

Abstract
Plasmodium falciparum malaria, an infectious disease caused by a parasitic protozoan, claims the lives of nearly a million children each year in Africa alone and is a top public health concern. Evidence is accumulating that resistance to artemisinin derivatives, the frontline therapy for the asexual blood stage of the infection, is developing in southeast Asia. Renewed initiatives to eliminate malaria will benefit from an expanded repertoire of antimalarials, including new drugs that kill circulating P. falciparum gametocytes, thereby preventing transmission. Our current understanding of the biology of asexual blood-stage parasites and gametocytes and the ability to culture them in vitro lends optimism that high-throughput screenings of large chemical libraries will produce a new generation of antimalarial drugs. There is also a need for new therapies to reduce the high mortality of severe malaria. An understanding of the pathophysiology of severe disease may identify rational targets for drugs that improve survival.
Introduction
Malaria is a devastating disease, killing nearly a million children each year in Africa alone. There exists a real and urgent need for new antimalarial drugs that can meet the threat of acquired resistance to artemisinin derivatives, treat severe malaria to reduce death and complications and kill gametocytes to block transmission. Malaria is caused by Apicomplexan pathogens of the genus Plasmodium, the most deadly of which, P. falciparum, predominates in Africa. Infection by this malarial parasite usually results in an uncomplicated, mild febrile disease in which intermittent episodes of fever and peaks of parasitemia are controlled by the body's immune defenses and eventually eliminated. In some cases, however, the disease becomes severe and may lead to death. Young children are particularly susceptible to severe malaria in endemic areas of Africa, where it is estimated that nearly a quarter of all childhood deaths are caused by malaria.
There is always a need for new antimalarial drugs because we never know when P. falciparum will become resistant to the drugs that are presently available. P. falciparum resistance to the synthetic antimalarial drug chloroquine was a problem for Vietnam and US soldiers in Vietnam. The spread of chloroquine resistance heralded increased mortality and spurred the rush to find a replacement for chloroquine. The development of today's greatest antimalarial drug resulted from the Vietnam War when Mao Zedong assigned many institutions and over 600 scientists to search for new antimalarial drugs. In the early 1970s, the isolation of artemisinin from the ancient Chinese remedy qinghaosu brought new and urgently needed treatments in the form of artemisinin combination therapies (ACTs). Although ACTs are now highly effective, history has taught us that we must continue the search for new drugs in the event that artemisinin resistance develops.
Severe malaria in children encompasses three overlapping syndromes: cerebral malaria, metabolic acidosis and severe anemia (Fig. 1). Today's frontline antimalarial drugs target the asexual blood stage of the infection that causes the symptoms of disease. These drugs improve host survival by reducing parasite loads, disease symptoms and the risk of progression to severe disease. Despite treatment with the most rapidly acting antimalarial drugs, artemisinin derivatives, cerebral malaria and metabolic acidosis have a mortality of 15–20%, and survivors may suffer from persistent neurological sequelae. As our understanding of the underlying causes of severe malaria grows, it may be possible to develop therapies that reverse the complications of malaria infections and lessen the risk of disability or death. Therapies of such nature would modify the physiologic state of the infected human, for example, by targeting the responses of the vascular endothelium to the adhesion of infected erythrocytes with the goal of extending survival and allowing more time for antiparasitic therapies to work.
Figure 1.
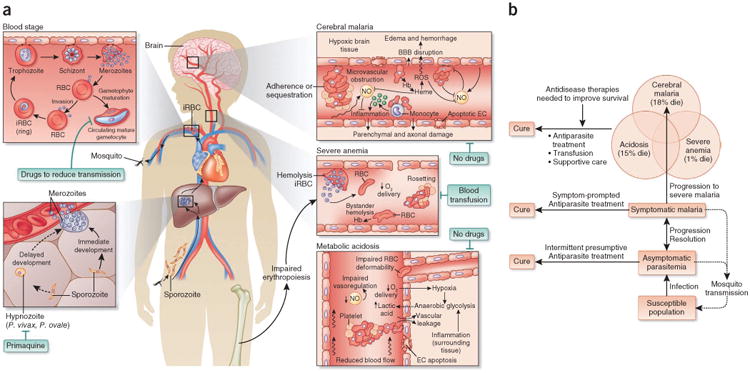
Severe malaria in children. (a) Life cycle and pathogenesis of malaria. Malaria infections begin with the injection of parasite sporozoites by infected mosquitoes during a blood meal. Sporozoites invade hepatocytes and proliferate into merozoites. One P. falciparum sporozoite develops into 40,000 merozoites per liver cell over 6 d. During P. vivax and Plasmodium ovale infection, some sporozoites also differentiate into hypnozoites that remain dormant in the liver for months to years before undergoing division and development into merozoites. Only one drug family, the 8-aminoquinolines such as primaquine, kills hypnozoites. However, the 8-aminoquinolines are toxic in glucose-6-phosphate dehydrogenase (G6PD)-deficient humans, a common deficiency in malaria-endemic regions of the world. Consequently, elimination of P. vivax and P. ovale may require new antihypnozoite drugs that can be safely administered to a population in which G6PD deficiency is prevalent. The blood stage of malaria begins when hepatic merozoites invade erythrocytes. Within 12 h of invasion, the parasite remodels the red blood cell (RBC), facilitating the growth of the parasite and transporting PfEMP1 to the erythrocyte membrane. Infected RBCs (iRBCs) bind to endothelium through PfEMP1 primarily to avoid clearance by the spleen. Sequestration of infected RBCs injures endothelial cells (ECs) and disrupts blood flow, causing tissue hypoxia and lactic acidosis. These mechanisms contribute to organ-specific syndromes such as cerebral malaria and placental malaria when sequestration occurs in the brain or placenta. Hemolysis of infected and bystander (uninfected) RBCs causes anemia that may be exacerbated by impaired erythropoiesis. Hemolysis also contributes to endothelial injury and dysfunction as free hemoglobin (Hb) catalyzes oxidative damage and consumes nitric oxide (NO), a regulator of endothelial cells. Merozoites develop in the sequestered RBCs, and the rupture of infected erythrocytes causes fever and rigors. Most merozoites invade uninfected RBCs and circulate as ring-stage parasites, but a small fraction of merozoites develop into male and female gametocytes that infect mosquitoes when taken up during a blood meal. Gametocytes continue to circulate after treatment at the asexual blood stages; therefore, safe drugs to kill circulating gametocytes would help in P. falciparum elimination. ROS, reactive oxygen species; BBB, blood-brain barrier. (b) Progression of malaria in a susceptible population and opportunities for treatment. Approximately 2 billion people live in areas where malaria is transmitted. In regions where malaria is endemic, asymptomatic parasitemia is common and contributes to transmission. Intermittent presumptive treatment given to a population helps to eliminate parasites from asymptomatic carriers. Of the ∼500 million symptomatic cases of malaria globally each year, only about 1% progress to severe malaria. The major severe malaria syndromes are cerebral malaria, acidosis (respiratory distress) and severe anemia. Effective antidisease therapies that can be combined with parasite-killing drugs are needed to improve survival from severe malaria.
Where will new drugs to counter resistance and new therapies to improve survival come from? The involvement of Medicines for Malaria Ventures, the industry and scientists supported by governments and foundations have worked on drug discovery since the end of the Vietnam War. Antimalarial drugs will be discovered through random screening of small-molecule or natural-product libraries against either whole organisms or specific parasite molecules (Box 1). Indeed, ACT, the currently recommended treatment for malaria, was discovered by a screen of Chinese herbal medicines used to treat fevers. Antimalarial drugs may also be derived from known compounds, as exemplified by the development of atebrine, chloroquine and primaquine from efforts to find a synthetic substitute for the natural antimalarial, quinine. As our knowledge of the parasite and its life cycle expands (Fig. 1), it will be possible to target parasite molecules that are unique to it and necessary for its survival (Box 2). Particularly helpful will be drugs against gametocytes that persist after antimalarial treatment clears asexual blood-stage parasites. Antigametocyte drugs can disrupt transmission and have an important role in programs to eliminate malaria. The development of therapies to improve outcome from malarial infection may arise from a growing understanding of the molecular basis of the host-parasite interactions that cause severe disease.
Box 1. Discovery of antimalarial drugs: new methods of screening chemical compounds for antimalarial drugs available to the malaria community.
High-throughput screening of large numbers of chemical compounds have identified many leads for potential antimalarial drugs27,51,126–131. Screens can be classified by their use of whole-cell screening or target-based approaches. In whole-cell screening, parasite inhibition is monitored by measures of growth and proliferation, usually without knowledge of the molecular targets of the compounds that are being screened. For example, using a single concentration of 10 μM of each compound and radioactive labeling, Chong et al.126 screened 2,687 compounds and identified 87 existing drugs that resulted in >50% inhibition; a nonsedating antihistamine, astemizole, and its principal human metabolite were found to be promising new inhibitors of chloroquine-sensitive and multidrug-resistant parasites127. More recently, many more promising compounds have been identified through screening large collections of small molecules using similar approaches27,128–131, including a spiroindolone (NITD609) that kills erythrocytic-stage parasites with nanomolar potency130.
A current focus of antimalarial drug discovery is to screen for small molecules that are active against gametocytes and can block malaria transmission. The only drugs completely effective against P. falciparum gametocytes are 8-aminoquinolines; these drugs cause hemolysis that is especially severe in people with Mediterranean G6PD deficiency. Artemisinin derivatives are poorly effective against mature stage V P. falciparum gametocytes. In addition to spiroindolones132, other promising compounds have been screened and found to be active against gametocytes133–135 and have the potential to block parasite transmission. A combination of quantitative high-throughput screening and genome-wide association and linkage analyses has also allowed for the identification of potential molecular targets or molecules involved in transport of the small molecules27,131.
Another approach commonly used for drug screening involves assays that detect inhibitors of a specific parasite target, such as a kinase or cell-surface ligand for merozoite invasion of erythrocytes136,137. Although only a limited number of kinases or receptors have been studied in malaria parasites so far, some recent studies have shown that these molecules are promising targets for inhibitors blocking parasite development, invasion and transmission136. Other promising leads have emerged from target-based screens for specific inhibitors of P. falciparum dihydroorotate dehydrogenase138 and enzymes of the P. falciparum respiratory chain, cytochrome bc1 and PfNDH2 (ref. 139).
Box 2. Examples of specific drug targets: the apicoplast and the nutrient channel.
The apicoplast is an organelle in Apicomplexa derived from the chloroplast and from photosynthesis in its ancient ancestor. Recent studies demonstrated that the apicoplast has only one crucial function during the asexual blood stage, namely isoprenoid precursor synthesis. Yeh and DeRisi140 showed that the elimination of the apicoplast of P. falciparum by treatment with fosmidomycin, which blocks the conversion of 1-deoxy-D-xylulose-5-phosphate (DOXP) to methylerythritol phosphate, killed P. falciparum. The parasites without an apicoplast survived in media supplemented with isopentenyl pyrophosphate (IPP). Although both IPP and dimethylallyl diphosphate (DMAPP) are required for isoprenoid synthesis, IPP alone was sufficient to supplement parasites lacking apicoplasts, indicating that there must be an IPP isomerase to convert IPP to DMAPP, although this enzyme has not yet been identified. The elimination of the apicoplast in fosmidomycin-treated IPP-supplemented P. falciparum has been proven by the absence of the apicoplast genome and by the absence of the four-membrane apicoplast organelle. This study demonstrates that the pathway from DOXP to IPP is the crucial function of the apicoplast during the asexual blood stage and that enzymes in this pathway141 would be excellent targets for small-molecule inhibitors and should be synergistic with fosmidomycin.
