

Abstract
Ecological understanding of disease risk, emergence, and dynamics and of the efficacy of control strategies relies heavily on efficient tools for microorganism identification and characterization. Misdetection, such as the misclassification of infected hosts as healthy, can strongly bias estimates of disease prevalence and lead to inaccurate conclusions. In natural plant ecosystems, interest in assessing microbial dynamics is increasing exponentially, but guidelines for detection of microorganisms in wild plants remain limited, particularly so for plant viruses. To address this gap, we explored issues and solutions associated with virus detection by serological and molecular methods in noncrop plant species as applied to the globally important Barley yellow dwarf virus PAV (Luteoviridae), which infects wild native plants as well as crops. With enzyme-linked immunosorbent assays (ELISA), we demonstrate how virus detection in a perennial wild plant species may be much greater in stems than in leaves, although leaves are most commonly sampled, and may also vary among tillers within an individual, thereby highlighting the importance of designing effective sampling strategies. With reverse transcription-PCR (RT-PCR), we demonstrate how inhibitors in tissues of perennial wild hosts can suppress virus detection but can be overcome with methods and products that improve isolation and amplification of nucleic acids. These examples demonstrate the paramount importance of testing and validating survey designs and virus detection methods for noncrop plant communities to ensure accurate ecological surveys and reliable assumptions about virus dynamics in wild hosts.
INTRODUCTION
Understanding pathogen dynamics and implementing pathogen management strategies require consistent and reliable detection of microorganisms in hosts, reservoirs, and dispersal agents (1–5). Because loss of crop production due to microbial plant pathogens poses serious threats to food security and human health (United Nations Food and Agriculture Organization [www.fao.org]) (6), much work has been invested in developing means of identifying and quantifying pathogen infection within agronomic species. A recent and growing exploration of disease ecology in unmanaged ecosystems is driving interest in assessing the infection status of wild plants (3, 7–11). However, detection methods that are efficient for detecting pathogens in crop species may be less reliable for wild plants, which are structurally and biochemically different. In this study, we examine challenges for accurately assessing wild plants for virus infection, demonstrate the importance of testing differences in virus signal among plant tissue types, and illustrate how inhibitors in wild plants may suppress virus signal unless corrective measures are taken.
Detecting virus infection in plants can be difficult due to the general unreliability of visual detection methods and the variability in symptom expression within plant hosts (12, 13). Virus detection was substantially improved by the development of serological (e.g., enzyme-linked immunosorbent assay [ELISA]) (14) and molecular (e.g., PCR) (15) assays, which became mainstays in plant pathology and dramatically increased the feasibility and scope of studies in disease ecology and epidemiology (2, 12, 13). The effectiveness of these techniques, as for any diagnostic method, depends strongly on their accuracy. For example, it is essential to control the rate of false positives, in which healthy plants are incorrectly identified as infected. False positives can occur because of cross-contamination among samples and because detection reagents react with background material (i.e., exhibit low specificity) (16–18), leading to overestimates of virus prevalence. It is equally important to control the rate of false negatives in which infected plants are incorrectly identified as uninfected, leading to underestimates of virus prevalence (17, 19, 20).
When virus detection methods developed for crop plants are applied to wild plants, it is particularly important to assess the rate of false negatives. Wild plant communities exhibit much more diversity than crop plants do in genotype, morphology (e.g., number of tillers in grasses), and age distribution (21, 22). In crop and model plants, it is recognized that the distribution of viruses within plants is not uniform and may vary over time and with leaf position and age (23, 24). In contrast, within wild plant hosts, the question of a potential variability in distribution of viruses has generally been overlooked. Virus prevalence could be underestimated if within-host virus distribution is not uniform in wild plants and if the sampling effort is insufficient to capture infection within individual hosts, i.e., through targeting tissue types and structural units that would yield a too low detection signal. A particularly interesting question lies in the potential difference in virus signals between stems and leaves, the latter being more commonly sampled but not necessarily more effective in yielding high virus signal, as well as differences in virus detection between multiple tillers of the same host.
A second essential issue in evaluating rates of false negatives in wild plant species is the nature of the host plant's physiology and biochemistry (25, 26) and the potential for biochemical inhibition of detection assays. An ELISA, a serological assay in which plant extracts are present only at low concentrations, is not notably sensitive to inhibitors that might be found in wild plant extracts (Agdia, Inc.). However, PCR (and reverse transcription-PCR [RT-PCR]) can be substantially inhibited by multiple substances, including environmental molecules (e.g., phenolic compounds, humic acids, and heavy metals), cell constituents, and reagents used to extract nucleic acids (e.g., phenol, EDTA, ethanol, and isopropanol) (19, 27, 28). If retained during sample processing, inhibitors may produce false negatives by interfering with the release of target nucleic acids during extraction, by precipitating or degrading target nucleic acids, or by inactivating the reverse transcriptase or DNA polymerase (27–29). The extent of inhibitors in samples has been extensively considered in clinical, forensic, food, and environmental microbiology but so far rarely addressed in plant disease ecology and epidemiology (5, 19, 27–32). Inhibitory effects could be particularly problematic and produce misleading results (i) in studies of environmental effects (e.g., heavy metals) on disease prevalence or (ii) in comparisons of prevalences among plant species that differ notably in inhibitory potentials, such as comparisons of crop and wild plants and of annual and perennial plants.
In molecular assays, the investigator typically faces the challenging dilemmas of (i) extracting virus particles and/or nucleic acids from host materials while preserving their integrity, (ii) diluting or removing potential inhibitors without removing the viral signal itself, which may be of very low titer, (iii) detecting viruses when inhibitors are retained in plant extracts, and (iv) ensuring that each of these steps can be performed equally efficiently for all host species studied. Numerous approaches and products for addressing these issues have been developed and commercialized, but rarely have they been tested on wild plant species. Nucleic acid extraction methods for downstream PCR-based diagnostics include cetyltrimethylammonium bromide (CTAB)-based methods and phenol-chloroform protocols (33), which are regarded as being among the best for nucleic acid extraction, even from low-titer virus species, although their efficacy varies among plant species (34–36). Alternatively, column-based extraction methods may produce cleaner plant extracts (37), but there is some risk of loss of microbial signal. While PCR is theoretically more sensitive than ELISA (38, 39), it may be more susceptible to biochemical interference. Nucleic acids and RT-PCR enzymatic reagents can be highly sensitive to inhibitors, whereas intact virions, the typical target of antibodies in ELISAs, are less sensitive to inhibitors naturally occurring in various plant species (27–29). In addition, methods and materials used to extract nucleic acids produce extracts with variable nucleic acid purity levels and concentrations that give variable downstream results in PCR (5, 19, 27, 28, 40).
