

Abstract
Paenibacillus sp. strain JDR-2 (Paenibacillus JDR-2) secretes a multimodular cell-associated glycoside hydrolase family 10 (GH10) endoxylanase (XynA10A1) that catalyzes the depolymerization of methylglucuronoxylan (MeGXn) and rapidly assimilates the products of depolymerization. Efficient utilization of MeGXn has been postulated to result from the coupling of the processes of exocellular depolymerization and assimilation of oligosaccharide products, followed by intracellular metabolism. Growth and substrate utilization patterns with barley glucan and laminarin similar to those observed with MeGXn as a substrate suggest similar processes for 1,3-1,4-β-glucan and 1,3-β-glucan depolymerization and product assimilation. The Paenibacillus JDR-2 genome includes a cluster of genes encoding a secreted multimodular GH16 β-glucanase (Bgl16A1) containing surface layer homology (SLH) domains, a secreted GH16 β-glucanase with only a catalytic domain (Bgl16A2), transporter proteins, and transcriptional regulators. Recombinant Bgl16A1 and Bgl16A2 catalyze the formation of trisaccharides, tetrasaccharides, and larger oligosaccharides from barley glucan and of mono-, di-, tri-, and tetrasaccharides and larger oligosaccharides from laminarin. The lack of accumulation of depolymerization products during growth and a marked preference for polymeric glucan over depolymerization products support a process coupling extracellular depolymerization, assimilation, and intracellular metabolism for β-glucans similar to that ascribed to the GH10/GH67 xylan utilization system in Paenibacillus JDR-2. Coordinate expression of genes encoding GH16 β-glucanases, transporters, and transcriptional regulators supports their role as a regulon for the utilization of soluble β-glucans. As in the case of the xylan utilization regulons, this soluble β-glucan regulon provides advantages in the growth rate and yields on polymeric substrates and may be exploited for the efficient conversion of plant-derived polysaccharides to targeted products.
INTRODUCTION
The bioconversion of plant biomass to biofuels and chemicals depends on the saccharification of plant polysaccharides to fermentable hexoses and pentoses. Processes currently employed for the utilization of insoluble lignocellulosic biomass depend on thermochemical pretreatment followed by enzymatic saccharification to liberate the glucose from cellulose, the pentoses xylose and arabinose, and hemicelluloses (1, 2). The structural properties of cellulose define its role in plant cell walls and, through its interactions with hemicellulosic polysaccharides and lignin, its macrostructural properties related to plant development (3). The intrinsically insoluble 1,4-β-glucans that make up cellulose associate through hydrogen bonding to provide structures recalcitrant to enzymatic processing and present a technical challenge to the cost-effective processing of plant biomass to targeted products. The hemicellulosic methylglucuronoxylans (MeGXn) in dicots and methylglucuronoarabinoxylans (MeGAXn) in monocots may be solubilized by alkaline pretreatment followed by enzyme-mediated saccharification or may be directly hydrolyzed by a dilute acid to release fermentable pentoses (4). The cost of enzymes for the processing of cellulose as well as hemicellulose is a major factor in the development of economically acceptable protocols for the bioconversion of lignocellulosic biomass. A strategy for lowering this cost is the development of biocatalysts that produce the enzymes required for saccharification and ferment the saccharides released to targeted products. Such biocatalysts must secrete enzymes to release oligosaccharides, which would be imported, intracellularly converted to free sugars, and then fermented to a biofuel (e.g., ethanol or butanol) or a chemical feedstock (e.g., lactic or succinic acid). This strategy has been developed for the consolidated bioprocessing of cellulose by Clostridium species, in which cellulolytic activities provided by the cell-associated cellulosomes generate cellodextrins that are imported for glucose release and fermentation (5).
Systems for the consolidated processing of hemicelluloses may focus on the acidic xylans solubilized by alkaline pretreatment of lignocellulosic biomass to release MeGXn from dicots and MeGAXn from monocots. The glycoside hydrolase family 10 (GH10)/GH67 system defined in Paenibacillus sp. strain JDR2 (Paenibacillus JDR-2) includes a secreted cell-associated GH10 endoxylanase that generates xylooligosaccharides and the acidic aldouronate methylglucuronoxylotriose (MeGX3), ABC transporters, transcriptional regulators, and intracellular enzymes, including a GH67 α-glucuronidase, to release xylose. The secreted xylanase includes four carbohydrate binding modules (CBM) (1 CBM9 domain, putatively binding cellulose, and 3 CBM22 domains, putatively binding xylan) and three surface layer homology (SLH) domains for anchoring to the cell surface. During growth on MeGXn or MeGAXn, the absence of accumulated neutral xylooligosaccharides or acidic aldouronic xylooligosaccharides in the medium and the coordinate expression of genes comprising a xylan utilization regulon encoding these proteins support a process in which extracellular depolymerization, import of oligosaccharides, and intracellular metabolism are thermodynamically, if not mechanistically, coupled (6–9).
In the poaceous cereal crops, 1,3-1,4-β-glucans may make up a significant portion of the grain, where they may play a structural role and a potential storage role in plant development (10). As with the xylans of the hemicellulose fractions of biomass, these are relatively soluble and accessible to enzyme-mediated depolymerization. These polysaccharides have value as dietary fiber for humans and are of nutritional value for ruminants and other animals (11).
From the sequenced genome of Paenibacillus JDR-2, a gene (bgl16A1) encoding a secreted GH16 β-endoglucanase containing five CBM domains and 3 SLH domains and an adjacent gene (bgl16A2) encoding a secreted GH16 β-endoglucanase with only the catalytic domain were identified. In this study, recombinant β-glucanase 16A1 (rBgl16A1) is proposed to be a 1,3-β-glucanase (EC 3.2.1.39) with activity on laminarin as well as some activity on barley glucan. Recombinant β-glucanase 16A2 (rBgl16A2) is proposed to be a 1,3(4)-β-glucanase (EC 3.2.1.6) with activity on barley glucan as well as some activity on laminarin. These two enzymes generated similar oligosaccharide products from laminarin and barley glucan. The preferential growth rates on both polysaccharides relative to those on cellobiose and glucose, and the coordinate expression of a cluster of genes encoding Bgl16A1, Bgl16A2, an ABC transporter, and transcriptional regulators, support the presence of a regulon for the efficient utilization of soluble glucan, similar to the xylan utilization regulon described previously (7). To the extent that 1,3-1,4-β-glucans make up a significant portion of the grains of poaceous cereal crops, including barley, wheat, and oats, the β-glucanase regulon in Paenibacillus JDR-2 may serve as a model for the development of systems for the bioconversion to biofuels and chemical feedstocks of those portions of commodity crops that are in surplus relative to their current applications. The ability to process laminarin efficiently also provides an opportunity to process a surplus from industries utilizing brown algae as a resource.
MATERIALS AND METHODS
Growth and cultures of Paenibacillus sp. strain JDR-2.