Infected erythrocyte membranes have nutrient channels with increased permeability to a range of solutes, including anions, organic cations, sugars, amino acids, purines and vitamins. A recent study demonstrated that a single parasite-encoded molecule was responsible in part for the permeability changes in the infected erythrocyte142. One inhibitor identified by high-throughput screening was highly specific for solute uptake by the P falciparum clone Dd2 and had little or no activity against the P. falciparum clone HB3. The progeny clones from a Dd2 × HB3 genetic cross were used to map the locus to a single region on chromosome 3 of the parasite; complementation and allelic exchange implicated two related genes, clag3.1 and clag3.2. These findings are strong evidence for a parasite-encoded nutrient uptake channel. In addition, other parasite molecules and perhaps host erythrocyte molecules may be involved in channel activity. Although parasite clone–specific inhibitors were crucial for gene identification, high-throughput screening of them showed that most inhibitors target conserved parts of the channel; some inhibitors have preserved activity against the channel from divergent P. falciparum clones. The unusual nature of the parasite channel has no function or molecular homology to channels in humans. Different parasite channel inhibitors show a correlation of blocking the channel and parasite growth inhibition. Thus, blocking the channel would lead to parasite death.
In this review, we will discuss recent developments in studies of the molecular basis of disease, parasite development and host-cell invasion and point to potential parasite targets that may be explored for drug development and improved treatment of children with severe disease. We will focus on the discovery of drugs for use against P. falciparum in endemic countries and not on the development of drugs for tourists or the military. Readers who are interested in the broader aspects of antimalarial drug development, current drugs in the pipeline, efforts to develop drugs against relapses of Plasmodium vivax and malaria elimination are referred to some excellent review articles on the subjects1–3.
The digestive vacuole and continuing drug discovery
About two-thirds of the hemoglobin content of P. falciparum–infected erythrocytes is consumed in the digestive vacuole to provide the maturing parasite with amino acids and guard against premature rupture. Plasmodium parasites digest hemoglobin by an array of functionally redundant peptidases, making proteases a poor drug target4. A consequence of hemoglobin digestion is the release of heme as Fe(ii)–protoporphyrin IX (Fe(ii)PPIX) that quickly oxidizes to Fe(iii)PPIX (hematin), a product that poisons the parasite by membrane damage and lysis (Fig. 2)5. Malaria parasites avoid heme toxicity by converting dimers of hematin into inert crystals (called hemozoin) and perhaps also by heme degradation through peroxidative or glutathione-mediated pathways6. Drugs that interfere with crystal formation have been some of the most successful in malaria therapy. Among these is chloroquine, an antimalarial drug that was used for hundreds of millions of treatments annually before chloroquine resistance developed.
Figure 2.
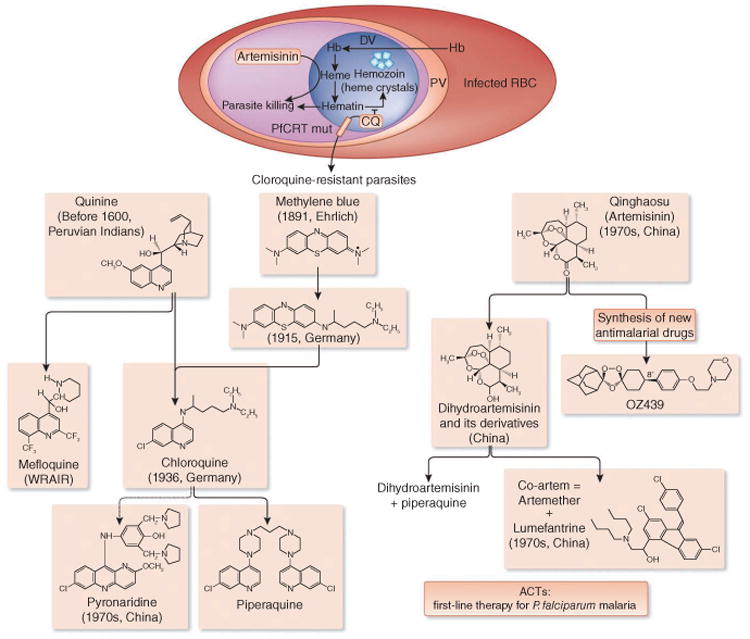
Quinine and artemisinin discoveries led to the development of many synthetic antimalarial drugs. In the digestive vacuole (DV) of the intraerythrocytic forms of the parasite, hemoglobin (Hb) is digested, and hematin is released, which is detrimental to the parasite. The parasite can reduce the harmful effects of hematin by converting it into hemozoin; however, this reaction is inhibited by chloroquine (CQ). Heme activates artemisinin activity, resulting in parasite killing. RBC, red blood cell; Mut, mutant; PV, parasitophorous vacuole.
Resistance is conferred by mutations in a conserved molecule termed the P. falciparum chloroquine resistance transporter (PfCRT) that is expressed on the digestive vacuole membrane7. Drug efflux by the mutated molecule represents an expansion of the normal physiological function of PfCRT, which may include transport of amino acids or short polypeptides from the digested hemoglobin8. Mutations or changes in the expression of a P. falciparum P-glycoprotein homolog (PfPgh1) encoded by the P. falciparum multidrug resistance 1 gene (pfmdr1) that also resides on the digestive vacuole membrane9 can modulate chloroquine resistance in PfCRT-mutant parasites and, along with PfCRT and other transport molecules, alter the responses to other antimalarial drugs, including mefloquine, quinine, lumefantrine, halofantrine and artemisinin10.
The concentration of chloroquine that kills chloroquine-resistant P. falciparum parasites in vitro can be reduced by verapamil and other pharmacologically and structurally diverse agents, including tricyclic antidepressants, antihistamines, phenothiazine, some plant alkaloids and primaquine. This effect on chloroquine-resistant but not chloroquine-sensitive parasites is referred to as chemosensitization. These agents generally include a pharmacophore of one or more aromatic rings plus a protonated nitrogen that competes with chloroquine in the PfCRT pore, with the extent of chemosensitization depending on the mutations present11. Differential degrees of resistance to the active metabolites of chloroquine (monodesethylchloroquine), amodiaquine (monodesethylamodiaquine) and other quinoline drugs are similarly linked to variations in the mutations of PfCRT12,13. The structural specificity implied by these observations coincides with reports that a number of compounds related to chloroquine but not efficiently effluxed by PfCRT can be remarkably effective against chloroquine-resistant as well as chloroquine-sensitive P. falciparum. Examples include such well-known antimalarial drugs as pyronaridine and piperaquine (Fig. 1), compounds with 4-amino side chain variations on the 7-chloro-4-aminoquinoline moiety of chloroquine and certain other 3- or 7-substituted 4-aminoquinolines14–18. The risk of arrhythmia, as indicated by hERG (human ether-a-go-go related gene product) channel assays, remains a concern for the 4-aminoquinoline compounds. These risks must be considered in light of the safety record of chloroquine, evidence that the ratio of hERG half-maximum inhibitory concentration (IC50) to free drug concentration may not be a reliable predictor of human risk and the suggestion that 4-aminoquinoline candidates with higher hERG IC50 values have lower risk of arrhythmia in comparison to chloroquine18,19.
Although chemosensitizing agents are not sufficiently active at concentrations that can be safely and effectively used in humans20, their resistance-reversing activity has inspired the synthesis of chloroquine derivatives that incorporate various chemosensitizer moieties at the 4-amino position. Examples of side chains in these ‘reversed chloroquine’ compounds include dibemethins, imipramine and other pharmacophores predicted to confer reversal activity21,22. Ferroquine, a potent new organometallic chloroquine analog that carries a ferrocenyl group in the side chain23, also seems to fall within this theoretical framework. Successful phase 1 trials of ferroquine24 and AQ-13, a short-chain analog of chloroquine25, have raised hope that these and other 4-aminoquinolines will be successful in clinical development and join pyronaridine and piperaquine as alternative drugs for use against chloroquine-resistant malaria.
Various studies have identified a number of antimalarial compounds that show greater activity against chloroquine-resistant than chloroquine-sensitive P. falciparum and have IC50 values that are linked to PfCRT isotype26,27. Combinations of various antimalarial pairs may also act in synergy or in antagonism, depending on the exact mutations encoded by the PfCRT allele12,27. These observations raise the possibility of drug combinations that together may combat malaria drug resistance by selection pressure against mutant forms of PfCRT. Examples of suppressive drug combinations have been reported in studies of Escherichia coli and Staphylococcus aureus populations in which strongly antagonistic pairs of drugs reduced the potential for evolving resistance far more efficiently than pairs of synergistic drugs28,29.
Products of hemoglobin digestion are also involved in the action of artemisinin-class antimalarials on the erythrocytic stages of Plasmodium spp.30. The artemisinins, developed from the ancient Chinese herbal remedy qinghaosu, carry a superoxide pharmacophore with a peroxide bridge that is cleaved and activated by ferrous heme or free ferrous iron. Highly reactive carbon-centered radicals from this activation kill the parasite by processes that may include damage to cellular lipids and digestive vacuole membranes31, inactivation of Plasmodium proteins, alkylation of heme and interference with heme sequestration into crystals32. The effect on intraerythrocytic stages is broad, including strong action on rings stages as well as trophozoites and early schizonts33.
Artemisinin-based treatments with artesunate, artemether or dihydroartemisinin are now the first-line recommendation for P. falciparum malaria in most endemic areas and are increasingly used against the problem of chloroquine-resistant P. vivax malaria34,35. Artemisinin treatment reduces parasitemia very rapidly, with a parasite reduction of about 10,000-fold per cycle in vitro. When given as monotherapy, a 7-d regimen was found to be required to achieve acceptable cure rates, but some recrudescences by day 28 still occurred36. These recrudescences, the difficulty of patient adherence to the dosing regimen and the short in vivo half-life of the active dihydroartemisinin metabolite (t1/2 < 1 h) led to early realization of the importance of artemisinin therapy being partnered with a more slowly eliminated antimalarial drug. Because combinations can fail in regions where malaria parasites are resistant to the partner drugs, artemisinin-combination therapies require continuous monitoring for efficacy, and the appropriate choices of alternative drug combinations must be available when failure rates become unacceptable.
Prolonged parasite clearance times (PCTs) have recently been reported from western Cambodia37,38, raising concerns that P. falciparum parasites in the region are becoming resistant to artemisinin and its derivatives39. However, the parasites that show a prolonged PCT do not show correlated increases of IC50 values to dihydroartemisinin in vitro37,40. Multiple host and parasite factors are involved in PCT. Recrudescences in association with prolonged PCTs have been documented after 7-d treatment courses of artesunate41 (Box 3). Evaluations of patients in Cambodia infected with identical or nonidentical parasite genotypes showed that prolonged PCTs could be attributed to heritable parasite factors (∼60% of PCT variation)42. Evidence from DNA microarray studies suggests that prolonged PCTs may be associated with reduced activity of basic metabolic and cellular maturation pathways in the parasites43. More recently, genome surveys have suggested that differences in a number of genome regions may be associated with prolonged PCT after artemisinin treatment, including the possibility of a selective sweep on P. falciparum chromosome 13 (ref. 44).