To facilitate studies of disease ecology in natural plant systems, we here focus on essential steps to ensure that virus detection methods for wild plants are reliable for different plant species. As a model system, we conducted this work with the Barley and Cereal yellow dwarf virus (B/CYDV)-grass host system. These positive-sense single-stranded RNA (+ssRNA) viruses are globally distributed viruses and transmitted obligately from plant to plant via aphid vectors (Aphididae) in a persistent, circulative, and nonpropagative manner (41, 42). B/CYDVs are host generalists that infect at least 150 species in the Poaceae (grass) family (43). Infection can cause dwarfing, yellowing, reddening, and reduction in host fecundity and life span. B/CYDVs have caused significant agricultural losses worldwide, contributed to a dramatic plant species shift in natural California grasslands, and could potentially alter the efficacy of biofuel production (44–47). At least 25 aphid species transmit B/CYDVs (41, 48). In both crop and wild hosts, Barley yellow dwarf virus PAV (BYDV-PAV) is one of the most frequently detected B/CYDVs and is transmitted primarily by two aphid species, Rhopalosiphum padi and Sitobion avenae (49, 50). Infection is systemic within plants but restricted to host phloem cells. Consequently, these viruses are low-titer pathogens in many plant hosts, which makes them more challenging to detect than less restricted microorganism species and thus invaluable subjects with which to examine the causes and means of reducing false negatives in detection (51, 52). Thus, these viruses serve as a “worst-case” scenario, providing an excellent case study for considering the multiple biological and biochemical causes of reduced virus detection in this and other host-pathogen systems.
In this study, we demonstrate the importance of assessing (i) within-plant variability in virus distribution and (ii) the extent of host tissue inhibition of diagnostic assays in any survey of virus infection in wild plants. Specifically, we ask the questions listed below.
To what extent does virus detection vary within individual hosts? Is virus detection in leaves (commonly sampled) similar to that in stems (less frequently sampled)? To avoid false negatives, it is critical to predetermine which host tissue types will yield the highest detection signals, particularly if tissue is limited, and to assess potential variability in detection signals among individual host structural units.
To what extent can inhibitors in wild plants reduce RT-PCR sensitivity? Can inhibition be overcome by optimization of extraction methods or adoption of easy-to-use postextraction steps? Estimates of virus prevalence and disease risk according to host identity could be confounded with host species variations in inhibitor content and false-negative detection rates.
How best can inhibitors be reduced without overly large loss of a low-titer virus? In addition to pure nucleic acid extracts (i.e., free of inhibitors), accurate pathogen detection relies on an optimum nucleic acid concentration isolated from samples.
MATERIALS AND METHODS
Assessing patterns of virus signal within individual wild plants: example of ELISA applied to switchgrass. (i) Experimental approach.
To highlight the need for careful sampling design in studies of wild plants, we quantified within-plant heterogeneity of virus signal within mature native perennial plants of field-grown size. We used a triple antibody sandwich (TAS) ELISA to test samples from various tissue types of well-developed switchgrass plants (Panicum virgatum cv. Shawnee; Poaceae; perennial) in which experimental BYDV-PAV infection had been confirmed by ELISA 3 months earlier when the plants were young. The aim was to compare virus signal strengths among multiple tillers within individual hosts, among leaves in different positions, and between leaves and stem sections, the latter of which are often undersampled in surveys.
Panicum virgatum cv. Shawnee seedlings were started in SureMix Perlite potting medium (Michigan Grower Products, Inc., Galesburg, MI, USA) in 38-well large plug trays (X-38ST; Landmark Plastic, Akron, OH, USA) under natural winter light in a greenhouse at Michigan State University (temperature range, 12 to 24°C). Once a week, seedlings were fertilized with liquid fertilizer (Peters Professional Allrounder 20-20-20 + TE; Scotts International B. V.) at a constant application rate of 1 g liter−1. Seedlings grew slowly and were transplanted 51 days after seeding to individual plastic pots (1,573-cm3 volume) filled with SureMix and slow-release fertilizer (4.6 g of fertilizer per 28.3 liter of potting material; Osmocote); fertilization with liquid fertilizer continued. For virus inoculation, nonviruliferous R. padi aphids were allowed to acquire Michigan-origin BYDV-PAV by feeding on infected tissue for 24 h and then were transferred to large caged oat plants in a high-light growth chamber where a population of viruliferous aphids built up over 2 weeks. Oat plants and aphid subsamples tested positive for BYDV-PAV with RT-PCR (data not shown). Inoculations occurred at 87 days after seeding, when seedlings were less than 30 cm tall. To inoculate each plant, 15 to 20 viruliferous adult wingless aphids (R. padi) were placed into a 15-ml clear polypropylene conical tube (Becton Dickinson Labware, France) and transferred onto the second fully expanded leaf on the main stem. Tubes were plugged with cotton wool to prevent aphid escape. After a 48-h inoculation access period, tubes and aphids were removed, and all plants were sprayed with a systemic aphicide. The virus status of all seedlings was then assessed 65 days after inoculation using a TAS-ELISA on 0.5-g samples of young foliar tissue (Agdia, Inc., Elkhart, IN, USA). Shawnee seedlings with confirmed BYDV-PAV infection were transplanted into 6-liter pots and grown to maturity in an air-conditioned 20°C greenhouse with natural light and irrigation.