Fresh Paenibacillus JDR-2 colonies were obtained by streaking a 1:1 dilution of a glycerol stock of Paenibacillus JDR-2, passage 20, onto a 1.5% (wt/vol) agar plate containing 0.1% (wt/vol) yeast extract (YE), 0.5% (wt/vol) oat spelt xylan, and Zucker Hankin (ZH) salts medium (6, 12). One Paenibacillus JDR-2 colony was picked and was inoculated into 300 μl of 0.2% (wt/vol) yeast extract in ZH salts medium. This culture was incubated at 30°C with shaking to the mid-log-growth phase and was then added to 5.4 ml of 0.2% (wt/vol) yeast extract in ZH salts. At mid-log growth, 250 μl of this culture was inoculated into 5 ml of culture media containing yeast extract (0.01%) and various carbohydrate sources (0.2% [wt/vol]) in ZH salts. Duplicate cultures were incubated at 30°C with shaking. Aliquots of 150 μl, removed from each culture during the course of bacterial growth, were centrifuged for 2 min at 12,000 × g. The supernatants were removed and saved, and the cell pellets were rinsed with 150 μl water, recentrifuged, and saved. The total carbohydrate content in the medium was determined by the phenol-sulfuric acid method (13). The protein content of the cell pellets was determined by the Bradford assay (14). Thin-layer chromatography (TLC) for the detection of substrate processing was performed as described previously (6).
For RNA extraction studies, a single Paenibacillus JDR-2 colony picked from an agar plate of 0.5% (wt/vol) oat spelt xylan–0.1% YE–ZH salts was dispersed into 5 ml of 0.2% (wt/vol) yeast extract in 1× ZH salts, and the culture was incubated at 30°C with shaking at 220 rpm. At mid-log growth, 150 μl of the culture was inoculated into 5 ml of culture media containing different carbohydrate sources. To ensure adequate levels of RNA for isolation, cultures contained ZH salts medium with 0.4% (wt/vol) YE with or without 0.4% (wt/vol) of the carbohydrate source. At the mid-log-growth phase, 4 ml of culture was removed, the cells were pelleted, and RNA was prepared using the RNAqueous kit from Ambion (catalog no. AM1912; Life Technologies). The RNA was twice treated with Turbo DNase (catalog no. AM2238; Life Technologies) to remove genomic DNA. The absence of genomic DNA was confirmed by performing control reverse transcription (RT)-PCRs with no added RNA.
qRT-PCR.
Quantitative RT-PCR (qRT-PCR) was performed as described previously (7). Quantitative RT-PCRs were set up using the iScript One-Step RT-PCR kit with SYBR green (catalog no. 170-8892; Bio-Rad). The protocol was as follows: 1 cycle at 50°C for 10 min; 1 cycle at 96°C for 5 min; 40 cycles at 95°C for 10 s and 60°C for 30 s; 1 cycle at 95°C for 1 min and 55°C for 1 min; and a melt curve analysis for 10 s starting at 55°C with a 0.5°C increment to 95°C.
Quantitative RT-PCR data were analyzed by using the absolute quantification method as described in reference 15. Briefly, a known amount of Paenibacillus JDR-2 genomic DNA was amplified by PCR using primers designed to amplify 200- to 250-bp segments of the genes of interest. Test PCR protocols were as follows: 1 cycle at 95°C for 1.5 min; 40 cycles at 95°C for 10 s, 58°C for 20 s, and 72°C for 20 s; 95°C for 1 min; 55°C for 1 min; and a melt curve analysis with 95°C denaturation for 1 min, 55°C annealing for 1 min, and 80 cycles of 0.5°C increments (10 s each) beginning at 55°C. Tenfold dilutions of genomic molecules, ranging from 102 to 107 molecules, were added as the template to the reaction mixtures, and the threshold cycle (CT) values for the detection of products were plotted against the logarithmic values of the initial numbers of copies of the template added. Amplification efficiency (E) was calculated from the slope of the curve using the formula E = 10−1/slope, and the percentage of efficiency was calculated as (E − 1) × 100%. Several sets of primer pairs for each gene under study were designed and were evaluated to provide 90 to 105% efficiency. A standard curve of 90 to 100% efficiency was thus generated for each optimal primer pair for each gene. Linear regression (y = mx + b) was used to calculate the number of molecules of RNA present in the sample, where y is the CT value, m is the slope, x is the logarithmic value of the number of molecules of RNA, and b is the intercept.
Molecular cloning of Bgl16A1CD and Bgl16A2.
The catalytic domain of β-glucanase 16A1 (Bgl16A1CD) was cloned by PCR amplification of the fragment of bgl16A1 from nucleotide 865 to nucleotide 2028 of the coding sequence, encompassing the peptide from amino acid residue 289 to amino acid residue 676 of the protein as the catalytic domain without the carbohydrate binding modules and the SLH domains that function in cell association. The amplified product was ligated into the NdeI and BamHI restriction sites of the pET15b vector (catalog no. 69661; Novagen). The β-glucanase catalytic domain identified by NCBI annotation spans the region from amino acid residue 390 to amino acid residue 622. Likewise, β-glucanase 16A2 (Bgl16A2) including the catalytic domain without the secretion sequence was cloned by PCR amplification of the fragment of bgl16A2 from nucleotide 76 to nucleotide 714 of the coding sequence, encompassing the peptide from amino acid 26 to amino acid 237, and the amplified product was ligated into the NdeI and BamHI restriction sites of the pET15b vector.
Escherichia coli Rosetta 2 cells were transformed with the ligation products, and the transformants were selected in Luria broth (LB) containing ampicillin and chloramphenicol. The Rosetta 2 strains containing the Bgl16A1 or Bgl16A2 protein expression vector were grown in 500-ml cultures of LB containing ampicillin (100 μg/ml) and chloramphenicol (15 μg/ml) in 2.8-liter Fernbach flasks at 37°C with shaking at 250 rpm to an optical density at 600 nm (OD600) of 0.8 (as determined with a Beckman DU640 spectrophotometer). Isopropyl β-d-1-thiogalactopyranoside was added to a final concentration of 0.1 mM. After 4 h of incubation at room temperature with shaking, cells were harvested by centrifugation (10,000 × g, 10 min, 4°C), washed twice with 25 ml of 20 mM sodium phosphate buffer (pH 7.4), and resuspended in 20 ml of the same buffer. Cells were passed through a French pressure cell at 16,000 lb/in2. The crude extract was clarified by centrifugation (30,000 × g, 45 min), and the supernatant was filtered through a 0.22-μm filter. The filtered protein solution was loaded onto a HiTrap chelating column in the Ni form (5 ml; GE Life Sciences). After washing with 10 column volumes of phosphate buffer with 0.5 M NaCl, followed by 10 column volumes of phosphate buffer with 50 mM imidazole and 0.5 M NaCl, His-tagged Bgl16A1CD and Bgl16A2 proteins were eluted with 0.5 M imidazole in phosphate buffer with 0.5 M NaCl. Imidazole was removed from the sample by use of a PD-10 column (GE Life Sciences) eluted with 0.05 M sodium acetate (pH 6.0). Purified proteins were analyzed by SDS-PAGE. For evaluation of the effect of the His tag on catalytic activities, the proteins encoded by recombinant bgl genes that contained an N-terminal thrombin cleavage site were treated with thrombin (Thrombin Cleavage Capture kit; catalog no. 69022-3) from Novagen to remove the His tags. After the cleavage reaction, biotinylated thrombin was removed with streptavidin agarose and the target protein recovered by spin filtration. Preparations were evaluated for purity by SDS-PAGE, and their enzyme activities on barley glucan (BG) and laminarin (LN) were compared with those of Bgl16A1CD and Bgl16A2 containing His tags.