Box 3. Is recrudescence after artemisinin treatment linked to a dormancy phenomenon?
Dormancy, a quiescent state in which a small fraction of inactive parasites are unaffected by drug treatment, is proposed to account for some observed discrepancies between in vivo and in vitro responses to drugs. Although dormancy during chloroquine treatment was not demonstrated in laboratory studies, results from sorbitol-treated, pyrimethamine-treated or mefloquine-treated parasite cultures suggested that small numbers of inactive parasites, perhaps lag-phase ring forms, could survive these exposures and later reactivate with the same characteristics of drug sensitivity as the original population143,144. Other reports have described possible cell-cycle delays and recrudescence without innate changes in drug sensitivity after treatment with atovaquone, atovaquone plus proguanil or mefloquine145,146. Dormancy has also been proposed from mathematical modeling of artesunate treatment results147, and several studies have identified dormancy and a ring-stage quiescence mechanism of survival during exposure to artemisinin drugs148–151. These and additional observations152–154 have raised important questions regarding the nature of the relationship between a dormancy phenomenon by which no more than 1–2% of parasites recover147,149 and a mechanism that reduces clearance rates by doubling or tripling the PCTs to over 100 h after the initiation of artemisinin drug treatment38. Evidence from DNA microarray studies suggests that prolonged PCTs are associated with reduced activity of basic metabolic and cellular maturation pathways43; data implicating these same pathways in quiescent parasites would provide evidence for common underlying mechanisms in prolonged PCTs and dormancy.
Because the artemisinin derivatives act for only a short time and require frequent dosing, searches for other artemisinin derivatives and synthetic analogs with improved pharmaceutical properties over artemisinin have received considerable attention (Fig. 2). Among these are completely synthetic, low-cost peroxides, including 1,2,4-trioxanes, 1,2,4-trioxolanes and 1,2,4,5-tetraoxanes, some of which are currently in clinical trials or are new candidates for preclinical development45,46. A particularly promising series of ozonide compounds from spiroadamantine-shielded 1,2,4-trioxolanes has yielded compounds that are well tolerated with good oral bioavailability, outstanding efficacy and pharmacokinetic half-lives that are much longer than those of artemisinin derivatives47. OZ439, a particularly encouraging candidate currently in phase 2 trials supported by Medicines for Malaria Ventures, showed a half-life of ∼30 h in preclinical tests and produced full cures after one-time oral administration in mice infected with Plasmodium berghei, raising the prospect that OZ439 (in combination with a suitable partner) may be effective for single-dose treatment for malaria48. As OZ439 moves into phase 3 trials, it will be important to establish the effect of its much longer half-life compared to that of dihydroartemisinin on parasites with prolonged PCTs and evaluate the impact of this longer period of continuous drug exposure on recrudescences after treatment.
Inhibitors targeting parasite kinases
Kinases are traditionally the targets of drug screens because these molecules are involved in various important biological pathways, and their activities can be readily assayed. Additionally, large numbers of known or potential kinase inhibitors are available in the small-molecule libraries of pharmaceutical companies. This principle can be applied to screening for antimalarial drugs, although malaria parasites have a much smaller number of kinases in their genomes49,50. The divergences between Plasmodium protein kinases and those of their mammalian hosts suggest that the parasite kinases can be attractive targets for new antimalarials50. Examples include P. falciparum MAPK2 (PfMAPK2), which is member of MAPK family but has unique characteristics, the FIKK kinases that are found only in Apicomplexa and the calcium-dependent protein kinases (CDPKs) that are present exclusively in plants and alveolates49. Additionally, different kinases are expressed at different parasite developmental stages, which can be explored for drugs of different purposes, such as transmission blocking.
One example of parasite kinases used in screening of small molecules for inhibitors is P. falciparum CDPK1 (PfCDPK1). In a recent study, ∼20,000 compounds from a kinase-directed heterocyclic library were screened for their ability to inhibit recombinant PfCDPK1, which seems to be essential for the asexual stages51. Recombinant PfCDPK1 was expressed as a GST fusion protein, and biotinylated casein kinase II peptide was used as a substrate in an assay measuring the ability of PfCDPK1 to catalyze the transfer of the γ-phosphate group from (γ-33P)ATP to the biotinylated substrate peptide. Approximately 50 compounds that could inhibit the kinase activity by >80% at a concentration of 1 μM were identified51, including a compound called purfalcamine that was found to bind PfCDPK1 in parasite lysates and block the development of late schizonts. In another study, histidine-tagged PfCDPK1 was expressed in E. coli and used in screens against 54,733 compounds, leading to the identification of 70 compounds with submicromolar activities52. Cyclin-dependent kinases (CDKs) that control cell-cycle progression have also been investigated as the targets of antimalarial drugs. Several homologs of human CDKs have been characterized from P. falciparum, including PfPK5 (a homolog of human CDK1) and Pfmrk (a homolog of human CDK7)49,53,54. Both PfPK5 and Pfmrk were identified as potential targets for the development of antimalarial agents53–55. Screening of extracts from marine sponges for inhibitors of a P. falciparum never in mitosis Aspergillus (NIMA)-related protein kinase (Pfnek-1) also led to the identification of several molecules with activities against Plasmodium parasites in vitro and in vivo56,57. Another potential kinase target is the P. falciparum cyclic GMP (cGMP)-dependent protein kinase (PKG), which has been shown to be essential for xanthurenic acid–induced and zaprinast-induced gametogenesis58. PKG, xanthurenic acid and cGMP were all found to be components of the gametocyte activation pathway in P. falciparum. In a follow-up study, the same group examined the role of PfPKG in the asexual blood stage of the parasite life cycle using a PKG inhibitor, trisubstituted pyrrole. Addition of this compound prevented synchronized schizonts-infected erythrocytes from releasing merozoites. Using genetically manipulated P. falciparum parasites expressing a PfPKG allele that was insensitive to the compound, they also showed that the mutant parasites were able to complete schizogony in the presence of the compound but not in the presence of the broad-spectrum protein kinase inhibitor staurosporine, suggesting that PfPKG is the primary target of the compound during schizogony. These results showed that cGMP signaling is a key regulator of both the sexual and asexual stages, which can be explored for potential drug targets for both therapeutic and transmission-blocking activities.
Merozoite invasion of erythrocytes: in search of drug targets
Mature merozoites are released from infected erythrocytes in the blood stream by rupturing the erythrocyte membrane. The freed merozoites then rapidly invade uninfected erythrocytes. There is extensive redundancy of the use of ligands by P. falciparum for invasion, and, consequently, no erythrocyte type has been identified in endemic areas refractory to invasion as a result of the loss of a receptor for P. falciparum. This is in contrast to P. vivax, where the loss of an erythrocyte receptor, the Duffy blood group system, led to refractoriness.
Invasion of erythrocytes by merozoites involves five discrete steps (Fig. 3). The ligands in the first step, the binding of any part of the merozoite to erythrocytes, are unknown and are thus not currently drug targets. In the second step in invasion, the merozoite orients its apical end to the erythrocyte in a process that involves two families of P. falciparum ligands that bind receptors on the erythrocyte surface, namely the four Duffy binding-like (DBL) ligands and the five reticulocyte homology ligands (Fig. 3; step 2, reorientation). Possibly because of the redundancy in P. falciparum DBL and reticulocyte homology ligands, all these ligands can be knocked out with one exception, reticulocyte homology 5 (RH5), making it a possible drug target59,60. RH5 has no transmembrane domain and binds another soluble protein with ten epidermal growth factor-like domains (PFC1045c) that also has no transmembrane-like domains61. In addition, the erythrocyte receptor for RH5 was found to be CD147 (also known as Oka or basigin)62. In this study, soluble basigin and antibodies to basigin were found to block parasite invasion of erythrocytes. The essential nature of RH5 in the face of so much redundancy in ligand-receptor interactions raises the possibility that the interactions of RH5 could be blocked using small molecules.
Figure 3.
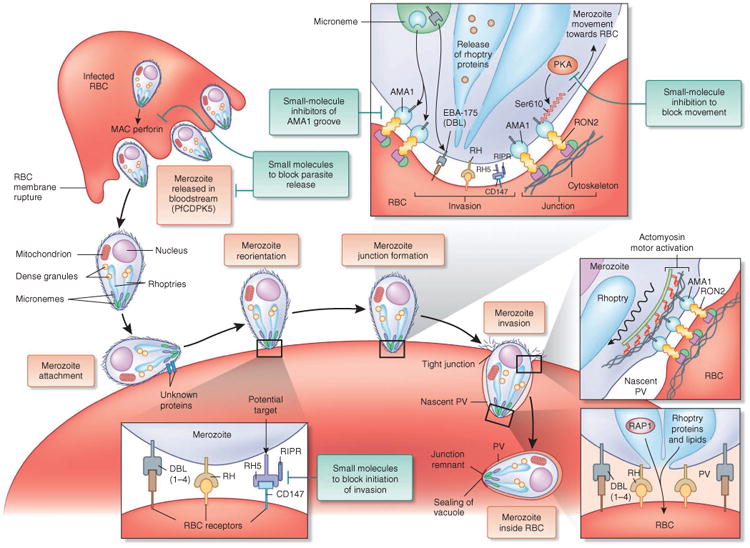
Merozoite invasion of the erythrocyte involves five steps, including the movement of erythrocyte membrane past the junction to form the parasitophorous vacuole (PV). RBC, red blood cell; RH, reticulocyte homology ligand; RIPR, RH5 interacting protein.
In the third step in invasion, apical membrane antigen 1 (AMA1) moves from micronemes to the merozoite surface and binds a segment of rhoptry neck protein 2 (RON2). AMA1 has a groove formed by two PAN/apple domains in AMA1 (ref. 63). The RON proteins are secreted into the erythrocyte membrane and accumulate on the cytoplasmic face, whereas a segment of RON2 remains outside the erythrocyte membrane to bind AMA1 (refs. 64,65). The junction that must be formed to bring the merozoite into the erythrocyte requires the binding of AMA1 to RON2 (Fig. 3; step 3, junction formation). No junction is formed and no invasion occurs if the AMA1 binding to RON2 is blocked by soluble RON2 peptide or antibodies to the AMA1 groove66. The RON2-AMA1 junction would be an ideal target for small-molecule inhibitors because RON2 and AMA1 are both parasite molecules that must fit together for effective junction formation. Small-molecule inhibitors of protein-protein interactions are potential targets for drug development where there is a pocket (groove) for binding small molecules67.
The internal signals for the release of microneme and rhoptry contents are still not completely understood. Binding of glycophorin A to its ligand, erythrocyte-binding antigen 175 (EBA-175), on the merozoite surface leads to the release of rhoptry proteins68. However, the EBA-175–glycophorin A interaction cannot be an absolute requirement, as parasites lacking EBA-175 still are able to invade erythrocytes.
