroxidase deficiency by increasing respiration via key mitochondrial proteins. Proc Natl Acad Sci USA 112, 10685–10690, 10.1073/pnas.1505315112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavelka N. et al. Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature 468, 321–325, 10.1038/nature09529 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan A. W. et al. Aneuploidy as a mechanism for stress-induced liver adaptation. The Journal of clinical investigation 122, 3307–3315, 10.1172/JCI64026 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan A. W. et al. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature 467, 707–710, 10.1038/nature09414 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox D. T., Gall J. G. & Spradling A. C. Error-prone polyploid mitosis during normal Drosophila development. Genes Dev 24, 2294–2302, 10.1101/gad.1952710 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenfelder K. P. et al. Indispensable pre-mitotic endocycles promote aneuploidy in the Drosophila rectum. Development 141, 3551–3560, 10.1242/dev.109850 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson J. M. & Cimini D. Cancer karyotypes: survival of the fittest. Frontiers in oncology 3, 148, 10.3389/fonc.2013.00148 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver B. A. & Cleveland D. W. Does aneuploidy cause cancer ? Curr Opin Cell Biol 18, 658–667, 10.1016/j.ceb.2006.10.002 (2006). [DOI] [PubMed] [Google Scholar]
- Lengauer C., Kinzler K. W. & Vogelstein B. Genetic instability in colorectal cancers. Nature 386, 623–627, 10.1038/386623a0 (1997). [DOI] [PubMed] [Google Scholar]
- Thompson S. L. & Compton D. A. Examining the link between chromosomal instability and aneuploidy in human cells. J Cell Biol 180, 665–672, 10.1083/jcb.200712029 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upender M. B. et al. Chromosome transfer induced aneuploidy results in complex dysregulation of the cellular transcriptome in immortalized and cancer cells. Cancer Res 64, 6941–6949, 10.1158/0008-5472.CAN-04-0474 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson J. M. et al. Chromosome mis-segregation and cytokinesis failure in trisomic human cells. eLife 4, 10.7554/eLife.05068 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ried T. et al. The consequences of chromosomal aneuploidy on the transcriptome of cancer cells. Biochim Biophys Acta 1819, 784–793, 10.1016/j.bbagrm.2012.02.020 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardi G. et al. Karyotypic characterization of colorectal adenocarcinomas. Genes Chromosomes Cancer 12, 97–109 (1995). [DOI] [PubMed] [Google Scholar]
- Shin S. I., Freedman V. H., Risser R. & Pollack R. Tumorigenicity of virus-transformed cells in nude mice is correlated specifically with anchorage independent growth in vitro. Proc Natl Acad Sci USA 72, 4435–4439 (1975). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D. & Weinberg R. A. The hallmarks of cancer. Cell 100, 57–70 (2000). [DOI] [PubMed] [Google Scholar]
- Zheng L., Kelly C. J. & Colgan S. P. Physiologic hypoxia and oxygen homeostasis in the healthy intestine. A Review in the Theme: Cellular Responses to Hypoxia. Am J Physiol Cell Physiol 309, C350–360, 10.1152/ajpcell.00191.2015 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G. et al. Targeting the adaptability of heterogeneous aneuploids. Cell 160, 771–784, 10.1016/j.cell.2015.01.026 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly P. et al. Characterization of aneuploid populations with trisomy 7 and 20 derived from diploid human colonic epithelial cells. Neoplasia 13, 348–357 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C. et al. Chromosome instability, chromosome transcriptome, and clonal evolution of tumor cell populations. Proc Natl Acad Sci USA 104, 8995–9000, 10.1073/pnas.0700631104 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingele S. et al. Global analysis of genome, transcriptome and proteome reveals the response to aneuploidy in human cells. Molecular systems biology 8, 608, 10.1038/msb.2012.40 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habermann J. K. et al. Stage-specific alterations of the genome, transcriptome, and proteome during colorectal carcinogenesis. Genes Chromosomes Cancer 46, 10–26, 10.1002/gcc.20382 (2007). [DOI] [PubMed] [Google Scholar]