Comparison of BglA1CD and Bgl16A2 activities.
Activities for the generation of reducing termini as a measure of glycosidic bond cleavage were determined using glucose as a standard (6). Reaction mixtures containing 0.05 M sodium acetate buffer (pH 6.0), 0.2% BG or LN, and the enzyme were incubated at 30°C, and samples were removed for assay every 5 min. With linear rates of activity over 30 min or more and rates proportional to the amount of enzyme added, the conditions provided zero-order kinetics approximating substrate saturation, with rates approaching Vmax. One unit of activity generates the equivalent of 1 μmol of glucose-reducing termini per min at 30°C. Protein content was determined by the bicinchoninic acid (BCA) assay as described previously (8). Specific activities are presented as units per milligram of protein. For the determination of products of digestion, 0.01 U of each His-tagged enzyme (0.01 and 0.1 U for Bgl16A2 with laminarin) was incubated for 24 h at 30°C in 1.0-ml reaction mixtures containing 0.5% BG or LN in 0.05 M sodium acetate buffer. Samples (0.01 ml) were spotted onto silica gel G plates, developed two times in ethyl acetate–acetic acid–water (2:1:1), and treated for detection as described previously (8).
RESULTS
Gene neighborhood of GH16 endoglucanase genes.
Recent whole-genome sequencing of Paenibacillus JDR-2 made it possible to locate resident glycoside hydrolase genes (26). In that study on glucan utilization, two adjacent GH16 endoglucanase genes, flanked by three genes that encode an ABC transporter and a pair of transcriptional regulators, were found in a cassette (Fig. 1A). The bgl16A1 gene encodes a large multimodular protein that begins with a secretion signal peptide at the N terminus, followed sequentially by three S-layer homology domains, a GH16 endoglucanase catalytic domain, and five carbohydrate binding domains at the carboxyl terminus. The bgl16A2 gene encodes a smaller protein containing only an amino-terminal secretion signal peptide followed by a catalytic domain (Fig. 1B). The modular composition of the Bgl16A1 protein including 3 SLH domains suggests a similarity to the secreted Xyn10A1 protein, which has been implicated in the efficient utilization of methylglucuronoxylans (6–8).
FIG 1.
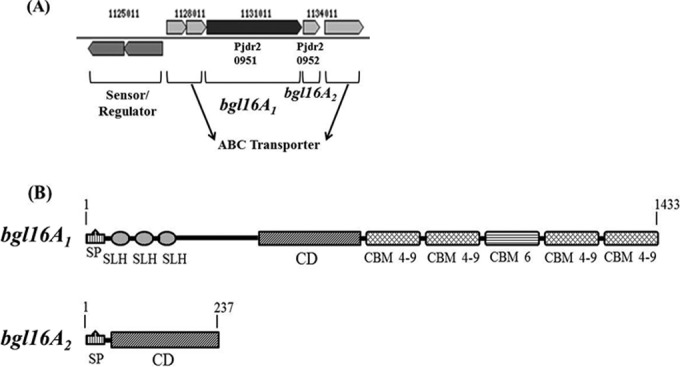
GH16 glucanase gene neighborhood and conserved domain organizations. (A) Gene neighborhood of bgl16A1 and bgl16A2. (B) Domain architecture of Bgl16A1. Amino acid residue numbers are given above the gene maps. SP, signal peptide; SLH, S-layer homology domain; CD, catalytic domain; CBM 4-9, carbohydrate binding domain, families 4 and 9; CBM 6, carbohydrate binding domain, family 6.
Utilization of soluble β-glucans by Paenibacillus JDR-2.
Figure 2A shows the growth of Paenibacillus JDR-2 in 0.2% (wt/vol) barley glucan, laminarin, cellobiose, or glucose. In barley glucan, Paenibacillus JDR-2 grew rapidly, reaching an optical density at 600 nm (OD600) of 0.86 after 20 h. In laminarin, growth was less rapid, reaching an OD600 of 0.72 after 25 h. However, growth in cellobiose and glucose was much slower and less robust, reaching an OD600 of 0.53 for each of these substrates after 31 h of growth. Figure 2B shows the remaining substrates and their depolymerized products in the media at different phases in the growth processes shown in Fig. 2A. In media containing polymeric substrates at the exponential-growth phase (Fig. 2B), oligomers released from the depolymerization of barley glucan (lane 7) and laminarin (lane 17) were observed along with small amounts of cellobiose, laminaribiose, and glucose. At the end of the declining growth phase (Fig. 2B, lanes 9 and 19), all carbohydrate molecules had been consumed. Oligosaccharides accumulated at higher levels in the mid-log-growth phase with barley glucan (13 h) than with laminarin (16 h). In media containing cellobiose, there was a steady but slow decrease in the amount of the substrate with time (Fig. 2B, lanes 10 to 14).
FIG 2.

Growth measured by turbidity and the utilization of different carbohydrate substrates by Paenibacillus JDR-2. (A) Duplicate 5-ml cultures of 0.01% yeast extract in Zucker Hankin salts medium supplemented with 0.2% carbohydrate (□, barley glucan; △, laminarin; ○, cellobiose; ♢, glucose; ×, no carbohydrate) were inoculated with 250 μl of Paenibacillus JDR-2 cells (OD600, 0.2) and were incubated at 30°C with shaking at 220 rpm. (B) TLC analysis of carbohydrate contents in the growth media of Paenibacillus JDR-2 cultures. Standards spotted were glucose (G) (4 μg), laminarin (LN) (4 μg; spotted with G), barley glucan (BG) (2 μg), cellobiose (CB) (4 μg), and laminaribiose (LB) (4 μg). Samples (18 μl) in media cleared by centrifugation were spotted for each time point (36 μg of the carbohydrate substrate at time zero). The developing solvent was chloroform–acetic acid–water (6:7:1).