AMA1 has a crucial serine on its cytoplasmic tail (Ser610) that if mutated to an alanine (S610A) cannot function in invasion. Ser610 is phosphorylated by the parasite protein kinase A (PKA, also known as PFI1685w), a cyclic AMP–activated kinase69. Although crucial for invasion, the function of this phosphorylation event and the timing of changes in the level of phosphorylation are unknown. One speculation is that it may signal the actin-myosin motor to begin bringing the merozoite into the erythrocyte. The binding of aldolase to the cytoplasmic tail of AMA1 that connects it to the actin-myosin motor is unaffected by the S610A mutation. Although the role of this phosphorylation in invasion is unknown, PKA might be a target for small-molecule inhibition.
In the fourth step of invasion after the formation of the junction, unknown factors stimulate the actin-myosin motor to power the junction to move from the anterior to the posterior end of the merozoite, bringing the merozoite into the erythrocyte (Fig. 3; step 4, invasion). Simultaneously with the movement of the junction, a vacuole forms around the merozoites derived from the erythrocyte membrane, flowing past the junction as the parasite moves into the erythrocyte70. Little is known of the signals for the release of parasite proteins from organelles and the factors involved in activating the motor. It has been observed that one of the rhoptry proteins, rhoptry associated protein 1 (RAP1), is located within the invaginated membrane71, but proof that it is the cause of the invagination is lacking. The fifth step of invasion, the resealing of the vacuole and the erythrocyte membrane (Fig. 3; step 5, resealing), is crucial for parasite survival, as anything that blocks resealing, such as a residual body attached to the merozoite, leads to erythrocyte lysis. The remnants of the moving junction remain on the outside of the vacuolar membrane70.
Besides invasion, release from the erythrocyte is an essential process and, consequently, a potential drug target. The process of erythrocyte membrane rupture is less well understood at a molecular level than is erythrocyte invasion. Nonetheless, several factors required for merozoite release have been identified, including a calcium-dependent protein kinase, PfCDPK5. PfCDPK5 conditionally deficient parasites mature into fully viable merozoites in the infected erythrocyte but are unable to leave the vacuole and thus are not released from the infected erythrocyte72. PfCDPK5 evolved from a plant kinase and may be selectively targeted by small-molecule inhibitors that may be less likely to bind human kinases. A Plasmodium membrane attack complex (MAC)/perforin domain-may be another potential target, as a MAC/perforin domain has been shown to be involved in the release of Toxoplasma gondii from its vacuole and host cell73 and may have a similar role in Plasmodium escape from the infected erythrocyte.
Endothelium-targeted strategies to improve survival
Antiparasitic drugs are the cornerstone of malaria treatment; however, even the most efficacious parasite-killing drug, artesunate, could not prevent deaths among children with severe complications of malaria, such as severe anemia (1% mortality)74, acidosis (15% mortality) or coma (18% mortality)35. Thus, even rapid and effective parasite killing is insufficient to prevent death in severe cases. Although blood transfusion reduces mortality in children with severe anemia and respiratory distress, there are no specific therapies for acidosis or coma (Fig. 1b).
Metabolic acidosis is present in 40–60% of cases of severe malaria, often presenting as respiratory distress. The blood concentration of lactic acid, a product of anaerobic glycolysis from inadequately perfused host tissues, predicts death from severe malaria. Unlike septic shock, where lactic acid production is associated with hypotension, hypovolemia or both, most patients with malaria-associated lactic acidosis are not substantially volume depleted75, and intravenous fluid bolus may be harmful76.
Impaired consciousness, measured by the Blantyre coma score, is consistently associated with increased risk of lasting neurological injury and death. The prognostic value of the coma score is enhanced when combined with visual examination of the retina. Papilledema and retinal hemorrhage are each associated with increased risk of death77, and retinopathy is associated with postmortem counts of adherent infected erythrocytes, thrombosis and hemorrhage in cerebral microvessels78. Microvascular obstruction may be a pathogenic mechanism shared by cerebral malaria and acidosis, the two severe malarial syndromes that cause the most deaths.
Disruption of microvascular blood flow has been directly observed in human clinical studies of severe malaria79. Beyond mechanically obstructing small vessels, adherent infected erythrocytes transduce pathologic endothelial activation signals that promote adhesion, coagulation and inflammation, disrupt endothelial barrier integrity and impair vasoregulation (Fig. 4)80–82. Endothelial dysfunction is exacerbated by malaria-induced decreases in nitric oxide, a signaling molecule that normally maintains endothelial homeostasis83. To improve survival from severe malaria, therapeutics that can modulate key endothelial functions need to be tested in combination with parasite-killing drugs.
Figure 4.
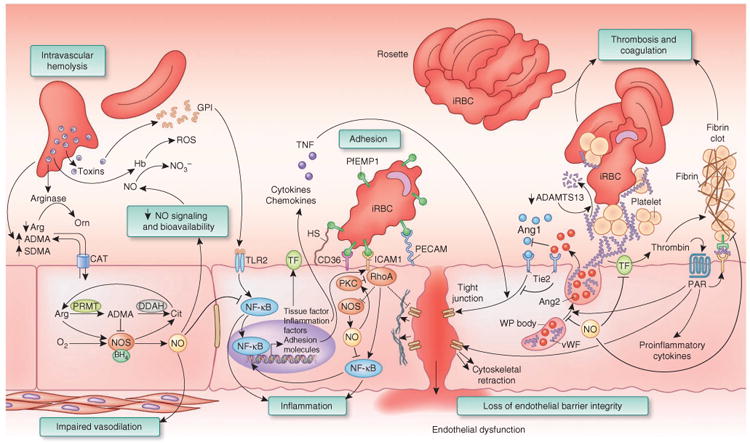
Malaria infection disrupts nitric oxide metabolism and causes harmful endothelial activation. Intravascular hemolysis limits nitric oxide (NO) bioavailability: destruction of the erythrocyte releases hemoglobin, arginase and ADMA into plasma. Hemoglobin (Hb) reacts with endothelial nitric oxide and converts it to biologically inactive nitrate (NO3–), diminishing nitric oxide signaling. Hemoglobin can also catalyze the production of reactive oxygen species (ROS). Arginase is released from erythrocytes and metabolizes arginine to ornithine (Orn), limiting the arginine that is available to NOS. Erythrocytes have high concentrations of ADMA incorporated in proteins; hemolysis and proteolysis releases free ADMA into plasma. Impaired nitric oxide synthesis: NOS catalyzes the generation of nitric oxide from the substrates oxygen (O2), Arg and NADPH. Tetrahydrobiopterin (BH4) is an essential NOS cofactor. In the absence of either arginine or BH4, NOS functions as an oxidase, generating superoxide from NADPH and molecular oxygen. The product citrulline (Cit) can be recycled to arginine by argininosuccinate synthase (ASS1) and argininosuccinate lyase (ASL). ADMA is a NOS inhibitor that is generated by the methylation of arginine in polypeptides by protein arginine methyltransferase (PRMT) followed by proteolysis to release free ADMA. DDAH regulates nitric oxide synthesis by metabolizing ADMA. Arginine, ADMA and symmetrical dimethylarginine (SDMA) cross the endothelial cell membrane by a cationic amino acid transporter (CAT). Inflammation: glycosylphosphatidylinositol (GPI) from malaria parasites can trigger inflammation through Toll-like receptor 2 (TLR2) signaling, leading to NF-κB activation and transcription of inflammatory cytokines (such as TNF), adhesion molecules (such as ICAM1) and procoagulant molecules (such as tissue factor, TF). Nitric oxide can downregulate NF-κB to exert anti-inflammatory effects. Adhesion: infected erythrocytes (iRBCs) bind to endothelial cells through parasite-encoded PfEMP1 and endothelial receptors such as ICAM1 and heparan sulfate (HS). Binding to ICAM1 triggers RhoA and Rho kinase, leading to cytoskeletal rearrangements that cause cell retraction and disrupt cell-cell junctions. Rho kinase also activates NF-κB to exert proinflammatory, proadhesive and procoagulant effects. Rho kinase downregulates NOS activity, but nitric oxide can suppress RhoA activation by nitrosylation of protein kinase C (PKC). Loss of barrier integrity: tight junctions between endothelial cells are maintained by signaling through the Ang1–endothelial-specific receptor tyrosine kinase (Tie2) axis. Ang2 binds to Tie2 but does not transduce a signal, thereby interrupting constitutive Ang1-Tie2 signaling. In the setting of proinflammatory cytokines such as TNF, this leads to loss of integrity of the endothelial cell layer. Weibel-Palade (WP) body exocytosis: Weibel-Palade bodies contain vWF multimers that can bind to circulating platelets and trigger thrombosis. vWF multimers are cleaved by the vWF protease ADAMTS13 to limit the extent of thrombosis. Patients with severe malaria have reduced ADAMTS13 activity and an overabundance of vWF multimers. Tethered vWF multimers can bind to platelets and form a bridge to infected erythrocytes. Thrombosis and coagulation: adherent infected erythrocytes trigger a display of tissue factor on the endothelial cell apical surface and recruit circulating coagulation factors to activate thrombin. Thrombin generates fibrin to form a blood clot that can block the lumen of small vessels and stop blood flow. Thrombin also activates protease-activated receptors (PARs) that couple with multiple G proteins to cause cytoskeletal retraction and expression of inflammatory cytokines and adhesion molecules.
Endothelium-targeted strategies to improve survival
Targeting parasite adhesion to the vascular endothelium
P. falciparum expresses highly variable proteins on the surface of erythrocytes called P. falciparum erythrocyte membrane proteins 1 (PfEMP1s). PfEMP1s bind to protein and carbohydrate ligands on the surfaces of endothelial cells (including CD36, intercellular adhesion molecule 1 (ICAM1), platelet/endothelial cell adhesion molecule (PECAM), CR1, heparan sulfate and chondroitin sulfate A), allowing infected erythrocytes to attach, sequester and avoid clearance in the spleen. Clinical episodes of severe malaria have recently been associated with expression of var genes that encode specific PfEMP1s84. The same var genes were independently observed to bind brain endothelium in vitro85,86. Once the crucial var ligand and its endothelial receptor have been identified, high-throughput screening could be used to identify small molecules that block infected erythrocytes from binding to or activating microvascular endothelium through this pathway. An existing drug, levamisole, interrupts CD36-dependent binding by inhibiting the dephosphorylation of CD36 that is required for high-affinity binding.
Interrupting malaria-mediated blood coagulation and thrombosis
Systematic postmortem examinations have demonstrated thrombosis and hemorrhage where infected erythrocytes adhere to cerebral microvascular endothelium78. Adherent infected erythrocytes trigger coagulation by inducing tissue factor and activating thrombin on the apical surface of endothelial cells80. Thrombin not only catalyzes fibrin deposition in blood vessels but also amplifies inflammation and disrupts endothelial barrier integrity through protease-activated receptor signaling87. Heparin, which activates antithrombin III, was tested in several small studies of severe malaria but had no effect on outcome88. Activated protein C, an endogenous anticoagulant that is diminished in severe malaria89, has been administered therapeutically but has never been evaluated in a controlled study. Defibrotide, a DNA aptamer, blocks endothelial tissue factor induction by infected erythrocytes in vitro and shows favorable anti-inflammatory properties in vivo90 but has yet to be tested in human malaria.