- Gemoll T. et al. Chromosomal aneuploidy affects the global proteome equilibrium of colorectal cancer cells. Analytical cellular pathology 36, 149–161, 10.3233/ACP-140088 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheltzer J. M. et al. Aneuploidy drives genomic instability in yeast. Science 333, 1026–1030, 10.1126/science.1206412 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Z. et al. Aneuploidy underlies a multicellular phenotypic switch. Proc Natl Acad Sci USA 110, 12367–12372, 10.1073/pnas.1301047110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Pavelka N., Bradford W. D., Rancati G. & Li R. Karyotypic determinants of chromosome instability in aneuploid budding yeast. PLoS genetics 8, e1002719, 10.1371/journal.pgen.1002719 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmecki A., Gerami-Nejad M., Paulson C., Forche A. & Berman J. An isochromosome confers drug resistance in vivo by amplification of two genes, ERG11 and TAC1. Molecular microbiology 68, 624–641, 10.1111/j.1365-2958.2008.06176.x (2008). [DOI] [PubMed] [Google Scholar]
- Biron-Shental T., Liberman M., Sharvit M., Sukenik-Halevy R. & Amiel A. Amniocytes from aneuploidy embryos have enhanced random aneuploidy and signs of senescence—Can these findings be related to medical problems? Gene, 10.1016/j.gene.2015.02.075 (2015). [DOI] [PubMed] [Google Scholar]
- Duesberg P., Rausch C., Rasnick D. & Hehlmann R. Genetic instability of cancer cells is proportional to their degree of aneuploidy. Proc Natl Acad Sci USA 95, 13692–13697 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reish O. et al. Sporadic aneuploidy in PHA-stimulated lymphocytes of Turner’s syndrome patients. Chromosome Res 14, 527–534, 10.1007/s10577-006-1050-9 (2006). [DOI] [PubMed] [Google Scholar]
- Reish O., Regev M., Kanesky A., Girafi S. & Mashevich M. Sporadic aneuploidy in PHA-stimulated lymphocytes of trisomies 21, 18, and 13. Cytogenet Genome Res 133, 184–189, 10.1159/000323504 (2011). [DOI] [PubMed] [Google Scholar]
- Nicholson J. M. & Cimini D. Link between Aneuploidy and Chromosome Instability. International review of cell and molecular biology 315, 299–317, 10.1016/bs.ircmb.2014.11.002 (2015). [DOI] [PubMed] [Google Scholar]
- Ben-David U. et al. Aneuploidy induces profound changes in gene expression, proliferation and tumorigenicity of human pluripotent stem cells. Nat Commun 5, 4825, 10.1038/ncomms5825 (2014). [DOI] [PubMed] [Google Scholar]
- Dephoure N. et al. Quantitative proteomic analysis reveals posttranslational responses to aneuploidy in yeast. eLife 3, e03023, 10.7554/eLife.03023 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheltzer J. M., Torres E. M., Dunham M. J. & Amon A. Transcriptional consequences of aneuploidy. Proc Natl Acad Sci USA 109, 12644–12649, 10.1073/pnas.1209227109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dürrbaum M. et al. Unique features of the transcriptional response to model aneuploidy in human cells. BMC Genomics 15, 139, 10.1186/1471-2164-15-139 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewhurst S. M. et al. Tolerance of whole-genome doubling propagates chromosomal instability and accelerates cancer genome evolution. Cancer discovery 4, 175–185, 10.1158/2159-8290.CD-13-0285 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsova A. Y. et al. Chromosomal instability, tolerance of mitotic errors and multidrug resistance are promoted by tetraploidization in human cells. Cell Cycle 14, 2810–2820, 10.1080/15384101.2015.1068482 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengauer C., Kinzler K. W. & Vogelstein B. Genetic instabilities in human cancers. Nature 396, 643–649 (1998). [DOI] [PubMed] [Google Scholar]
- Weaver B. A. & Cleveland D. W. Aneuploidy: instigator and inhibitor of tumorigenesis. Cancer Res 67, 10103–10105, 10.1158/0008-5472.CAN-07-2266 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter S. L., Eklund A. C., Kohane I. S., Harris L. N. & Szallasi Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat Genet 38, 1043–1048, 10.1038/ng1861 (2006). [DOI] [PubMed] [Google Scholar]
- Walther A., Houlston R. & Tomlinson I. Association between chromosomal instability and prognosis in colorectal cancer: a meta-analysis. Gut 57, 941–950, 10.1136/gut.2007.135004 (2008). [DOI] [PubMed] [Google Scholar]
- Lee A. J. et al. Chromosomal instability confers intrinsic multidrug resistance. Cancer Res 71, 1858–1870, 10.1158/0008-5472.CAN-10-3604 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanton C. et al. Chromosomal instability determines taxane response. Proc Natl Acad Sci USA 106, 8671–8676, 10.1073/pnas.0811835106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hojsgaard S., Halekoh U. & J Y. The R package geepack for generalized estimating equations. J Stat Softw 15, 1–11 (2006). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.