These bacterial growth trends are reflected in the temporal relationships between the consumption of carbohydrates and the corresponding increase in bacterial cell mass, shown in Fig. 3. In media containing polymeric barley glucan or laminarin, maximal growth yield was achieved at 20 h, with maximal bacterial cell protein produced: 0.23 mg/ml with BG (Fig. 3A) and 0.13 mg/ml with LN (Fig. 3B). At this time point, a residual amount of 20% barley glucan or 40% laminarin remained in the medium. These residual carbohydrate contents consist of the input substrate (barley glucan or laminarin) and polymeric breakdown products of the oligosaccharides cellobiose and glucose (Fig. 2B, lanes 8 and 18). In cellobiose or glucose, maximal growth was reached later, at 25 h, with a maximal level of 0.12 mg/ml protein synthesized (Fig. 3C and D). At the point of maximal growth yield, a residual amount of 40% cellobiose or 38% glucose remained in the medium, most of which was subsequently consumed. The maximal rates of increase in turbidity (Fig. 2A) and the level of cell protein (Fig. 3) track the rate of utilization of total carbohydrate as a measure of the depolymerization of barley glucan and laminarin and the assimilation of depolymerization products (Table 1). The increase in the protein content as a measure of biomass is likely a more reliable measure of this relationship, in which the assimilation and metabolism of saccharides derived from polysaccharide depolymerization are significantly more efficient than for the disaccharide cellobiose and the monosaccharide glucose.
FIG 3.

Growth (amount of protein, measured in milligrams per milliliter [□]) and utilization of barley glucan (A), laminarin (B), cellobiose (C), or glucose (D) (measured in glucose equivalents [△]). Data points with error bars represent the averages for duplicate cultures.
TABLE 1.
Comparison of growth rates with different substrates, determined as increases in turbidity and protein content and rates of substrate utilizationa
| Substrate | ΔOD600/hb | ΔProtein (mg/ml/h)c | ΔCH2O/hd |
|---|---|---|---|
| Barley glucan | 0.116 (4.83) | 0.023 (3.2) | 0.154 (3.08) |
| Laminarin | 0.058 (2.0) | 0.014 (2.0) | 0.111 (2.22) |
| Cellobiose | 0.024 (1.0) | 0.011 (1.4) | 0.071 (1.42) |
| Glucose | 0.024 (1.0) | 0.0071 (1.0) | 0.050 (1.0) |
Numbers in parentheses in each column are the fold increase over the value with glucose.
Rate of maximum increase in turbidity, measured as the increase in the optical density at 600 nm per hour (Fig. 2A).
Rate of maximum increase in the protein content of the cell per 1.0 ml of culture (Fig. 3).
Rate of maximum decrease in total carbohydrate (CH2O) levels per 1.0 ml of culture (Fig. 3).
Enzymatic activities of Bgl16A1 and Bgl16A2.
To determine which genes were responsible for the depolymerization and utilization of barley glucan (BG) and laminarin (LN), the sequence of bgl16A1 encoding the catalytic domain (Bgl16A1CD) of native multimodular Bgl16A1 and the sequence of bgl16A2 encoding the complete Bgl16A2 protein minus the signal sequence were cloned into an E. coli expression vector, and the purified recombinant proteins were tested for their enzymatic activities on barley glucan and laminarin. Figure 4 shows that both the His-tagged recombinant form of Bgl16A1CD (molecular size by gene translation, 45.6 kDa) and that of Bgl16A2 (molecular size by translation, 24.3 kDa) show single bands on SDS-PAGE, of 45 and 24 kDa, respectively. After treatment with thrombin, the protein bands for both enzymes are shifted to a slightly lower molecular size, reflecting the removal of the His tag-containing peptide (with a molecular size of 1.9 kDa). Table 2 compares the activities of rBgl16A1CD and rBgl16A2 with and without His tags for the release of reducing termini from BG and LN. Both enzymes show similar activities with BG or LN before and after the removal of the His tag, indicating that the His tag does not significantly interfere with the activities of these enzymes with respect to the release of reducing termini. Bgl16A1CD shows similar, relatively low levels of activity with BG and LN, whereas Bgl16A2 shows much stronger activity with BG as the substrate than with LN.
FIG 4.
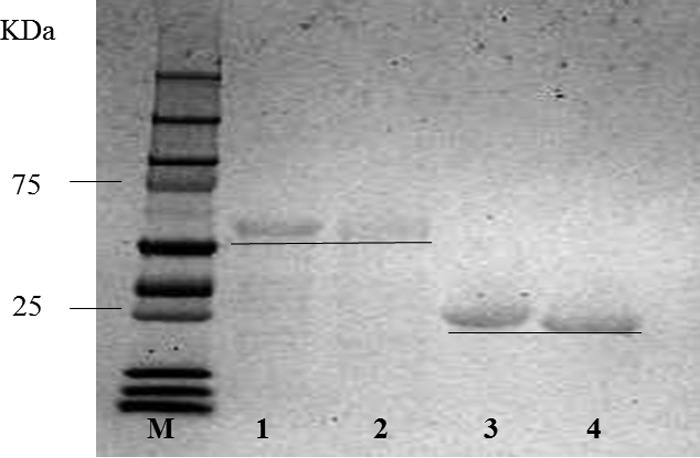
SDS-PAGE analysis of preparations of rBglA16A1CD and rBcl16A2 before and after the removal of His tags with thrombin as described in Materials and Methods. Lane M, molecular size standards; lane 1, rBgl16A1CD with a His tag; lane 2, rBgl16A1CD without a His tag; lane 3, rBgl16A2 with a His tag; lane 4, rBgl16A2 without a His tag.
TABLE 2.
Comparison of specific enzyme activities of recombinant forms of Bgl16A1CD and Bgl16A2 with and without His tags, with barley glucan or laminarin as the substrate
| Substrate | Sp act (U/mg of protein)a (±SE) |
|||
|---|---|---|---|---|
| Bgl16A1CD |
Bgl16A2 |
|||
| With His tag | Without His tag | With His tag | Without His tag | |
| Barley glucan | 31.5 (±0.7) | 28.0 (±1.6) | 686 (±18.5) | 421 (±23) |
| Laminarin | 6.10 (±0.3) | 6.90 (±0.3) | 1.1 (±0.7) | 1.9 (±0.1) |
Determined by the release of reducing termini with constant rates of release over 30 min, as described in Materials and Methods. One unit of activity is defined as the release of the equivalent of 1 μmol of glucose-reducing termini/min at 30°C. Standard errors are based on duplicate assays.
rBgl16A1CD and rBgl16A2 generated the same products from barley glucan, including prominent levels of oligosaccharides with degrees of depolymerization of 3, 4, 5, 6, or more and traces of di- and monosaccharides (Fig. 5, lanes 5 to 7). On the other hand, rBgl16A1 digested laminarin to release two prominent oligosaccharide products as well as laminaribiose and glucose, and unresolved larger products (Fig. 5 lane 9). Based on the intensities of resolved products, the rBgl16A1 catalytic domain and rBgl16A2 generate similar levels of the same products from barley glucan (Fig. 5, lanes 5 to 7). With laminarin as the substrate, 0.01 U rBgl16A2 generated barely detectable quantities of products similar to some of those generated by rBgl16A1 (Fig. 5, lane 10). With 0.1 U of Bgl16A2, the 24-h digestion pattern obtained with laminarin was similar to that obtained with 0.01 U of Bgl16A1CD (Fig. 5, lane 12).The combined activities of rBgl16A1CD and rBgl16A2 (Fig. 5, lane 11) on laminarin resemble the activity obtained with rBgl16A1 alone.