Microvascular obstruction can also be caused by deficiency of the von Willebrand Factor (vWF) protease ADAMTS13 (ref. 91), resulting in long vWF multimers that tether platelets and infected erythrocytes to endothelium92. The number and length of vWF multimers could be decreased by restoring the deficient activity of the vWF protease ADAMTS13 (ref. 91) with plasma transfusion or by inhibiting exocytosis of the endothelial Weibel-Palade bodies that release vWF multimers93.
Preserving endothelial barrier integrity
Loss of endothelial barrier integrity may be an important step in the progression to fatal cerebral malaria. The angiopoietin (Ang) proteins regulate endothelial barrier integrity and are associated with retinopathy and death from cerebral malaria94,95. In the setting of tumor necrosis factor (TNF) stimulation, Ang2 permits breakdown of endothelial barrier integrity and expression of endothelial adhesion molecules such as ICAM1. The exocytosis of Ang2 from endothelial Weibel-Palade bodies might contribute to the vascular leak, inflammation and cerebral edema that are associated with cerebral malaria, whereas upregulation of ICAM1 would increase the adhesion of infected erythrocytes. Endothelium-targeted therapies that suppress Weibel-Palade body exocytosis might interrupt the pathogenic autocrine activity of Ang2.
Erythropoietin is secreted primarily by renal peritubular cells in response to hypoxia. In addition to promoting erythropoiesis, erythropoietin exerts neuroprotective effects through the heterodimer of the erythropoietin receptor and CD131. Erythropoietin decreases the expression of inflammatory cytokines, limits astrocyte water uptake and prevents neuronal apoptosis. Among Kenyan children with cerebral malaria, high amounts of erythropoietin were associated with protection from neurological sequelae and death96. In a mouse model of cerebral malaria, erythropoietin decreased the expression of inflammatory cytokines in the brain, reduced the number of perivascular hemorrhages and improved neurological function and survival97. Erythropoietin was recently shown to be safe in children with malaria, but efficacy studies have not been performed.
Endothelial nitric oxide is diminished in severe malaria
Vascular endothelium regulates adhesion, coagulation, inflammation, barrier integrity and smooth muscle tone through nitric oxide. Diminished nitric oxide bioavailability may exacerbate endothelial dysfunction and contribute to the pathogenesis of severe malaria. Indonesian adults with severe malaria had decreased endothelial nitric oxide bioavailability, as demonstrated by impaired vasodilation after 5 minutes of brachial artery occlusion83. This vasodilation index correlated with the plasma concentration of arginine and the nitric oxide synthase (NOS) substrate and inversely correlated with the concentration of methylarginine, an endogenous NOS inhibitor98. Malian children with severe malarial anemia showed evidence of pulmonary vasoconstriction, consistent with low nitric oxide bioavailability99. In both studies, the functional activity of nitric oxide was inversely correlated with the concentration of plasma hemoglobin, a nitric oxide scavenger.
Intravascular hemolysis releases hemoglobin into the plasma, where it can interrupt endothelial nitric oxide signaling. Haptoglobin binds to hemoglobin in plasma, and the haptoglobin-hemoglobin complex is removed from circulation by CD163 on macrophages and monocytes. Intracellularly, heme oxygenase-1 (HO1) metabolizes heme into biliverdin, releasing carbon monoxide and ferrous iron. Although carbon monoxide has anti-inflammatory properties, the net effect of HO1 activity in humans may be proinflammatory because free iron potentiates the neutrophil oxidative burst. In Gambian children, HMOX1 alleles with high inducible expression conferred susceptibility to severe malaria100.
In addition to nitric oxide scavenging and inflammation, hemolysis causes depletion of the NOS substrate arginine by releasing arginase from erythrocytes and inducing expression of host arginase101. Arginine depletion not only diminishes nitric oxide synthesis but also causes NOS to function as an oxidase, generating superoxide radicals instead of nitric oxide from molecular oxygen.
Intravascular hemolysis followed by proteolysis of erythrocyte proteins releases asymmetric dimethylarginine (ADMA) into plasma that can overwhelm host mechanisms of ADMA clearance and cause endothelial dysfunction102,103. By inhibiting NOS, ADMA activates endothelial Rho kinase104 (a molecular switch also activated by the adherence of infected erythrocytes81), which increases expression of NF-κB, secretion of TNF, display of procoagulant tissue factor and expression of the infected erythrocyte receptor, ICAM1. ADMA decreases endothelial cell motility, disrupts endothelial barrier integrity and inhibits vascular repair of endothelial disruptions. Dimethylarginine dimethylaminohydrolase (DDAH) indirectly regulates nitric oxide synthesis by metabolizing ADMA105. DNA sequence polymorphism in the gene DDAH1 was associated with increased susceptibility to severe malaria in a recent genome-wide association study of Gambian children106.
Restoring endothelial nitric oxide bioavailability
The emerging evidence for decreased nitric oxide bioavailability in the pathogenesis of severe malaria has stimulated interest in restoring nitric oxide signaling as an endothelium-targeted adjunctive therapy. Arginine supplementation resulted in increased exhaled nitric oxide and nitric oxide–mediated vasodilation in adult patients with moderately severe malaria; however, these patients had normal nitric oxide production before treatment83. Arginine supplementation might be less effective at restoring NOS-mediated nitric oxide production in severely ill patients in which arginase, ADMA, NOS glutathionylation or tetrahydrobiopterin deficiency may limit NOS activity. Furthermore, plasma hemoglobin99,107,108 or reactive oxygen species109 limit the half-life of nitric oxide.
Restoration of nitric oxide bioavailability may require a NOS-independent approach. Nitric oxide gas can be delivered directly to the pulmonary circulation by inhalation, where it exerts local pulmonary arterial vasodilator effects. Inhaled nitric oxide can be transported in blood as S-nitrosothiols, nitrosylhemoglobin or nitrite, where it can act at distant sites. Administration of exogenous nitric oxide inhibits NF-κB–mediated expression of inflammatory cytokines, adhesion molecules and tissue factor110,111 and decreases platelet activation and adhesion of leukocytes and infected erythrocytes93,112,113. Furthermore, nitric oxide preserves endothelial barrier integrity and prevents pathologic thrombosis by inhibiting Weibel-Palade body exocytosis of Ang2 and vWF93. There are currently two controlled trials of inhaled nitric oxide ongoing in African children with malaria.
An alternative is to generate nitric oxide in blood and tissues by the nitrite-heme–nitric oxide pathway, a physiologic mechanism that regulates hypoxia-mediated vasodilation in humans114. Nitrite can be reduced to nitric oxide by deoxyhemoglobin in blood115, as well as by hemeglobins116, xanthine oxidase and aldehyde oxidase117 in tissues. Nitrite improves nitric oxide bioavailability in animal models of hemolysis118, hemoglobin-based oxygen carrier transfusion119, ischemia-reperfusion injury120, lipopolysaccharide-induced inflammation121 and argininosuccinate lyase deficiency122, conditions that have features in common with malaria. Nitrite improved survival in a mouse model of severe malaria123. In humans, nitrite has been safely administered to healthy volunteers114 and patients with sickle cell–induced hemolysis in which nitrite improves regional blood flow124. The low cost, room-temperature stability and oral bioavailability of nitrite would make it feasible for use in resource-limited health care settings.
The way forward
Each sick child represents our failure to prevent malaria, and each death represents our failure to treat soon enough. Despite having the ‘wonder drug’, artemisinin derivatives, we must always have new drugs in reserve. We have outlined promising targets for drug discovery, such as parasite metabolism, parasite signaling and host responses. These and other targets will require the expertise of industry or public and private collaboration to see them to completion, and whole-cell screens (infected erythrocytes and gametocytes) for drug discovery are important. New drugs against stage V P. falciparum gametocytes and P. vivax hypnozoites are needed immediately for malarial elimination. Vaccines may ultimately offer the most cost-effective tool for disease prevention, but they are not currently available. Likewise, vector research will be essential for the development of new insecticides to replace the failing synthetic pyrethroids125 and to create mosquitoes refractory to malaria transmission. Ultimately, elimination of malaria will require advancements in drug development, vaccines and vector biology. Until malaria elimination has been achieved, drug treatment will remain crucial to prevent complications and death from malaria. Drugs in the development pipeline will always be needed as parasites acquire resistance to each new generation of antimalarial drugs.
Acknowledgments
This review was supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, US National Institutes of Health. We thank S.K. Pierce, S. Desai and C. Pola for critical comments and figure design.
Footnotes
Competing Financial Interests: The authors declare no competing financial interests.