After 3 months of growth, all tillers of all plants were scored for red or yellow discoloration that is sometimes indicative of BYDV infection. Five tillers of specified types were harvested from each of four randomly selected plants: one young tiller (none of the young tillers displayed symptoms of infection at the time of sampling), two mature green tillers showing symptoms, and two symptomless mature green tillers. Young tillers were tested for the presence of virus in the first fully expanded leaf and in the stem internode associated with that leaf. For the older tillers, we harvested tissue from three fully expanded leaves toward the top of the tiller and from the three stem sections adjacent to each leaf. A total of 26 samples were thus collected per plant.
Then, we performed TAS-ELISA on these mature individuals. Fresh tissue, 0.21 or 0.5 g (according to tiller size), was ground with either 700 or 1,000 μl of general extraction reagent with Tween (Agdia, Inc., Elkhart, IN, USA) in Universal U-form heavy-duty plastic extraction bags with a synthetic intermediate layer (article no. 480100; Bioreba AG, Reinach, Switzerland) with a Bioreba Homex tissue homogenizer. Extracts were tested with reagents and antibodies produced to detect intact BYDV-PAV virions (ELISA Reagent Set SRA 27500; Agdia, Elkhart, IN, USA), according to the manufacturer's instructions. The absorbance (A405) in each well of the ELISA plates was read at 405 nm using a Vmax kinetic microplate reader (Molecular Devices, LLC, Sunnyvale, CA, USA). Samples were distributed across two different ELISA plates (Nunc; Thermo Scientific, Rochester, NY, USA) arbitrarily designated A and B in order to test all samples during the same ELISA.
(ii) Statistical analyses.
All statistical analyses throughout this work were performed using the nlme package of R, version 2.15.2 (R Foundation for Statistical Computing, Vienna, Austria) (53).
The response variable was the ratio of the absorbance (A405) obtained for each test sample to the A405 of the negative uninfected control (i.e., A405 sample/A405 negative control; henceforth, A405 ratio). When necessary, the A405 ratio was log transformed to meet assumptions of normality. Untransformed values of the A405 ratio of ≥2 were considered positive for infection. The choice of this threshold was validated by performance of these antibodies in separate work on uninfected switchgrass, which found that among 95 mock-inoculated switchgrass samples, 95% exhibited A405 ratios of 1.2 or less; the highest value, in a single individual, was 1.5 (data not shown). In the work reported here, the average A405 ratios of tillers considered negative and positive for virus detection were <1.18 and >7.8, respectively. First, we used linear regression to test for differences in A405 ratios among plants. Next, we tested for variation in virus detection as follows: (a) among different tillers within a plant (tillers were identified by arbitrary numbers or by type: young or older tiller with or without symptoms); (b) between stem and leaf tissue from the same tiller; (c) among leaves from the same tiller; and (d) among pieces of stem from different parts of the same tiller. We used a linear mixed-effects model (54) and controlled for variation in the A405 ratio at higher organization levels (tiller and/or plant). Weight of the fresh plant tissue, extraction reagent volume, and the identification of the ELISA plate (A and B) used for each sample were included as covariates.
Assessing effects of RT-PCR inhibitors in plant RNA extracts.
We conducted a mixing test in which we extracted nucleic acids (i) from leaf tissue of cultivated oat (Avena sativa cv. California Red; Poaceae; annual) infected with BYDV-PAV, from which a strong positive signal is easily recovered, and (ii) from this same infected oat tissue mixed 2:1 with uninfected switchgrass (Panicum virgatum; Poaceae; perennial) suspected to contain inhibitors. Two samples of each tissue type separately ground in liquid nitrogen were extracted by each of two commonly used extraction processes: (i) TRI Reagent from Molecular Research Center, Inc. (75 mg of tissue/sample), and (ii) a Spectrum Plant Total RNA kit from Sigma-Aldrich (100 mg of tissue/sample). We used a previously tested RT-PCR protocol to detect virus infection (55).
Comparison and modification of phenol-chloroform extraction methods to remove inhibitors. (i) Experimental approach.
We compared the effectiveness of three different extraction reagents, TRIzol (Invitrogen), TRI Reagent (Sigma-Aldrich), and Plant RNA Reagent (Invitrogen), when they were utilized according to the manufacturers' instructions (regular protocol) and when they were modified three ways to improve the isolation of inhibitor-free nucleic acids (modified 1, 2, and 3). In all methods, 50 mg of frozen plant tissue was ground using copper beads and a bead beater (Biospec Products). Modification 1 consisted of addition of linear polyacrylamide (10 μl per sample, 25 mg/ml; Sigma-Aldrich) as a carrier of nucleic acids, which can increase the concentration of extracted RNA, just before the addition of chloroform for phase separation. Modification 2 was inclusion of a second chloroform extraction to increase the purity of RNA extracts (32). Modification 3 was a combination of both. After extraction, recovered pellets were dissolved in 50 μl of RNase-free water, and RNA concentration (nanograms/microliter) and purity (A260/A280 ratio) were quantified using a NanoDrop spectrophotometer (Thermo Scientific). RNA extracts were stored at −20°C until use. To evaluate the influence of any inhibitors, these RNA extracts were tested using a published “regular” RT-PCR protocol (56) that includes only specific primers for BYDV-PAV (PAVR1, ATTGTGAAGGAATTAATGTA; PAVL1, AGAGGAGGGGCAAATCCTGT; 10 μM) (57).
We tested these approaches using uninfected foliar tissue from three different grass species: A. sativa (Poaceae; annual crop), Koeleria macrantha (Poaceae; wild perennial), and Andropogon gerardii (Poaceae; wild perennial). The experimental design was fully factorial: 3 extraction reagents × 4 extraction procedures × 3 plant species × 5 replicates = 180 experimental units (i.e., samples). Prior to RT-PCR, we mixed 0.9 μg of RNA from each of the 180 extractions from uninfected leaves with 0.1 μg of RNA from BYDV-PAV-infected A. sativa leaves (henceforth referred to as 9:1 mixed samples) to test the influence of any inhibitors in the RNA extracts on downstream diagnostics. Such postextraction mixing permits effective balancing of extract proportions.
(ii) Statistical analyses.
To evaluate the efficiency of extraction methods, we used a generalized linear model (53) with a logistic regression to test for the effect of plant species (A. sativa, A. gerardii, and K. macrantha), extraction reagent (TRIzol, TRI Reagent, and Plant RNA Reagent) and extraction procedure (regular and modified 1, 2, and 3) on the efficiency of detecting BYDV-PAV in 9:1 mixed RNA samples.