FIG 5.
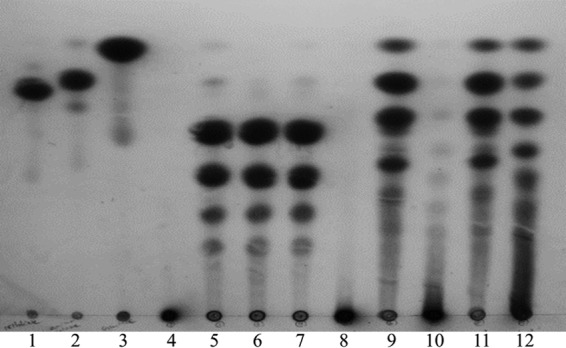
TLC products generated by rBgl16A1 and rBgl16A2. Reaction mixtures (100 μl) containing 0.01 U of purified enzymes (determined by the release of reducing termini from BG and LN separately) and either 0.5% barley glucan or 0.5% laminarin in 50 mM sodium acetate (pH 6.0) were incubated at 37°C for 24 h. rBgl16A2 was also added at 0.1 U for the digestion of LN. Samples (10 μl) were spotted onto TLC plates. The plates were developed twice with ethyl acetate–acetic acid–water (2:1:1). Samples of barley glucan (BG) (lanes 4 to 7) and laminarin (LN) (lanes 8 to 11) included no enzyme (lanes 4 and 8), rBgl16A1CD (lanes 5 and 9), 0.01 U rBgl16A2 (lanes 6 and 10), both enzymes (lanes 7 and 11), and 0.1 U rBgl16A2 with LN (lane 12). Samples containing 10 nmol of cellobiose (lane 1), laminaribiose (lane 2), or glucose (lane 3) were run as standards.
qRT-PCR analysis of β-glucanase gene expression.
As described above (Fig. 1), the bgl16A1 and bgl16A2 genes are found in an operon flanked by genes encoding transcriptional regulators and transporter proteins. In consideration of the rapid uptake and growth of Paenibacillus JDR-2 in barley glucan and laminarin, compared to the utilization of cellobiose (Fig. 2 and 3) derived from the action of both Bgl16A1CD and BglA2 in barley glucan (Fig. 5), qRT-PCR analyses of the effects of growth with these different substrates on the expression of these genes were performed. Laminaribiose was also evaluated as an expected product of the action of both enzymes on laminarin (Fig. 2B and 5). The level of bgl16A1 transcription was higher when Paenibacillus JDR-2 was grown in barley glucan (445-fold) or laminarin (131-fold) than that obtained when the organism was grown in yeast extract alone. bgl16A1 expression was reduced when Paenibacillus JDR-2 was grown in cellobiose (0.3-fold) and was unchanged in glucose (1.1-fold) (Fig. 6B). Similarly, the level of bgl16A2 transcription was increased in barley glucan (54-fold) and laminarin (14-fold) but was reduced in either cellobiose (0.2-fold) or glucose (0.2-fold) from that found when Paenibacillus JDR-2 was grown in yeast extract alone (Fig. 6B). As shown in Fig. 6B, sbp(0953) expression increased 1,342-fold or 824-fold with growth in barley glucan or laminarin, respectively, and was reduced half or more with growth in cellobiose (0.4-fold) or glucose (0.1-fold) from that with growth in yeast extract alone. The expression of the araC(0947) gene was increased slightly by growth in barley glucan (5.5-fold) or laminarin (2.5-fold) and was unchanged with growth in cellobiose (1.0-fold) or glucose (0.9-fold) (Fig. 6B). The pyruvate kinase gene pk(4477), a housekeeping gene analyzed as a control, was evenly expressed (0.9- to 1.2-fold) with growth in the different substrates as well as in yeast extract alone (Fig. 6B), in contrast to the genes in the glucanase cassette. Growth in laminaribiose increased the expression of bgl16A1 (84.9-fold), bgl16A2 (31.2-fold), and sbp(0953) (622-fold) (Fig. 6B), an expression profile similar to that elicited by polymeric barley glucan and laminarin rather than to that elicited by cellobiose or glucose.
FIG 6.

RNA (100 ng) prepared from cells grown in 5-ml cultures containing either 0.4% yeast extract (YE) alone or 0.4% yeast extract and 0.4% carbohydrate substrate (BG, barley glucan; LN, laminarin; LB, laminaribiose; CB, cellobiose; G, glucose) in Zucker-Hankin medium was added to 8 μl of a 2× SYBR green mixture with deoxynucleoside triphosphates and 0.5 μM (each) forward and reverse primers in a final reaction mixture volume of 16 μl. One unit of reverse transcriptase was added as needed. PCR conditions were those described in Materials and Methods. (A) Numbers of RNA molecules detected in cell cultures grown in the following carbohydrate substrates: YE without an additional carbon source (open bars), YE with BG (dark crosshatched bars), YE with LN (light crosshatched bars), YE with LB (stippled bars), YE with CB (horizontally striped bars), and YE with G (dark shaded bars). Bars represent averages for duplicate PCRs with the same RNA preparations; error bars define the ranges of values. (B) Ratio of the number of expressed RNA molecules of each gene indicated in a culture grown in yeast extract with BG, LN, LB, CB, or G to that of the same gene in a culture grown in yeast extract only. (C) Cassette comprising the glucanase genes.
DISCUSSION
As shown in Fig. 2A, Paenibacillus JDR-2 exhibits a general trend of rapid and robust growth in polymeric glucans and slower, reduced growth in the disaccharide cellobiose and the monosaccharide glucose. Growth in barley glucan proceeds through a phase of generation of oligosaccharides followed by their nearly complete consumption (Fig. 2B, lanes 5 to 9). The residual amount of oligosaccharides (Fig. 2B, lane 8) at the peak of growth at 20 h (Fig. 2A, YE+BG) corresponds to the remaining 20% of the substrate initially added and represents products from the dual hydrolytic actions of secreted Bgl16A1 and Bgl16A2 on the barley glucan detected in the medium. Barley glucan, a polymer of β-1,4-linked d-glucose residues, is interspersed with β-1,3 linkages. The largest portion of this substrate has β-1,3 linkages every 3 residues, with >80% containing a spacing of ≤4 residues (16, 17). When incubated in an enzymatic reaction mixture with rBgl16A1CD and rBgl16A2, this polymer is hydrolyzed to trimeric and larger oligosaccharides (Fig. 5). Bgl16A1 is a multimodular protein with carbohydrate binding domains that secure the barley glucan molecules to the enzyme and SLH domains that are known to anchor the enzyme to the bacterial cell surfaces (6, 18). The structural features of Bgl16A1 would then sequester the enzyme-polysaccharide complex to the cell surface, allowing the released oligosaccharide products to be transported rapidly into the cell. Bgl16A2 contains only the catalytic domain, and the oligosaccharide products produced by Bgl16A2 may disperse freely into the culture medium. It is likely the oligosaccharides generated from barley glucan that are detected at 20 h (Fig. 2B) are mainly the products of Bgl16A2.