References
- 1.Delves M, et al. The activities of current antimalarial drugs on the life cycle stages of Plasmodium: a comparative study with human and rodent parasites. PLoS Med. 2012;9:e1001169. doi: 10.1371/journal.pmed.1001169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wells TN, Burrows JN, Baird JK. Targeting the hypnozoite reservoir of Plasmodium vivax: the hidden obstacle to malaria elimination. Trends Parasitol. 2010;26:145–151. doi: 10.1016/j.pt.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 3.Burrows JN, Chibale K, Wells TN. The state of the art in anti-malarial drug discovery and development. Curr Top Med Chem. 2011;11:1226–1254. doi: 10.2174/156802611795429194. [DOI] [PubMed] [Google Scholar]
- 4.Drew ME, et al. Plasmodium food vacuole plasmepsins are activated by falcipains. J Biol Chem. 2008;283:12870–12876. doi: 10.1074/jbc.M708949200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fitch CD, et al. Lysis of Plasmodium falciparum by ferriprotoporphyrin IX and a chloroquine-ferriprotoporphyrin IX complex. Antimicrob Agents Chemother. 1982;21:819–822. doi: 10.1128/aac.21.5.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lehane AM, McDevitt CA, Kirk K, Fidock DA. Degrees of chloroquine resistance in Plasmodium—is the redox system involved? Int J Parasitol Drugs Drug Resist. 2012;2:47–57. doi: 10.1016/j.ijpddr.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fidock DA, et al. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol Cell. 2000;6:861–871. doi: 10.1016/s1097-2765(05)00077-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martin RE, et al. Chloroquine transport via the malaria parasite's chloroquine resistance transporter. Science. 2009;325:1680–1682. doi: 10.1126/science.1175667. [DOI] [PubMed] [Google Scholar]
- 9.Cowman AF, Karcz S, Galatis D, Culvenor JG. A P-glycoprotein homologue of Plasmodium falciparum is localized on the digestive vacuole. J Cell Biol. 1991;113:1033–1042. doi: 10.1083/jcb.113.5.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mu J, et al. Multiple transporters associated with malaria parasite responses to chloroquine and quinine. Mol Microbiol. 2003;49:977–989. doi: 10.1046/j.1365-2958.2003.03627.x. [DOI] [PubMed] [Google Scholar]
- 11.van Schalkwyk DA, Egan TJ. Quinoline-resistance reversing agents for the malaria parasite Plasmodium falciparum. Drug Resist Updat. 2006;9:211–226. doi: 10.1016/j.drup.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 12.Cooper RA, et al. Mutations in transmembrane domains 1, 4 and 9 of the Plasmodium falciparum chloroquine resistance transporter alter susceptibility to chloroquine, quinine and quinidine. Mol Microbiol. 2007;63:270–282. doi: 10.1111/j.1365-2958.2006.05511.x. [DOI] [PubMed] [Google Scholar]
- 13.Sá JM, et al. Geographic patterns of Plasmodium falciparum drug resistance distinguished by differential responses to amodiaquine and chloroquine. Proc Natl Acad Sci USA. 2009;106:18883–18889. doi: 10.1073/pnas.0911317106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De D, Krogstad FM, Cogswell FB, Krogstad DJ. Aminoquinolines that circumvent resistance in Plasmodium falciparum in vitro. Am J Trop Med Hyg. 1996;55:579–583. doi: 10.4269/ajtmh.1996.55.579. [DOI] [PubMed] [Google Scholar]
- 15.Hawley SR, et al. Manipulation of the N-alkyl substituent in amodiaquine to overcome the verapamil-sensitive chloroquine resistance component. Antimicrob Agents Chemother. 1996;40:2345–2349. doi: 10.1128/aac.40.10.2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Madrid PB, Liou AP, DeRisi JL, Guy RK. Incorporation of an intramolecular hydrogen-bonding motif in the side chain of 4-aminoquinolines enhances activity against drug-resistant P. falciparum. J Med Chem. 2006;49:4535–4543. doi: 10.1021/jm0600951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hwang JY, et al. Synthesis and evaluation of 7-substituted 4-aminoquinoline analogues for antimalarial activity. J Med Chem. 2011;54:7084–7093. doi: 10.1021/jm200636z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pou S, et al. Sontochin as a guide to the development of drugs against chloroquine-resistant malaria. Antimicrob Agents Chemother. 2012;56:3475–3480. doi: 10.1128/AAC.00100-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O'Neill PM, et al. Candidate selection and preclinical evaluation of N-tert-butyl isoquine (GSK369796), an affordable and effective 4-aminoquinoline antimalarial for the 21st century. J Med Chem. 2009;52:1408–1415. doi: 10.1021/jm8012618. [DOI] [PubMed] [Google Scholar]
- 20.Sowunmi A, et al. Predictors of the failure of treatment with chloroquine plus chlorpheniramine, in children with acute, uncomplicated, Plasmodium falciparum malaria. Ann Trop Med Parasitol. 2005;99:331–338. doi: 10.1179/136485905X36226. [DOI] [PubMed] [Google Scholar]
- 21.Zishiri VK, et al. Quinoline antimalarials containing a dibemethin group are active against chloroquinone-resistant Plasmodium falciparum and inhibit chloroquine transport via the P. falciparum chloroquine-resistance transporter (PfCRT) J Med Chem. 2011;54:6956–6968. doi: 10.1021/jm2009698. [DOI] [PubMed] [Google Scholar]
- 22.Burgess SJ, et al. A chloroquine-like molecule designed to reverse resistance in Plasmodium falciparum. J Med Chem. 2006;49:5623–5625. doi: 10.1021/jm060399n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dive D, Biot C. Ferrocene conjugates of chloroquine and other antimalarials: the development of ferroquine, a new antimalarial. ChemMedChem. 2008;3:383–391. doi: 10.1002/cmdc.200700127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mombo-Ngoma G, et al. Phase I randomized dose-ascending placebo-controlled trials of ferroquine–a candidate anti-malarial drug–in adults with asymptomatic Plasmodium falciparum infection. Malar J. 2011;10:53. doi: 10.1186/1475-2875-10-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mzayek F, et al. Randomized dose-ranging controlled trial of AQ-13, a candidate antimalarial, and chloroquine in healthy volunteers. PLoS Clin Trials. 2007;2:e6. doi: 10.1371/journal.pctr.0020006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goldberg DE, et al. Probing the chloroquine resistance locus of Plasmodium falciparum with a novel class of multidentate metal(III) coordination complexes. J Biol Chem. 1997;272:6567–6572. doi: 10.1074/jbc.272.10.6567. [DOI] [PubMed] [Google Scholar]
- 27.Yuan J, et al. Chemical genomic profiling for antimalarial therapies, response signatures, and molecular targets. Science. 2011;333:724–729. doi: 10.1126/science.1205216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Michel JB, Yeh PJ, Chait R, Moellering RC, Jr, Kishony R. Drug interactions modulate the potential for evolution of resistance. Proc Natl Acad Sci USA. 2008;105:14918–14923. doi: 10.1073/pnas.0800944105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chait R, Craney A, Kishony R. Antibiotic interactions that select against resistance. Nature. 2007;446:668–671. doi: 10.1038/nature05685. [DOI] [PubMed] [Google Scholar]
- 30.Klonis N, et al. Artemisinin activity against Plasmodium falciparum requires hemoglobin uptake and digestion. Proc Natl Acad Sci USA. 2011;108:11405–11410. doi: 10.1073/pnas.1104063108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hartwig CL, et al. Accumulation of artemisinin trioxane derivatives within neutral lipids of Plasmodium falciparum malaria parasites is endoperoxide-dependent. Biochem Pharmacol. 2009;77:322–336. doi: 10.1016/j.bcp.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kannan R, Sahal D, Chauhan VS. Heme-artemisinin adducts are crucial mediators of the ability of artemisinin to inhibit heme polymerization. Chem Biol. 2002;9:321–332. doi: 10.1016/s1074-5521(02)00117-5. [DOI] [PubMed] [Google Scholar]
- 33.White NJ. Qinghaosu (artemisinin): the price of success. Science. 2008;320:330–334. doi: 10.1126/science.1155165. [DOI] [PubMed] [Google Scholar]
- 34.Baird JK. Real-world therapies and the problem of vivax malaria. N Engl J Med. 2008;359:2601–2603. doi: 10.1056/NEJMe0808729. [DOI] [PubMed] [Google Scholar]
- 35.Dondorp AM, et al. Artesunate versus quinine in the treatment of severe falciparum malaria in African children (AQUAMAT): an open-label, randomised trial. Lancet. 2010;376:1647–1657. doi: 10.1016/S0140-6736(10)61924-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li GQ, Guo XB, Fu LC, Jian HX, Wang XH. Clinical trials of artemisinin and its derivatives in the treatment of malaria in China. Trans R Soc Trop Med Hyg. 1994;88(suppl. 1):S5–S6. doi: 10.1016/0035-9203(94)90460-x. [DOI] [PubMed] [Google Scholar]
- 37.Noedl H, et al. Evidence of artemisinin-resistant malaria in western Cambodia. N Engl J Med. 2008;359:2619–2620. doi: 10.1056/NEJMc0805011. [DOI] [PubMed] [Google Scholar]
- 38.Dondorp AM, et al. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2009;361:455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dondorp AM, et al. The threat of artemisinin-resistant malaria. N Engl J Med. 2011;365:1073–1075. doi: 10.1056/NEJMp1108322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Noedl H, Socheat D, Satimai W. Artemisinin-resistant malaria in Asia. N Engl J Med. 2009;361:540–541. doi: 10.1056/NEJMc0900231. [DOI] [PubMed] [Google Scholar]
- 41.Noedl H, et al. Artemisinin resistance in Cambodia: a clinical trial designed to address an emerging problem in Southeast Asia. Clin Infect Dis. 2010;51:e82–e89. doi: 10.1086/657120. [DOI] [PubMed] [Google Scholar]
- 42.Anderson TJ, et al. High heritability of malaria parasite clearance rate indicates a genetic basis for artemisinin resistance in western Cambodia. J Infect Dis. 2010;201:1326–1330. doi: 10.1086/651562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mok S, et al. Artemisinin resistance in Plasmodium falciparum is associated with an altered temporal pattern of transcription. BMC Genomics. 2011;12:391. doi: 10.1186/1471-2164-12-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheeseman IH, et al. A major genome region underlying artemisinin resistance in malaria. Science. 2012;336:79–82. doi: 10.1126/science.1215966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O'Neill PM, Posner GH. A medicinal chemistry perspective on artemisinin and related endoperoxides. J Med Chem. 2004;47:2945–2964. doi: 10.1021/jm030571c. [DOI] [PubMed] [Google Scholar]
- 46.Jefford CW. Synthetic peroxides as potent antimalarials. News and views. Curr Top Med Chem. 2012;12:373–399. doi: 10.2174/156802612799362940. [DOI] [PubMed] [Google Scholar]
- 47.Vennerstrom JL, et al. Identification of an antimalarial synthetic trioxolane drug development candidate. Nature. 2004;430:900–904. doi: 10.1038/nature02779. [DOI] [PubMed] [Google Scholar]
- 48.Charman SA, et al. Synthetic ozonide drug candidate OZ439 offers new hope for a single-dose cure of uncomplicated malaria. Proc Natl Acad Sci USA. 2011;108:4400–4405. doi: 10.1073/pnas.1015762108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Doerig C, et al. Malaria: targeting parasite and host cell kinomes. Biochim Biophys Acta. 2010;1804:604–612. doi: 10.1016/j.bbapap.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 50.Doerig C, et al. Protein kinases of malaria parasites: an update. Trends Parasitol. 2008;24:570–577. doi: 10.1016/j.pt.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 51.Kato N, et al. Gene expression signatures and small-molecule compounds link a protein kinase to Plasmodium falciparum motility. Nat Chem Biol. 2008;4:347–356. doi: 10.1038/nchembio.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lemercier G, et al. Identification and characterization of novel small molecules as potent inhibitors of the plasmodial calcium-dependent protein kinase 1. Biochemistry. 2009;48:6379–6389. doi: 10.1021/bi9005122. [DOI] [PubMed] [Google Scholar]
- 53.Le Roch K, et al. Activation of a Plasmodium falciparum cdc2-related kinase by heterologous p25 and cyclin H Functional characterization of a P. falciparum cyclin homologue. J Biol Chem. 2000;275:8952–8958. doi: 10.1074/jbc.275.12.8952. [DOI] [PubMed] [Google Scholar]