We used a generalized linear model with a logistic regression and binomial response variable to assess whether the efficiency of BYDV-PAV detection, and thus the presence of inhibitors, in 9:1 RNA samples can be predicted from the A260/A280 ratio of RNA extracted from uninfected leaves. RNA samples characterized by an A260/A280 ratio of ≈2 are considered pure, whereas an A260/A280 ratio of <2 indicates the presence of contaminants. We also tested for a relationship between concentration (nanograms/microliter) and purity (A260/A280) of RNA obtained for extracts from uninfected leaves. In both analyses, plant species, extraction reagent, and extraction procedure were included as covariates.
Postextraction methods for reducing inhibitor influence. (i) Experimental approach.
The same 180 experimental units (i.e., RNA extracts from uninfected leaves mixed 9:1 with RNA from BYDV-PAV-infected A. sativa leaves) that were tested using a regular RT-PCR protocol (56), as described above for the comparison and modification of phenol-chloroform extraction methods, were further analyzed with a modified RT-PCR protocol that includes postextraction methods for reducing inhibitor influence: (a) a 10× dilution of RNA extract prior to RT, (b) addition of a proteinaceous amplification facilitator, T4gp32 (28, 37, 58, 59), in both RT and PCR steps, and (c) the extension of the regular PCR step (see “Comparison and modification of phenol-chloroform extraction methods” above) (56) by adding five amplification cycles. Encoded by gene 32 of the bacteriophage T4, the T4gp32 protein appears to facilitate amplification by binding and precipitating inhibitory chemical compounds, serving as a target for proteinases, facilitating denaturation of double-stranded DNA, and protecting single-stranded DNA from degradation (37, 60).
In preliminary analyses, various methods (i.e., 10× dilution of RNA extract and addition of five amplification cycles to the PCR step) and putative amplification facilitator products (i.e., glycerol, dimethyl sulfoxide [DMSO], polyethylene glycol [PEG], bovine serum albumin [BSA], and T4gp32) (19, 20, 27, 28, 59) were tested alone and in combination, as well as at various concentrations (see Tables S1 and S2 in the supplemental material). For these preliminary analyses, samples from various plant grass species, including Bromus hordeaceus, Andropogon gerardii, Koeleria macrantha, Poa pratensis, and Schizachyrium scoparium, were tested according to a previously published protocol (56) except for the following modifications. A pair of primers modified based on primers Lu1 (61) and C1R3 (62) and designed to be generic (i.e., able to amplify all B/CYD virus species) was used. The reverse generic R1 primer (TGGTAGGACTTRAGTAYTCC) was used both in the RT (at 0.5 μM) and the PCR (at 0.02 μM) while the forward generic F4 primer (CGGACARTGGTTRTGG) was used (at 0.07 μM) in the PCR. After a 15-min 95°C activation period of the Taq polymerase, cDNA fragments were amplified following 35 cycles of denaturation (30 s at 95°C), annealing (45 s at 45°C), and extension (1 min at 72°C). After a final extension time of 10 min at 72°C, DNA products were stored at −20°C until use and later visualized on a SybrSafe (Invitrogen)-stained 2% (wt/vol) Agarose-1000 (Invitrogen) gel. The combination of a 10× dilution of RNA extract, the addition of T4gp32 in both RT and PCR steps (38 μg/ml; New England BioLabs), and the extension of the PCR step by five amplification cycles was the only method found to be efficient in relieving inhibition (see Tables S1 and S2 in the supplemental material) and was thus selected for further testing on a wider range of plant samples.
(ii) Statistical analyses.
A generalized linear model (53) with a logistic regression, similar to the one described above for the comparison and modification of phenol-chloroform extraction methods, was used to test for the effect of plant species, extraction reagent, and extraction procedure on the efficiency of detecting BYDV-PAV in 9:1 mixed RNA samples when the modified RT-PCR protocol was used.
Efficiency of nucleic acid recovery in RNA extraction.
To analyze the efficiency of each extraction reagent to recover nucleic acids from plant tissues, we mixed 10 mg of PAV-infected A. sativa leaves with 40 mg of uninfected plant tissue from one of three host species (A. sativa, A. gerardii, and K. macrantha) prior to RNA extraction. Five replicates of each mixture were then extracted using each of the regular and modified RNA extraction methods (see “Comparison and modification of phenol-chloroform extraction methods” above). Reverse transcription, PCR, and gel electrophoresis were performed according to the modified protocol (see “Postextraction methods for reducing inhibitor influence” above) to ensure that lack of detection could be ascribed to a low-efficiency RNA extraction and not to the presence of inhibitors.
RESULTS
Patterns of virus signal within individual wild plants: example of ELISA applied to switchgrass.
Dissection of mature native switchgrass experimentally infected as young plants found that while infection remained detectable by ELISA in all plants, the detection signals varied strongly with the portion sampled. Averaged across all 26 sampled portions of each plant, the A405 sample/A405 negative control was ≥2 for each of the four dissected plants, indicating positive detection, but varied from 6 to 24 among plants (P < 0.001) (Fig. 1a). The range of average ratios reflects differences in virus detection among tillers within different individuals. Two plants (plants 259 and 267 but not 262 and 263) (Fig. 1b) demonstrated among-tiller partitioning of virus signal, with significantly different average A405 ratios among tillers of each plant (the average value per tiller represents combined values of all leaves and stem portions sampled from that tiller) (P < 0.001). In plant 259, three tillers tested positive (A405 ratio of ≥2) and two tested negative (A405 ratio of <2); in plant 267, four tillers tested positive, and one tested negative. Across all plants, A405 ratios were lower in mature green tillers lacking virus symptoms than in either mature green tillers with symptoms or young tillers (all nonsymptomatic) (P < 0.001) (Fig. 1c). Finally, while not differing significantly among leaves (P = 0.47) or among stem pieces (P = 0.15) from the same tiller, A405 ratios were ≥3 times higher in extracts from stem tissue than in those from leaves (P < 0.001) (Fig. 1d).
FIG 1.