The growth of Paenibacillus JDR-2 is slightly less rapid and robust in laminarin than in barley glucan (Fig. 2A). At the peak of growth, at 25 h (Fig. 2A, LN with yeast extract), only small amounts of oligosaccharide products are detected in the medium (Fig. 2B, Laminarin). The laminarin from Laminaria digitata, used in this study, contains primarily β-1,3-linked glucose and some β-1,6 intrastrand linkages and branch points (19). When incubated with rBgl16A1CD, laminarin is hydrolyzed to monomeric, dimeric, and larger oligosaccharides (Fig. 5). In growing cultures, the process is similar to that described above: products released by the multimodular protein Bgl16A1 are rapidly transported into the cell, while products released by Bgl16A2 diffuse freely in the medium. However, since rBgl16A2 is only slightly active on laminarin compared to its activity on barley glucan (Table 2; Fig. 5), fewer oligosaccharide products should be freely released into the medium with laminarin as the substrate, as is seen in Fig. 2B.
Comparison of the Clustal W alignment of the catalytic residues of these two proteins with those in the Bacillus species aligned by Hahn et al. (20) and consideration of the detailed analyses of an 1,3-β-endoglucanase, LamA, in Paenibacillus sp. strain CCRC 17245 (21) and an 1,3(4)-β-endoglucanase in Paenibacillus sp. strain F-40 (22) lead us to the conclusion that Bgl16A1 is a laminarinase (1,3-β-endoglucanase) that also displays activity on barley glucan, while Bgl16A2 is a lichenase with 1,3(4)-β-endoglucanase activity. Bgl16A1 is similar to the 1,793-amino-acid multimodular endoglucanase LamA identified in Paenibacillus sp. strain CCRC 17245 (21). A recombinant catalytic domain from the gene encoding this enzyme showed specific activities of 1.4 μmol reducing sugar/min/nmol enzyme for LN and 0.4 μmol reducing sugar/min/nmol enzyme for BG, which translate to 31 U/mg for LN and 8.85 U/mg for BG. Bgl16A1CD shows a specific activity of 32 U/mg enzyme with BG as the substrate and 31 U/mg with laminarin. This recombinant catalytic domain (molecular size, 45 kDa) within the multimodular LamA enzyme from Paenibacillus sp. strain CCRC 17245 showed a specific activity 3.5-fold greater with laminarin as the substrate than with barley glucan, whereas Paenibacillus JDR-2 rBgl16A1CD showed the same specific activities for both substrates. The activities determined for the multimodular forms of LamA are as much as 2.7-fold greater than that for the recombinant catalytic domain (21).
LamA is slightly larger than Paenibacillus JDR-2 Bgl16A1, with an additional Fa5/8C domain (an analogue of the C-terminal domain of coagulation factor 5/8). Clustal W alignment of the 243 residues of Bgl16A1 and the 251 residues of LamA that constitute the catalytic domains shows 42% identity. The catalytic string of amino acid residues, sgEiDlmE–G (capital letters designate essential catalytic residues), present in Bgl16A1 is identical to that in LamA and agrees with the conserved amino acid region described by Hahn et al. (20). Hydrolysis of laminarin by Bgl16A1 resulted in a mixture of oligosaccharide products, of which laminaribiose and laminaritriose are the most prominent (Fig. 5). TLC analysis of this mixture shows the same products obtained by LamA hydrolysis of laminarin (21). On the other hand, Bgl16A2 is composed only of the endoglucanase catalytic domain. This protein shares 79% identity with the endoglucanase from Paenibacillus sp. strain F-40 (22), which was analyzed in depth and was determined to be a 1,3(4)-β-d-endoglucanase. The catalytic string of amino acid residues, wdEiDiE–G, present in the Bgl16A2 protein agrees with the conserved amino acid region described by Hahn et al. (20) as well as with that found in the endoglucanase of Paenibacillus sp. strain F-40. Our results show that rBgl16A2 actively depolymerized barley glucan but was less active at degrading laminarin, a characteristic also reported for the 1,3(4)-β-d-endoglucanase from Paenibacillus sp. strain F-40 mentioned above. From these comparisons, it may be concluded that Bgl16A1 is a homologue of LamA and, like LamA, is an enzyme that may be in the EC 3.2.1.39 and EC 3.2.1.6 families as annotated in CAZy, while Bgl16A2 is a homologue of the endoglucanase from Paenibacillus sp. strain F-40 and, like the latter, is an enzyme in the EC 3.2.1.6 family.
Bioinformatic analyses by BPROM software (SoftBerry) and ARNold software (http://rna.igmors.u-psud.fr/toolbox/arnold/index.php) identified putative promoters and terminators within the genome sequence that includes the bgl16A1 and bg16lA2 genes and adjacent genes encoding transporters and potential regulators (Fig. 7A). BPROM is a bacterial sigma 70 promoter recognition program (the recognition sites of transcription factor sigma 70 at −10 and −35 are considered to be very similar to those of sigma A in Bacillus subtilis [23]). ARNold finds rho-independent terminators in nucleic acid sequences by applying an algorithm honed by a training set of 1,200 terminator sequences from Bacillus subtilis and Escherichia coli. Within the intragenic sequence IGS219, which is immediately 5′ to the endoglucanase operon, two putative promoter sequences in opposite directions are found (Fig. 7B), which may conduct the expression of the genes downstream. Interestingly, a 17-bp palindromic sequence was also found within IGS219. This 17-bp sequence forms a hairpin with a GC-rich apex and an AT-rich stem with one mismatch, the same features present in the 18-bp nucleotide sequence (AATGAACGCGCGTACATT) within the celC promoter region of Clostridium thermocellum to which the repressor protein GlyR3 binds. The binding of GlyR3, identified by DNase I footprinting and electrophoretic mobility shift assay analyses, represses the transcription of the downstream genes of this operon, celC, glyR3, and licA. The presence of laminaribiose relieves this repression and allows transcription (24). In the present study, the same effect was observed: the presence of laminaribiose in the medium of Paenibacillus JDR-2 cultures induced the expression of the bgl16A1 and bgl16A2 genes. In addition, two cre catabolite response elements were identified within the nucleotide sequence of bgl16A1. These elements may account for the lack of induction of transcription of bgl16A1 and bgl16A2 when Paenibacillus JDR-2 was grown in glucose (Fig. 6B). The possible interplay of these control elements on the expression of the bgl16A1 and bgl16A2 genes is depicted in Fig. 8. This interpretation leads to the suggestion that the endoglucanase genes may be switched on when laminaribiose is present and glucose is absent in the growth environment.
FIG 7.