- 54.Xiao Z, Waters NC, Woodard CL, Li Z, Li PK. Design and synthesis of Pfmrk inhibitors as potential antimalarial agents. Bioorg Med Chem Lett. 2001;11:2875–2878. doi: 10.1016/s0960-894x(01)00578-9. [DOI] [PubMed] [Google Scholar]
- 55.Bouloc N, et al. Synthesis and in vitro evaluation of imidazopyridazines as novel inhibitors of the malarial kinase PfPK7. Bioorg Med Chem Lett. 2008;18:5294–5298. doi: 10.1016/j.bmcl.2008.08.043. [DOI] [PubMed] [Google Scholar]
- 56.Desoubzdanne D, et al. Alisiaquinones and alisiaquinol, dual inhibitors of Plasmodium falciparum enzyme targets from a New Caledonian deep water sponge. J Nat Prod. 2008;71:1189–1192. doi: 10.1021/np8000909. [DOI] [PubMed] [Google Scholar]
- 57.Laurent D, et al. Antimalarial potential of xestoquinone, a protein kinase inhibitor isolated from a Vanuatu marine sponge Xestospongia sp. Bioorg Med Chem. 2006;14:4477–4482. doi: 10.1016/j.bmc.2006.02.026. [DOI] [PubMed] [Google Scholar]
- 58.McRobert L, et al. Gametogenesis in malaria parasites is mediated by the cGMP-dependent protein kinase. PLoS Biol. 2008;6:e139. doi: 10.1371/journal.pbio.0060139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hayton K, et al. Erythrocyte binding protein PfRH5 polymorphisms determine species-specific pathways of Plasmodium falciparum invasion. Cell Host Microbe. 2008;4:40–51. doi: 10.1016/j.chom.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Baum J, et al. Reticulocyte-binding protein homologue 5—an essential adhesin involved in invasion of human erythrocytes by Plasmodium falciparum. Int J Parasitol. 2009;39:371–380. doi: 10.1016/j.ijpara.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 61.Chen L, et al. An EGF-like protein forms a complex with PfRh5 and is required for invasion of human erythrocytes by Plasmodium falciparum. PLoS Pathog. 2011;7:e1002199. doi: 10.1371/journal.ppat.1002199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Crosnier C, et al. Basigin is a receptor essential for erythrocyte invasion by Plasmodium falciparum. Nature. 2011;480:534–537. doi: 10.1038/nature10606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pizarro JC, et al. Crystal structure of the malaria vaccine candidate apical membrane antigen 1. Science. 2005;308:408–411. doi: 10.1126/science.1107449. [DOI] [PubMed] [Google Scholar]
- 64.Tyler JS, Boothroyd JC. The C-terminus of toxoplasma RON2 provides the crucial link between AMA1 and the host-associated invasion complex. PLoS Pathog. 2011;7:e1001282. doi: 10.1371/journal.ppat.1001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lamarque M, et al. The RON2–AMA1 interaction is a critical step in moving junction-dependent invasion by apicomplexan parasites. PLoS Pathog. 2011;7:e1001276. doi: 10.1371/journal.ppat.1001276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Srinivasan P, et al. Binding of Plasmodium merozoite proteins RON2 and AMA1 triggers commitment to invasion. Proc Natl Acad Sci USA. 2011;108:13275–13280. doi: 10.1073/pnas.1110303108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gao M, Skolnick J. The distribution of ligand-binding pockets around protein-protein interfaces suggests a general mechanism for pocket formation. Proc Natl Acad Sci USA. 2012;109:3784–3789. doi: 10.1073/pnas.1117768109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Singh S, Alam MM, Pal-Bhowmick I, Brzostowski JA, Chitnis CE. Distinct external signals trigger sequential release of apical organelles during erythrocyte invasion by malaria parasites. PLoS Pathog. 2010;6:e1000746. doi: 10.1371/journal.ppat.1000746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Leykauf K, et al. Protein kinase a dependent phosphorylation of apical membrane antigen 1 plays an important role in erythrocyte invasion by the malaria parasite. PLoS Pathog. 2010;6:e1000941. doi: 10.1371/journal.ppat.1000941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Aikawa M, Miller LH, Johnson J, Rabbege J. Erythrocyte entry by malarial parasites. A moving junction between erythrocyte and parasite. J Cell Biol. 1978;77:72–82. doi: 10.1083/jcb.77.1.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Riglar DT, et al. Super-resolution dissection of coordinated events during malaria parasite invasion of the human erythrocyte. Cell Host Microbe. 2011;9:9–20. doi: 10.1016/j.chom.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 72.Dvorin JD, et al. A plant-like kinase in Plasmodium falciparum regulates parasite egress from erythrocytes. Science. 2010;328:910–912. doi: 10.1126/science.1188191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kafsack BFC, et al. Rapid membrane disruption by a perforin-like protein facilitates parasite exit from host cells. Science. 2009;323:530–533. doi: 10.1126/science.1165740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Marsh K, et al. Indicators of life-threatening malaria in African children. N Engl J Med. 1995;332:1399–1404. doi: 10.1056/NEJM199505253322102. [DOI] [PubMed] [Google Scholar]
- 75.Planche T, et al. Assessment of volume depletion in children with malaria. PLoS Med. 2004;1:e18. doi: 10.1371/journal.pmed.0010018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Maitland K, et al. Mortality after fluid bolus in African children with severe infection. N Engl J Med. 2011;364:2483–2495. doi: 10.1056/NEJMoa1101549. [DOI] [PubMed] [Google Scholar]
- 77.Beare NA, et al. Prognostic significance and course of retinopathy in children with severe malaria. Arch Ophthalmol. 2004;122:1141–1147. doi: 10.1001/archopht.122.8.1141. [DOI] [PubMed] [Google Scholar]
- 78.Taylor TE, et al. Differentiating the pathologies of cerebral malaria by postmortem parasite counts. Nat Med. 2004;10:143–145. doi: 10.1038/nm986. [DOI] [PubMed] [Google Scholar]
- 79.Dondorp AM, et al. Direct in vivo assessment of microcirculatory dysfunction in severe falciparum malaria. J Infect Dis. 2008;197:79–84. doi: 10.1086/523762. [DOI] [PubMed] [Google Scholar]
- 80.Francischetti IMB, et al. Plasmodium falciparum-infected erythrocytes induce tissue factor expression in endothelial cells and support the assembly of multi-molecular coagulation complexes. J Thromb Haemost. 2007;5:155–165. doi: 10.1111/j.1538-7836.2006.02232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Taoufq Z, et al. Rho kinase inhibition in severe malaria: thwarting parasite-induced collateral damage to endothelia. J Infect Dis. 2008;197:1062–1073. doi: 10.1086/528988. [DOI] [PubMed] [Google Scholar]
- 82.Cabrales P, Zanini GM, Meays D, Frangos JA, Carvalho LJM. Murine cerebral malaria is associated with a vasospasm-like microcirculatory dysfunction, and survival upon rescue treatment is markedly increased by nimodipine. Am J Pathol. 2010;176:1306–1315. doi: 10.2353/ajpath.2010.090691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yeo TW, et al. Impaired nitric oxide bioavailability and l-arginine reversible endothelial dysfunction in adults with falciparum malaria. J Exp Med. 2007;204:2693–2704. doi: 10.1084/jem.20070819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lavstsen T, et al. Plasmodium falciparum erythrocyte membrane protein 1 domain cassettes 8 and 13 are associated with severe malaria in children. Proc Natl Acad Sci USA. 2012;109:E1791–E1800. doi: 10.1073/pnas.1120455109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Claessens A, et al. A subset of group A-like var genes encodes the malaria parasite ligands for binding to human brain endothelial cells. Proc Natl Acad Sci USA. 2012;109:E1772–E1781. doi: 10.1073/pnas.1120461109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Avril M, et al. A restricted subset of var genes mediates adherence of Plasmodium falciparum-infected erythrocytes to brain endothelial cells. Proc Natl Acad Sci USA. 2012;109:E1782–E1790. doi: 10.1073/pnas.1120534109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vouret-Craviari V, Grall D, Van Obberghen-Schilling E. Modulation of Rho GTPase activity in endothelial cells by selective proteinase-activated receptor (PAR) agonists. J Thromb Haemost. 2003;1:1103–1111. doi: 10.1046/j.1538-7836.2003.00238.x. [DOI] [PubMed] [Google Scholar]
- 88.Hemmer CJ, Kern P, Holst FG, Nawroth PP, Dietrich M. Neither heparin nor acetylsalicylic acid influence the clinical course in human Plasmodium falciparum malaria: a prospective randomized study. Am J Trop Med Hyg. 1991;45:608–612. doi: 10.4269/ajtmh.1991.45.608. [DOI] [PubMed] [Google Scholar]
- 89.Vogetseder A, Ospelt C, Reindl M, Schober M, Schmutzhard E. Time course of coagulation parameters, cytokines and adhesion molecules in Plasmodium falciparum malaria. Trop Med Int Health. 2004;9:767–773. doi: 10.1111/j.1365-3156.2004.01265.x. [DOI] [PubMed] [Google Scholar]
- 90.Francischetti IMB, et al. Defibrotide interferes with several steps of the coagulation-inflammation cycle and exhibits therapeutic potential to treat severe malaria. Arterioscler Thromb Vasc Biol. 2012;32:786–798. doi: 10.1161/ATVBAHA.111.240291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.de Mast Q, et al. ADAMTS13 deficiency with elevated levels of ultra-large and active von Willebrand factor in P. falciparum and P. vivax malaria. Am J Trop Med Hyg. 2009;80:492–498. [PubMed] [Google Scholar]
- 92.Bridges DJ, et al. Rapid activation of endothelial cells enables Plasmodium falciparum adhesion to platelet-decorated von Willebrand factor strings. Blood. 2010;115:1472–1474. doi: 10.1182/blood-2009-07-235150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Matsushita K, et al. Nitric oxide regulates exocytosis by S-nitrosylation of N-ethylmaleimide–sensitive factor. Cell. 2003;115:139–150. doi: 10.1016/s0092-8674(03)00803-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yeo TW, et al. Angiopoietin-2 is associated with decreased endothelial nitric oxide and poor clinical outcome in severe falciparum malaria. Proc Natl Acad Sci USA. 2008;105:17097–17102. doi: 10.1073/pnas.0805782105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Conroy AL, et al. Angiopoietin-2 levels are associated with retinopathy and predict mortality in Malawian children with cerebral malaria: a retrospective case-control study. Crit Care Med. 2012;40:952–959. doi: 10.1097/CCM.0b013e3182373157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Casals-Pascual C, et al. High levels of erythropoietin are associated with protection against neurological sequelae in African children with cerebral malaria. Proc Natl Acad Sci USA. 2008;105:2634–2639. doi: 10.1073/pnas.0709715105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kaiser K, et al. Recombinant human erythropoietin prevents the death of mice during cerebral malaria. J Infect Dis. 2006;193:987–995. doi: 10.1086/500844. [DOI] [PubMed] [Google Scholar]
- 98.Yeo TW, et al. Increased asymmetric dimethylarginine in severe falciparum malaria: association with impaired nitric oxide bioavailability and fatal outcome. PLoS Pathog. 2010;6:e1000868. doi: 10.1371/journal.ppat.1000868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Janka JJ, et al. Increased pulmonary pressures and myocardial wall stress in children with severe malaria. J Infect Dis. 2010;202:791–800. doi: 10.1086/655225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Walther M, et al. HMOX1 gene promoter alleles and high HO-1 levels are associated with severe malaria in Gambian children. PLoS Pathog. 2012;8:e1002579. doi: 10.1371/journal.ppat.1002579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Omodeo-Sale F, Cortelezzi L, Vommaro Z, Scaccabarozzi D, Dondorp A. Dysregulation of l-arginine metabolism and bioavailability associated to free plasma heme. Am J Physiol Cell Physiol. 2010;299:C148–C154. doi: 10.1152/ajpcell.00405.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kato GJ, et al. Endogenous nitric oxide synthase inhibitors in sickle cell disease: abnormal levels and correlations with pulmonary hypertension, desaturation, haemolysis, organ dysfunction and death. Br J Haematol. 2009;145:506–513. doi: 10.1111/j.1365-2141.2009.07658.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Davids M, et al. Role of the human erythrocyte in generation and storage of asymmetric dimethylarginine. Am J Physiol Heart Circ Physiol. 2012;302:H1762–H1770. doi: 10.1152/ajpheart.01205.2011. [DOI] [PubMed] [Google Scholar]