Ratio of absorbances (A405 sample/A405 negative control) obtained in ELISA for tested samples from Panicum virgatum plants. (a) Per-plant averages derived from 26 tissue samples per plant. (b) Per-tiller averages for each plant, derived for four mature green tillers (six tissue samples each) and one young tiller (two tissue samples). Empty circles, black circles, gray triangles, asterisks, and empty squares represent five different stems in each plant. (c) Averages per tiller class. Old NS (48 samples), mature green tiller with no symptoms of virus infection; Old S (48 samples), mature green tiller with symptoms; Young (8 samples), younger green tiller (all nonsymptomatic). (d) Averages derived for leaves (52 samples total) and stem portions of tillers (52 samples total). Error bars represent ±1 standard error of the means.
In testing the statistical significance of these biological factors, we used statistical models that appropriately accounted for variability from other sources. In evaluating these additional sources of variability, we found, for example, that the quantity of fresh tissue (0.2 to 0.5 g) used in each extraction positively affected detection in three models that tested for differences in detection among plants (P = 0.03) and across tillers identified by arbitrary names (P < 0.01) or by type (P = 0.03). Likewise, there were some plate-to-plate (batch) differences in absorbance values (P < 0.001) evident in one model that tested for differences in detection among tiller type (i.e., young or older tillers with or without symptoms), but we found that the volume of reagent (0.7 to 1 ml) used to process each sample in ELISAs did not influence virus detection when it was included as a covariate. The significant differences in absorbance (A405) values among plants and stems reported above were obtained from statistical models that accounted for the variability in the quantity of fresh tissue and batch differences in ELISA runs.
RT-PCR inhibitors in RNA extracts from wild plants.
To illustrate and address the effects of inhibitors in wild plants on virus detection by RT-PCR, we conducted mixing tests in which leaf tissue from uninfected native perennial switchgrass was mixed 1:2 with tissue from Avena sativa (oat) plants infected with BYDV-PAV and compared to the virus signal obtained from infected A. sativa alone. TRI Reagent RNA extraction (Molecular Research Center, Inc.) and RT-PCR of infected A. sativa alone produced clearly evident BYDV-PAV amplicons (Fig. 2, TO lanes). However, mixing leaf tissue from uninfected switchgrass with leaf tissue from infected A. sativa inhibited virus detection (Fig. 2, TM lanes). This inhibition was not observed when all samples were extracted using a Spectrum Plant Total RNA kit (Sigma-Aldrich). Both the infected A. sativa samples (SO lanes) or the mixed samples containing both infected oats and uninfected switchgrass (SM lanes) produced strong bands (Fig. 2).
FIG 2.

Agarose gel visualization of 832-bp BYDV-PAV PCR products from a switchgrass (Panicum virgatum) mixing experiment. Lane L, 1-kb Plus DNA ladder (Invitrogen). Samples coded with “T” prefixes were extracted with TRI Reagent, and those with “S” prefixes were extracted with a Sigma-Aldrich Spectrum Plant Total RNA kit. Suffix codes are as follows: N, negative (uninfected) tissue; O, infected oats; M, mix of infected oats and switchgrass (see Materials and Methods).
In the next set of mixing experiments, we evaluated the efficacy of three different extraction reagents—TRIzol (Invitrogen), TRI Reagent (Sigma-Aldrich), and Plant RNA Reagent (Invitrogen)—and four extraction procedures (regular and modified 1, 2, and 3) in detecting BYDV-PAV signal in three plants that we suspected to have different tendencies to inhibit RT-PCR: A. sativa (annual crop species, little inhibition), K. macrantha (wild perennial species, moderate inhibition), and A. gerardii (wild perennial species, strong inhibition). For these tests (see “Comparison and modification of phenol-chloroform extraction methods” above), RNA extracted by each method from uninfected leaves of each species was mixed 9:1 with RNA from BYDV-PAV-infected A. sativa leaves and then processed further according to treatment.
Overall, we found that choice of RNA extraction reagent was more important in determining detection rates than any of the three modifications to extraction procedures that we tested (addition of linear polyacrylamide, second chloroform extraction, or both). When RNA was extracted according to each of the three manufacturers' protocols with RT-PCR as per Lacroix et al. (56) (regular protocol), all positive controls (pure RNA extracts from BYDV-PAV-infected A. sativa leaf tissue) were correctly detected, but overall only 59% of the 180 9:1 mixed samples were positively detected. As predicted, virus detection was lower in wild species than in the crop species and fell with the supposed inhibitory tendencies of the plant species. Positives were detected in 88.1% of 9:1 mixed samples containing RNA from uninfected plants of the crop species A. sativa but in only 56.7% and 32.2% of 9:1 mixed samples of RNA from plants of the wild perennial species K. macrantha and A. gerardii, respectively (P < 0.001). RNA extraction reagents varied notably in effectiveness, with positives detected in 93.1% of samples extracted with Plant RNA Reagent but in only 43.3% with TRIzol and 41.7% with TRI Reagent (P < 0.001). There was also a plant species-reagent interaction evident (P < 0.001) (Fig. 3a). All three extraction reagents led to correct detection of most 9:1 mixed samples (85 to 90%) containing extracts obtained from uninfected and infected tissue of only A. sativa (Fig. 3a). However, with the two wild plant species, only Plant RNA Reagent allowed detection levels to remain high (89.5 to 100% across plant species) (Fig. 3a). TRIzol and TRI Reagent led to detection of virus in only 40% of 9:1 mixed samples containing extracts from K. macrantha and in none of the samples containing extracts from A. gerardii (Fig. 3a).
FIG 3.

Proportion of samples (RNA extracts from uninfected Avena sativa, Koeleria macrantha, and Andropogon gerardii leaves each mixed 9:1 with RNA extract from BYDV-PAV-infected A. sativa leaves) for which BYDV-PAV was positively detected using the regular (a) and modified (b) RT-PCR protocols (i.e., including a 10-fold dilution of RNA extract prior to RT, the addition of a proteinaceous amplification facilitator, T4gp32 in both RT and PCR, and the addition of five amplification cycles to the PCR step). Black, gray, and white circles indicate the Plant RNA Reagent, TRIzol, and TRI-Reagent, respectively, used to extract RNA from uninfected leaves. Each data point illustrates the proportion of positively detected 9:1 mixed RNA samples out of 20 tested samples. Error bars represent ±1 standard error of the means.