Transcription control elements within the bgl16A1–bgl16A2 cassette. (A) Bent arrows indicate putative transcription start sites; the inverted U at IGS219 indicates the location of the 17-bp palindromic sequence and GlyR3-like repressor binding site; lollipops denote putative transcription termination sites; and triangles mark the locations of intergenic sequences, e.g., IGS219. (B) Nucleotide sequence of IGS219 annotated for the identification of transcriptional regulation elements.
FIG 8.

Graphic representation of the relationship of transcription control elements and the expression of the bgl16A1 and bgl16A2 genes. The laminaribiose and glucose columns show the presence or absence of the available carbohydrate nutrient. The third column depicts the four transcription units of the two membrane proteins, including the ABC transporter, bgl16A1 and bgl16A2, and the transcriptional regulators, as well as the binding of repressor elements in this bgl16A1–bgl16A2 gene vicinity. The putative GlyR3-like repressor binds to the 17-bp palindromic sequence. CcpA is the catabolite repressor protein that binds to cre sites. The circled R represents the GlyR-3 repressor protein binding to DNA just before the first gene encoding ABC transporter proteins. The rightmost column shows the on/off state of gene expression.
Nucleotide sequence analysis of the intergenic sequence 5′ of sbp(0953), IGS248, shows the presence of a transcription termination hairpin followed by a promoter farther downstream. This finding is consistent with the results of this study, where transcription of sbp(0953) overcomes the dampening effect of polarity and can be induced manyfold more than bgl16A1 and bgl16A2. Higher induction of the transcription of substrate binding protein than of the transcription of the depolymerizing enzymes is a feature also found in other substrate utilization cassettes in Paenibacillus JDR-2 (7, 9). The expression of the downstream genes araC(0955), sbp(0956), and pel(0964) is unaffected by growth in barley glucan (results not shown), probably due to the presence of a stem-loop, rho-independent transcription terminator (ACCGCCTCTCATATGTAGAGGCGGT [underlined nucleotides indicate H bonding with each other to form a palindrome that results in termination independent of the rho termination factor; the termination site is shown in Fig. 7]) (ΔG, −14.40) found at IGS380.
Overall, these results are consistent with the interpretation that the endoglucanase genes bgl16A1 and bgl16A2, together with the ABC transporter-encoding genes sbp(0953), Pjdr2_0949, and Pjdr2_0950 and the transcription regulation unit comprising araC(0947) and Pjdr2_0948, form a cassette responsive to the different carbohydrate nutrients available. Significantly, the expression of the bgl16A1 and bgl16A2 genes is fine-tuned by the genetic elements within the DNA sequence of this cassette. The tandem arrangement of the two endoglucanase genes in Paenibacillus JDR-2 leads to simultaneous expression of these two enzymes. After synthesis, Bgl16A2 is likely secreted into the medium, not associated with the cell membrane, and would be expected to release the depolymerized glucan polymer products into the medium rather than close to the cell surface. Assimilation of these dispersed oligosaccharide molecules should be less efficient than that for oligosaccharides generated by cell-associated BglA1 on the cell surfaces, and their accumulation at higher levels in the mid-log-growth phase is observed, as described in Results. Based on sequenced genomes, other Paenibacillus spp. found to encode both bgl16A1 and bgl16A2 gene orthologues include Paenibacillus curdlanolyticus, Paenibacillus sp. strain FSL, Paenibacillus sp. strain PAMC, Paenibacillus sp. strain IHB, Paenibacillus borealis, and Paenibacillus mucilaginosus. Amino acid sequence identities between Paenibacillus JDR-2 Bgl16A1 and the homologues in these other Paenibacillus spp. range from 41 to 62%, while those between Paenibacillus JDR-2 Bgl16A2 and the homologues in these other species range from 65 to 86%. It is noteworthy that the tandem arrangement of bgl16A1 and bgl16A2 is found only in Paenibacillus JDR-2; in these other Paenibacillus spp., the homologues equivalent to bgl16A2 and sbp(0953) were instead placed immediately 5′ of the transcription regulation cassette.
Multimodular hydrolytic enzymes may confer evolutionary advantages. Cheng et al. (21) showed that the CBM domains in LamA substantially enhanced the hydrolytic activity of its catalytic module on insoluble complex substrates such as barley glucan, zymosan, curdlan, and pachyman. Their experimental findings are consistent with our proposal that through the CBM and SLH domains of Bgl16A1 help sequester both the enzyme and the polymeric substrate at the surface of the bacterial cell and thus are essential participants in the coupling system of the depolymerization/assimilation of large carbohydrate polymers. The utilization of substrates by this cassette of endoglucanase genes is similar to that encountered in the previous analyses of the xylan utilization regulon in the Paenibacillus JDR-2 genome (6–8). These findings suggest that the proposed coupling strategy is an efficient process that evolved in Paenibacillus JDR-2 and related species in that it provides a survival advantage through the conservation of ATP equivalents as well as the generation of oligosaccharides at the cell surface, allowing direct and efficient importation into the cell.
It has been noted that under conditions of oxygen limitation, Paenibacillus JDR-2 is able to produce low levels of succinate and acetate from MeGXn (6). Under similar conditions, Paenibacillus JDR-2 is also able to quantitatively ferment maltose to yield a mixture of lactate, acetate, and formate (N. Sawhney, unpublished results). Like Paenibacillus macerans, which has been shown to produce fermented products upon anaerobic growth (25), Paenibacillus JDR-2 contains the pyruvate decarboxylase lyase (pdl) gene required for this process. With the combination of the efficient coupling mechanism of the depolymerization/assimilation of multiple polymeric carbohydrate sources and the ability to ferment the metabolites intracellularly, Paenibacillus JDR-2 may be engineered as a biocatalyst for the efficient production of biofuels and/or chemical feedstocks from hemicellulosic resources.
ACKNOWLEDGMENTS
We acknowledge Lonnie O. Ingram, Keelnathan T. Shanmugam, Assistant Director of the Florida Center for Renewable Chemicals and Fuels Sheilachu P. Gomez, and other personnel in their laboratories as well as ours who provided assistance during the course of this study.
This project was supported in part by Biomass Research & Development Initiative competitive grant 2011-10006-30358 from the U.S. Department of Agriculture, National Institute of Food and Agriculture; the Florida Energy Systems Consortium, State University System of Florida, project 00077818; and the Florida Agricultural Experiment Station of the University of Florida Institute of Food and Agricultural Sciences.
Funding Statement
General support is provided by USDA Hatch Multistate Project 005137 through the University of Florida Institute for Food and Agricultural Sciences.