- 104.Wojciak-Stothard B, et al. The ADMA/DDAH pathway is a critical regulator of endothelial cell motility. J Cell Sci. 2007;120:929–942. doi: 10.1242/jcs.002212. [DOI] [PubMed] [Google Scholar]
- 105.Leiper J, et al. Disruption of methylarginine metabolism impairs vascular homeostasis. Nat Med. 2007;13:198–203. doi: 10.1038/nm1543. [DOI] [PubMed] [Google Scholar]
- 106.Jallow M, et al. Genome-wide and fine-resolution association analysis of malaria in West Africa. Nat Genet. 2009;41:657–665. doi: 10.1038/ng.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Reiter CD, et al. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med. 2002;8:1383–1389. doi: 10.1038/nm1202-799. [DOI] [PubMed] [Google Scholar]
- 108.Yeo TW, et al. Relationship of cell-free hemoglobin to impaired endothelial nitric oxide bioavailability and perfusion in severe falciparum malaria. J Infect Dis. 2009;200:1522–1529. doi: 10.1086/644641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Huie RE, Padmaja S. The reaction of NO with superoxide. Free Radic Res Commun. 1993;18:195–199. doi: 10.3109/10715769309145868. [DOI] [PubMed] [Google Scholar]
- 110.De Caterina R, et al. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J Clin Invest. 1995;96:60–68. doi: 10.1172/JCI118074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yang Y, Loscalzo J. Regulation of tissue factor expression in human microvascular endothelial cells by nitric oxide. Circulation. 2000;101:2144–2148. doi: 10.1161/01.cir.101.18.2144. [DOI] [PubMed] [Google Scholar]
- 112.Loscalzo J. Nitric oxide insufficiency, platelet activation, and arterial thrombosis. Circ Res. 2001;88:756–762. doi: 10.1161/hh0801.089861. [DOI] [PubMed] [Google Scholar]
- 113.Serirom S, et al. Anti-adhesive effect of nitric oxide on Plasmodium falciparum cytoadherence under flow. Am J Pathol. 2003;162:1651–1660. doi: 10.1016/S0002-9440(10)64299-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cosby K, et al. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat Med. 2003;9:1498–1505. doi: 10.1038/nm954. [DOI] [PubMed] [Google Scholar]
- 115.Huang Z, et al. Enzymatic function of hemoglobin as a nitrite reductase that produces NO under allosteric control. J Clin Invest. 2005;115:2099–2107. doi: 10.1172/JCI24650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gladwin MT, Kim-Shapiro D. The functional nitrite reductase activity of the heme-globins. Blood. 2008;112:2636–2647. doi: 10.1182/blood-2008-01-115261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Li H, Cui H, Kundu TK, Alzawahra W, Zweier JL. Nitric oxide production from nitrite occurs primarily in tissues not in the blood: critical role of xanthine oxidase and aldehyde oxidase. J Biol Chem. 2008;283:17855–17863. doi: 10.1074/jbc.M801785200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Minneci PC, et al. Nitrite reductase activity of hemoglobin as a systemic nitric oxide generator mechanism to detoxify plasma hemoglobin produced during hemolysis. Am J Physiol Heart Circ Physiol. 2008;295:H743–H754. doi: 10.1152/ajpheart.00151.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yu B, et al. Inhaled nitric oxide enables artificial blood transfusion without hypertension. Circulation. 2008;117:1982–1990. doi: 10.1161/CIRCULATIONAHA.107.729137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Duranski MR, et al. Cytoprotective effects of nitrite during in vivo ischemia-reperfusion of the heart and liver. J Clin Invest. 2005;115:1232–1240. doi: 10.1172/JCI22493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cauwels A, et al. Nitrite protects against morbidity and mortality associated with TNF- or LPS-induced shock in a soluble guanylate cyclase–dependent manner. J Exp Med. 2009;206:2915–2924. doi: 10.1084/jem.20091236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Erez A, et al. Requirement of argininosuccinate lyase for systemic nitric oxide production. Nat Med. 2011;17:1619–1626. doi: 10.1038/nm.2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Serghides L, et al. Inhaled nitric oxide reduces endothelial activation and parasite accumulation in the brain, and enhances survival in experimental cerebral malaria. PLoS ONE. 2011;6:e27714. doi: 10.1371/journal.pone.0027714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mack AK, et al. Sodium nitrite promotes regional blood flow in patients with sickle cell disease: a phase I/II study. Br J Haematol. 2008;142:971–978. doi: 10.1111/j.1365-2141.2008.07259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wondji CS, et al. Impact of pyrethroid resistance on operational malaria control in Malawi. Proc Natl Acad Sci USA. 2012;109:19063–19070. doi: 10.1073/pnas.1217229109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chong CR, Chen X, Shi L, Liu JO, Sullivan DJ., Jr A clinical drug library screen identifies astemizole as an antimalarial agent. Nat Chem Biol. 2006;2:415–416. doi: 10.1038/nchembio806. [DOI] [PubMed] [Google Scholar]
- 127.Weisman JL, et al. Searching for new antimalarial therapeutics amongst known drugs. Chem Biol Drug Des. 2006;67:409–416. doi: 10.1111/j.1747-0285.2006.00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Guiguemde WA, et al. Chemical genetics of Plasmodium falciparum. Nature. 2010;465:311–315. doi: 10.1038/nature09099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gamo FJ, et al. Thousands of chemical starting points for antimalarial lead identification. Nature. 2010;465:305–310. doi: 10.1038/nature09107. [DOI] [PubMed] [Google Scholar]
- 130.Rottmann M, et al. Spiroindolones, a potent compound class for the treatment of malaria. Science. 2010;329:1175–1180. doi: 10.1126/science.1193225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yuan J, et al. Genetic mapping of targets mediating differential chemical phenotypes in Plasmodium falciparum. Nat Chem Biol. 2009;5:765–771. doi: 10.1038/nchembio.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.van Pelt-Koops JC, et al. The spiroindolone drug candidate NITD609 potently inhibits gametocytogenesis and blocks Plasmodium falciparum transmission to anopheles mosquito vector. Antimicrob Agents Chemother. 2012;56:3544–3548. doi: 10.1128/AAC.06377-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Peatey CL, Spicer TP, Hodder PS, Trenholme KR, Gardiner DL. A high-throughput assay for the identification of drugs against late-stage Plasmodium falciparum gametocytes. Mol Biochem Parasitol. 2011;180:127–131. doi: 10.1016/j.molbiopara.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 134.Buchholz K, et al. A high-throughput screen targeting malaria transmission stages opens new avenues for drug development. J Infect Dis. 2011;203:1445–1453. doi: 10.1093/infdis/jir037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Eastman RT, et al. A class of tricyclic compounds blocking malaria oocyst development and transmission. Antimicrob Agents Chemother. 2013;57:425–435. doi: 10.1128/AAC.00920-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Burstein ES, et al. Integrative functional assays, chemical genomics and high throughput screening: harnessing signal transduction pathways to a common HTS readout. Curr Pharm Des. 2006;12:1717–1729. doi: 10.2174/138161206776873662. [DOI] [PubMed] [Google Scholar]
- 137.Goldstein DM, Gray NS, Zarrinkar PP. High-throughput kinase profiling as a platform for drug discovery. Nat Rev Drug Discov. 2008;7:391–397. doi: 10.1038/nrd2541. [DOI] [PubMed] [Google Scholar]
- 138.Coteron JM, et al. Structure-guided lead optimization of triazolopyrimidine-ring substituents identifies potent Plasmodium falciparum dihydroorotate dehydrogenase inhibitors with clinical candidate potential. J Med Chem. 2011;54:5540–5561. doi: 10.1021/jm200592f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Biagini GA, et al. Generation of quinolone antimalarials targeting the Plasmodium falciparum mitochondrial respiratory chain for the treatment and prophylaxis of malaria. Proc Natl Acad Sci USA. 2012;109:8298–8303. doi: 10.1073/pnas.1205651109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yeh E, DeRisi JL. Chemical rescue of malaria parasites lacking an apicoplast defines organelle function in blood-stage Plasmodium falciparum. PLoS Biol. 2011;9:e1001138. doi: 10.1371/journal.pbio.1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ralph SA, et al. Tropical infectious diseases: metabolic maps and functions of the Plasmodium falciparum apicoplast. Nat Rev Microbiol. 2004;2:203–216. doi: 10.1038/nrmicro843. [DOI] [PubMed] [Google Scholar]
- 142.Nguitragool W, et al. Malaria parasite clag3 genes determine channel-mediated nutrient uptake by infected red blood cells. Cell. 2011;145:665–677. doi: 10.1016/j.cell.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Nakazawa S, Kanbara H, Aikawa M. Plasmodium falciparum: recrudescence of parasites in culture. Exp Parasitol. 1995;81:556–563. doi: 10.1006/expr.1995.1149. [DOI] [PubMed] [Google Scholar]
- 144.Nakazawa S, Maoka T, Uemura H, Ito Y, Kanbara H. Malaria parasites giving rise to recrudescence in vitro. Antimicrob Agents Chemother. 2002;46:958–965. doi: 10.1128/AAC.46.4.958-965.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Thapar MM, Gil JP, Bjorkman A. In vitro recrudescence of Plasmodium falciparum parasites suppressed to dormant state by atovaquone alone and in combination with proguanil. Trans R Soc Trop Med Hyg. 2005;99:62–70. doi: 10.1016/j.trstmh.2004.01.016. [DOI] [PubMed] [Google Scholar]
- 146.Veiga MI, et al. Antimalarial exposure delays Plasmodium falciparum intra-erythrocytic cycle and drives drug transporter genes expression. PLoS ONE. 2010;5:e12408. doi: 10.1371/journal.pone.0012408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Hoshen MB, Na-Bangchang K, Stein WD, Ginsburg H. Mathematical modelling of the chemotherapy of Plasmodium falciparum malaria with artesunate: postulation of ‘dormancy’, a partial cytostatic effect of the drug, and its implication for treatment regimens. Parasitology. 2000;121:237–246. doi: 10.1017/s0031182099006332. [DOI] [PubMed] [Google Scholar]
- 148.Witkowski B, et al. Increased tolerance to artemisinin in Plasmodium falciparum is mediated by a quiescence mechanism. Antimicrob Agents Chemother. 2010;54:1872–1877. doi: 10.1128/AAC.01636-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Teuscher F, et al. Artemisinin-induced dormancy in Plasmodium falciparum: duration, recovery rates, and implications in treatment failure. J Infect Dis. 2010;202:1362–1368. doi: 10.1086/656476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Codd A, Teuscher F, Kyle DE, Cheng Q, Gatton ML. Artemisinin-induced parasite dormancy: a plausible mechanism for treatment failure. Malar J. 2011;10:56. doi: 10.1186/1475-2875-10-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Tucker MS, Mutka T, Sparks K, Patel J, Kyle DE. Phenotypic and genotypic analysis of in vitro–selected artemisinin-resistant progeny of Plasmodium falciparum. Antimicrob Agents Chemother. 2012;56:302–314. doi: 10.1128/AAC.05540-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Nosten F. Waking the sleeping beauty. J Infect Dis. 2010;202:1300–1301. doi: 10.1086/656478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Cheng Q, Kyle DE, Gatton ML. Artemisinin resistance in Plasmodium falciparum: a process linked to dormancy? Int J Parasitol Drugs Drug Resist. 2012;2:249–255. doi: 10.1016/j.ijpddr.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.LaCrue AN, Scheel M, Kennedy K, Kumar N, Kyle DE. Effects of artesunate on parasite recrudescence and dormancy in the rodent malaria model Plasmodium vinckei. PLoS ONE. 2011;6:e26689. doi: 10.1371/journal.pone.0026689. [DOI] [PMC free article] [PubMed] [Google Scholar]