In contrast to the large effect of RNA reagent, the three modifications to extraction procedures we tested (addition of linear polyacrylamide, second chloroform extraction, or both) did not significantly influence virus detection (P = 0.59) when the regular RT-PCR protocol was used (56). This result was consistent across extraction reagents (P = 0.07) and plant species (P = 0.66).
Postextraction methods for overcoming RT-PCR inhibition.
We next asked if modifications to the RT-PCR protocol could overcome effects of inhibitors from wild plants that were not removed during RNA extraction. Specifically, we tested a modified RT-PCR protocol that includes a 10× dilution of RNA extract prior to RT, addition of a proteinaceous amplification facilitator, T4gp32 (New England BioLabs) in both RT and PCR, and addition of five amplification cycles to the PCR step (see “Postextraction methods for reducing inhibitor influence” above). We tested this modified RT-PCR protocol on the same set of 9:1 mixed RNA extracts used to test the efficacy of different RNA reagents reported above and found that it did recover BYDV-PAV detection (Fig. 3b). With the extracts from the two wild plant species in which detection with TRI Reagent and TRIzol extractions had previously been poor (Fig. 3a), detection with the modified RT-PCR protocol was now 100% for samples containing RNA from K. macrantha and 91.5% for samples containing RNA extracts from the more challenging A. gerardii plant species. With the modified RT-PCR protocol, there was thus a significant plant species effect on virus detection (P < 0.01) but no significant effect of RNA extraction reagent (P = 0.77) (Fig. 3b) or RNA extraction protocol (P = 0.16). In other words, modification of the postextraction RT-PCR protocol steps compensated for poor removal of inhibitors at the earlier RNA extraction stage.
Predicting inhibition based on RNA extract concentration and purity.
We then asked how well the efficiency of virus detection with the unmodified (i.e., regular) RT-PCR protocol (56) (see also “Comparison and modification of phenol-chloroform extraction methods” above) could be predicted by common metrics of RNA extract quality typically obtained on a spectrophotometer: RNA concentration (nanograms/microliter), as estimated from A260, or RNA purity, as estimated by the A260/A280 ratio. Examination of extracts from the 9:1 mixed samples previously described found that purity (A260/A280 ratio) of RNA extracts significantly predicted virus detection rates (P < 0.001) (Fig. 4). Consistent with previous results, there was a significant plant species effect (P = 0.001) (Fig. 4) as well as a significant plant species-extraction reagent effect (P < 0.001) (Fig. 4) on the efficiency of BYDV detection (as detailed below). When we examined unmixed RNA extracts from oats and the two wild plant species, we further found that spectrophotometric estimates of RNA concentration fell as estimates of purity increased (P < 0.001) (Fig. 5), as detailed below, suggesting a trade-off. Significant differences were evident among plant species (P < 0.001) and extraction reagents (P < 0.001), with a plant species-extraction reagent interaction evident (P < 0.001) (Fig. 5).
FIG 4.
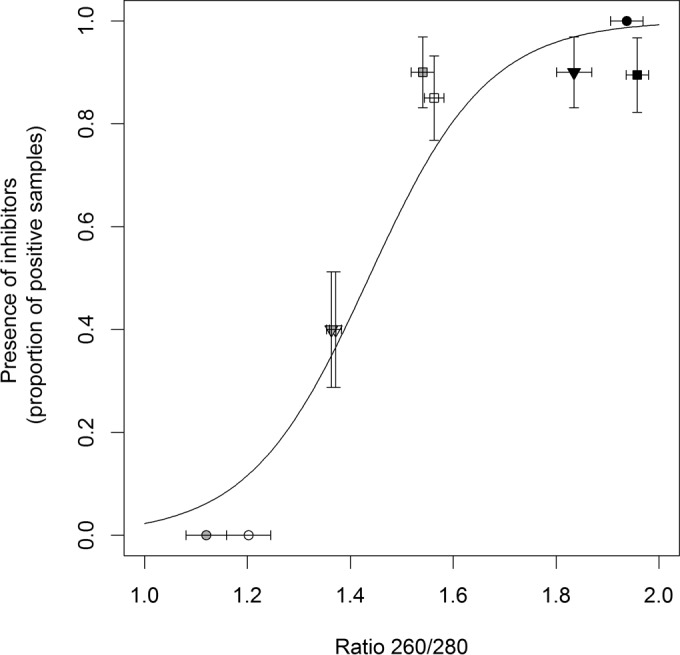
Proportion of samples (RNA extract from uninfected plant tissue mixed 9:1 with RNA extract from BYDV-PAV-infected A. sativa leaves) that led to positive detection of BYDV-PAV using a regular RT-PCR protocol as a function of the average A260/A280 ratios obtained for RNA extracted from uninfected leaves from A. sativa (squares), K. macrantha (triangles), and A. gerardii (circles) plants and using Plant RNA Reagent (black), TRIzol (gray), and TRI Reagent (white) extraction reagent. Each data point illustrates the proportion of positively detected 9:1 mixed RNA samples out of 20 tested samples. Error bars represent ±1 standard error of the means.
FIG 5.
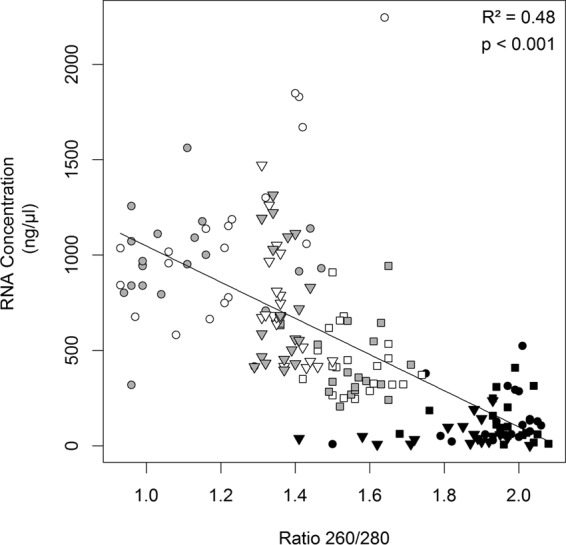
Concentration of RNA as a function of the A260/A280 ratio of each of 180 RNA samples extracted from uninfected A. sativa (squares), K. macrantha (triangles), and A. gerardii (circles) plant tissues using Plant RNA Reagent (black), TRIzol (gray), and TRI Reagent (white) extraction reagent. Error bars represent ±1 standard error of the means.