REFERENCES
- 1.Saha BC. 2003. Hemicellulose bioconversion. J Ind Microbiol Biotechnol 30:279–291. doi: 10.1007/s10295-003-0049-x. [DOI] [PubMed] [Google Scholar]
- 2.Preston JF, Hurlbert JC, Rice JD, Ragunathan A, St John FJ. 2003. Microbial strategies for the depolymerization of glucuronoxylan: leads to the biotechnological applications of endoxylanases. ACS Symp Ser 855:191–210. doi: 10.1021/bk-2003-0855.ch012. [DOI] [Google Scholar]
- 3.Scheller HV, Ulvskov P. 2010. Hemicelluloses. Annu Rev Plant Biol 61:263–289. doi: 10.1146/annurev-arplant-042809-112315. [DOI] [PubMed] [Google Scholar]
- 4.Kim Y, Mosier NS, Ladisch MR, Pallapolu VR, Lee YY, Garlock R, Balan V, Dale BE, Donohoe BS, Vinzant TB, Elander RT, Falls M, Sierra R, Holtzapple MT, Shi J, Ebrik MA, Redmond T, Yang B, Wyman CE, Warner RE. 2011. Comparative study on enzymatic digestibility of switchgrass varieties and harvests processed by leading pretreatment technologies. Bioresour Technol 102:11089–11096. doi: 10.1016/j.biortech.2011.06.054. [DOI] [PubMed] [Google Scholar]
- 5.Olson DG, McBride JE, Shaw AJ, Lynd LR. 2012. Recent progress in consolidated bioprocessing. Curr Opin Biotechnol 23:396–405. doi: 10.1016/j.copbio.2011.11.026. [DOI] [PubMed] [Google Scholar]
- 6.St John FJ, Rice JD, Preston JF. 2006. Paenibacillus sp. strain JDR-2 and XynA1: a novel system for methylglucuronoxylan utilization. Appl Environ Microbiol 72:1496–1506. doi: 10.1128/AEM.72.2.1496-1506.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chow V, Nong G, Preston J. 2007. Structure, function, and regulation of the aldouronate utilization gene cluster from Paenibacillus sp. strain JDR-2. J Bacteriol 189:8863–8870. doi: 10.1128/JB.01141-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nong G, Rice J, Chow V, Preston J. 2009. Aldouronate utilization in Paenibacillus sp. strain JDR-2: physiological and enzymatic evidence for coupling of extracellular depolymerization and intracellular metabolism. Appl Environ Microbiol 75:4410–4418. doi: 10.1128/AEM.02354-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sawhney N, Crooks C, St John F, Preston JF. 2015. Transcriptomic analysis of xylan utilization systems in Paenibacillus sp. strain JDR-2. Appl Environ Microbiol 81:1490–1501. doi: 10.1128/AEM.03523-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burton RA, Fincher GB. 2009. (1,3;1,4)-β-d-Glucans in cell walls of the Poaceae, lower plants, and fungi: a tale of two linkages. Mol Plant 2:873–882. doi: 10.1093/mp/ssp063. [DOI] [PubMed] [Google Scholar]
- 11.Vega-Sánchez ME, Verhertbruggen Y, Scheller HV, Ronald PC. 2013. Abundance of mixed linkage glucan in mature tissues and secondary cell walls of grasses. Plant Signal Behav 8:e23143. doi: 10.4161/psb.23143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zucker M, Hankin L. 1970. Regulation of pectate lyase synthesis in Pseudomonas fluorescens and Erwinia carotovora. J Bacteriol 104:13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dubois M, Gilles KA, Hamilton JK, Rebers PA, Smith F. 1956. Colorimetric method for determination of sugars and related substances. Anal Chem 28:350–356. doi: 10.1021/ac60111a017. [DOI] [Google Scholar]
- 14.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 15.Bio-Rad Laboratories, Inc. 2006. Real-time PCR applications guide: bulletin 5279, p 2–6, 34–37. Bio-Rad Laboratories, Inc, Hercules, CA. [Google Scholar]
- 16.Woodward JR, Fincher GB, Stone BA. 1983. Water-soluble (1→3), (1→4)-β-d-glucans from barley (Hordeum vulgare) endosperm. II. Fine structure. Carbohydr Polymers 3:207–225. doi: 10.1016/0144-8617(83)90019-X. [DOI] [Google Scholar]
- 17.Lazaridou A, Biliaderis CG. 2007. Molecular aspects of cereal β-glucan functionality: physical properties, technological applications and physiological effects. J Cereal Sci 46:101–118. doi: 10.1016/j.jcs.2007.05.003. [DOI] [Google Scholar]
- 18.Mesnage S, Fontaine T, Mignot T, Delepierre M, Mock M, Fouet A. 2000. Bacterial SLH domain proteins are non-covalently anchored to the cell surface via a conserved mechanism involving wall polysaccharide pyruvylation. EMBO J 19:4473–4484. doi: 10.1093/emboj/19.17.4473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Read SM, Currie G, Bacic A. 1996. Analysis of the structural heterogeneity of laminarin by electrospray-ionisation-mass spectrometry Carbohydr Res 281:187–201. [DOI] [PubMed] [Google Scholar]
- 20.Hahn M, Olsen O, Politz O, Borriss R, Heinemann U. 1995. Crystal structure and site-directed mutagenesis of Bacillus macerans endo-1,3-1,4-β-glucanase. J Biol Chem 270:3081–3088. doi: 10.1074/jbc.270.7.3081. [DOI] [PubMed] [Google Scholar]
- 21.Cheng YM, Hong TY, Liu CC, Meng M. 2009. Cloning and functional characterization of a complex endo-β-1,3-glucanase from Paenibacillus sp. Appl Microbiol Biotechnol 81:1051–1061. doi: 10.1007/s00253-008-1617-9. [DOI] [PubMed] [Google Scholar]
- 22.Yang P, Shi P, Wang Y, Bai Y, Meng K, Luo H, Yuan T, Yao B. 2007. Cloning and overexpression of a Paenibacillus β-glucanase in Pichia pastoris: purification and characterization of the recombinant enzyme. J Microbiol Biotechnol 17:58–66. [PubMed] [Google Scholar]
- 23.Whipple FW, Sonenshein AL. 1992. Mechanism of initiation of transcription by Bacillus subtilis RNA polymerase at several promoters. J Mol Biol 223:399–414. doi: 10.1016/0022-2836(92)90660-C. [DOI] [PubMed] [Google Scholar]
- 24.Newcomb M, Chen C-Y, Wu JHD. 2007. Induction of the celC operon of Clostridium thermocellum by laminaribiose. Proc Natl Acad Sci U S A 104:3747–3752. doi: 10.1073/pnas.0700087104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gupta A, Murarka A, Campbell P, Gonzalez R. 2009. Anaerobic fermentation of glycerol in Paenibacillus macerans: metabolic pathways and environmental determinants. Appl Environ Microbiol 75:5871–5883. doi: 10.1128/AEM.01246-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chow V, Nong G, St. John JF, Rice JD, Dickstein E, Chertkov O, Bruce D, Detter C, Brettin T, Han J, Woyke T, Pitluck S, Nolan M, Pati A, Martin J, Copeland A, Land ML, Goodwin L, Jones JB, Ingram LO, Shanmugam KT, Preston JF. 2012. Complete genome sequence of Paenibacillus sp. strain JDR-2. Stand Genomic Sci 6:1–10. doi: 10.4056/sigs.2374349. [DOI] [PMC free article] [PubMed] [Google Scholar]