Overall, the effectiveness of BYDV-PAV detection in 9:1 mixed samples was highest with RNA extracts that were isolated either from uninfected A. sativa leaves (across reagents) or with the Plant RNA Reagent (across plant species) (Fig. 4); these extracts were characterized by a high purity (i.e., A260/A280 ratio above ∼0.8) (Fig. 4) and a low RNA concentration (below ∼500 ng/μl) (Fig. 5). Intermediate-to-severe inhibition of BYDV-PAV detection efficiency in 9:1 mixed samples was observed with RNA extracts from uninfected K. macrantha and A. gerardii leaves that were isolated with TRIzol and TRI Reagent (Fig. 4); these extracts displayed a moderate to high reduction in RNA purity (A260/A280 ratio below ∼0.5) (Fig. 4) and increased estimated RNA concentration (above ∼500 ng/μl) (Fig. 5).
Recovery of nucleic acids in RNA extraction.
Finally, we tested how efficiently each extraction reagent (TRIzol [Invitrogen], TRI Reagent [Sigma-Aldrich], and Plant RNA Reagent [Invitrogen]) recovered virus nucleic acids from a mixture of plant tissues (10 mg of PAV-infected A. sativa leaves and 40 mg of uninfected plant tissue from one of three host species, A. sativa, A. gerardii, or K. macrantha) made prior to RNA extraction. BYDV-PAV was detected in most samples (80 to 100%), using the modified RT-PCR protocol (see “Postextraction methods for reducing inhibitor influence” above) to overcome the effect of inhibitors; these results were consistent across extraction reagents (93%, 93%, and 100% of detected samples when extracted with Plant RNA Reagent, TRI Reagent, and TRIzol, respectively) and plant species (86.7%, 96.3%, and 100% of detected K. macrantha, A. sativa, and A. gerardii, respectively).
DISCUSSION
Our results demonstrate that investigation of virus dynamics in wild plant species requires a clear understanding of plant structural and biochemical characteristics. The phloem-restricted nature of our model system (plant virus BYDV-PAV) and the wide use of ELISA and RT-PCR as detection methods make our findings particularly relevant to issues related to detection sensitivity in a variety of plant-microorganism systems. The methodological approaches we tested illustrate the importance of assessing and optimizing virus detection techniques for application in wild plant hosts. Such information is critically needed as the fields of ecology and epidemiology venture further into the study of plant-virus interactions within natural wild plant populations.
The heterogeneity in virus detection results observed across different structural units (i.e., among tillers and in stems versus leaves) of a single perennial species suggests that within-host virion distribution is variable. This phenomenon is consistent with variability in B/CYDV infections found previously in leaves of different ages and positions in annual crop species (24) and with findings for other microorganisms (23, 63, 64) but has not been well investigated in wild perennial plants. Moreover, our findings demonstrate the importance of sampling not only the leaves of wild plants but also the stems, in which virus signal may be markedly stronger.
Our findings further reinforce the inadvisability of relying on visual means of virus detection as symptom expression did not always predict infection status in our analysis of virus signal distribution within a native grass. Although we did not directly measure virus titer, the greater ELISA signals obtained from both young tillers and older symptomatic ones likely reflect higher virus accumulation in those tissues (23, 63, 64), as shown for the variability in B/CYDV concentration across different leaves of various ages and positions within individual annual plants (24). In addition, our study focused on a limited number of plant individuals and thus do not allow us to make inferences about the frequency of such phenomena in plant populations and communities. However, our study is based on an exhaustive sampling of different plant tissues and at various positions within the tested plants and reinforces, along with previously published results (24), the general importance of considering that the heterogeneous distribution and accumulation of virions within hosts could strongly bias estimates of prevalence. Particularly, relying on a single sample per plant, such as one leaf per plant (24) and one tiller per wild plant (as in this study), which often produces multiple tillers in contrast to crop plants that have been selected for a reduced lateral branching phenotype (22), could lead to false estimations of virus prevalence. Underestimating microorganism prevalence in nonagricultural populations of plants could strongly bias inferences on potential reservoirs of viruses, virus diversity, disease dynamics, and risks of disease emergence. However, this could be avoided by predetermining host tissue age and type that typically yield the highest detection signal or by developing a sampling protocol sufficient to compensate for variability in virus distribution among host structural units.
Our study also demonstrates the effects of inhibitory compounds within wild plants on the sensitivity of virus detection by PCR and highlights the considerable importance of attention to this issue in studies of wild vegetation. Several reagents used to extract nucleic acids from host tissues are known as potential inhibitors of enzymatic reactions occurring in PCR (19, 28, 37). However, our results demonstrated inhibition of virus detection based on extracts from three perennial wild species but little evidence of inhibition in an annual crop plant species. These results indicate that inhibition under our experimental conditions likely arises from differences among plants rather than as a response to reagents.
Our results also show that comparisons of estimates of virus prevalence and pathogen dynamics in natural unmanaged versus agricultural systems could be strongly biased because of variations in inhibitory potentials among plant species, such as in wild versus crop plants. Phenolic compounds constitute an example of phytochemicals that are known to mediate several plant-environment interactions, such as resistance to various pests and pathogens (65–67), and have also been described as inhibitors of microorganism detection via PCR (19, 28, 37). Species' allocation of resources to several fitness traits such as defense, growth, and reproduction have been shown to vary according to several trade-offs, including host life span and domestication history (68) (69–71). Across kingdoms, short-lived and domesticated species (e.g., annual crop plants) display relatively high reproductive output and population growth but invest less in defenses against pests and pathogens than closely related but longer-lived species (e.g., perennial wild plants) (72–75). Hence, if wild perennial plants invest more than annual crops in the production of certain phytochemicals, these wild perennial plants could display higher levels of both defenses against pests and pathogens and of inhibition induced in RT-PCR, which could bias disease ecology studies in natural versus crop plant settings. Testing this prediction would require a broader range of host species than the number of hosts used in this study but would open a fruitful avenue of research.
Our study also presents optimized methods for overcoming these issues that are applicable across a diversity of wild plant species. ELISA is a dependable method for detecting infection and is generally cost-efficient and relatively easy to implement in lab settings. In contrast, PCR requires more specific molecular skills and the use of more costly reagents and equipment. However, an ELISA can be limited by the availability of commercial antibodies, requires more tissue per plant than RT-PCR and so may be limited to larger plants or destructive harvests, and is generally less sensitive than PCR (38, 39). The reliability in virus detection via both ELISA and PCR could also be affected by variability in the sequence of virus genomes and associated encoded proteins (76, 77), highlighting the central importance of antibodies and primer design. PCR can be useful for detecting microorganisms in low-concentration samples but requires appropriate procedures to avoid inhibition. In particular, the nucleic acid isolation method is critical for success. It is influenced by multiple parameters, including the efficiency of isolating extracts that contain enough RNA/DNA but little inhibitor, as well as the time and cost per sample. As observed in our study, column-based kits (e.g., Spectrum Plant Total RNA; Sigma-Aldrich) can yield pure extracts of low concentration, are time efficient, but are more expensive than chloroform extraction procedures (40, 78). However, under our experimental conditions, we found that the Plant RNA Reagent (Invitrogen) is as cost-efficient but less time-consuming than the other tested phenol-chloroform procedures and also prevented inhibition of virus detection.
Our data also suggest that the use of postextraction methods for overcoming inhibition can be determined from the A260/A280 ratio indicative of RNA purity, which is particularly valuable when the extraction method does not efficiently remove inhibitors and in cases of the unexpected presence of inhibitory chemical compounds in plant tissue. We selected from an array of published and commercially available methods an efficient technique of RT-PCR optimization (i.e., a 10-fold dilution of RNA extracts, addition of an amplification facilitator T4gp32 [New England BioLabs], and addition of five amplification cycles to the PCR step) that overcame the effect of any inhibitors in plant extracts, consistent with previous work (58, 59). These results further confirm that diluting nucleic acid extracts reduces inhibition because it reduces concentrations of inhibitors, while detection of diluted nucleic acid extracts in PCR is supported by the addition of an amplification facilitator. In addition, the negative correlation found between RNA concentration and purity (A260/A280 ratio) indicates that nucleic acid concentration could be overestimated when plant extracts are impure although testing this observation would require further analyses (e.g., Northern blotting).
ELISA and PCR are two of the most important tools in the ecological and evolutionary investigation of disease. These methods have been developed for identifying and quantifying pathogens mainly in crop systems in which they are highly effective. Nonetheless, both methods have shortcomings that can lead to biased detection, especially in natural unmanaged systems unless virus detection techniques are optimized for the particularities of wild plant hosts. Our study shows that ecological understanding of disease risk and emergence, virus dynamics, and the efficacy of control strategies could be impeded if biases in virus detection are confounded with the questions of interest. For example, differences in detection rates across host species could be mistakenly attributed to variation in host susceptibilities and infection prevalences if sampling strategies and detection methods are not optimized to account for patterns of variation in virus titers among tissues of individual hosts (e.g., stems versus leaves and among tillers) just above or below detection thresholds, as well as of variation in inhibitor content according to host species (e.g., wild perennials versus annual crops). Here, we provide methodological guidelines for the extraction and amplification of virus nucleic acids that allow us to overcome many of these detection issues and that are reliable for plant tissues from both crop and wild species. These guidelines open the door for effective use of PCR and ELISA for detecting virus infection in wild plant hosts and contribute to the growing interest of studying plant virus life cycles and dynamics in natural wild plant populations.
Supplementary Material
ACKNOWLEDGMENTS
We thank Missy Rudeen, Alexis Rogers, Alisha Fischer, Taelor Haase, and many undergraduate students for help in the lab. We also thank Marty Dekkers for sharing expertise and advice on methods for virus detection.
We received support from the NSF program in Ecology and Evolution of Infectious Disease (grant DEB-1015805) to E.T.B. and E.W.S. and from the DOE Office of Science, BER, Great Lakes Bioenergy Center (DE-FC02-07ER64494), USDA NIFA Sustainable Biofuels Program award 2011-67009-30137, and MSU AgBioResearch funding to C.M.M.
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03538-15.
REFERENCES
- 1.Haydon DT, Cleaveland S, Taylor LH, Laurenson MK. 2002. Identifying reservoirs of infection: a conceptual and practical challenge. Emerg Infect Dis 8:1468–1473. doi: 10.3201/eid0812.010317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stewart JR, Gast RJ, Fujioka RS, Solo-Gabriele HM, Meschke JS, Amaral-Zettler LA, del Castillo E, Polz MF, Collier TK, Strom MS, Sinigalliano CD, Moeller PDR, Holland AF. 2008. The coastal environment and human health: microbial indicators, pathogens, sentinels and reservoirs. Environ Health 7(Suppl 2):S3. doi: 10.1186/1476-069X-7-S2-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Malmstrom CM, Melcher U, Bosque-Perez NA. 2011. The expanding field of plant virus ecology: historical foundations, knowledge gaps, and research directions. Virus Res 159:84–94. doi: 10.1016/j.virusres.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 4.Martinelli F, Scalenghe R, Davino S, Panno S, Scuderi G, Ruisi P, Villa P, Stroppiana D, Boschetti M, Goulart LR, Davis CE, Dandekar AM. 2015. Advanced methods of plant disease detection. A review. Agron Sustain Dev 35:1–25. doi: 10.1007/s13593-015-0314-1. [DOI] [Google Scholar]
- 5.Lopez MM, Llop P, Olmos A, Marco-Noales E, Cambra M, Bertolini E. 2009. Are molecular tools solving the challenges posed by detection of plant pathogenic bacteria and viruses? Curr Issues Mol Biol 11:13–45. [PubMed] [Google Scholar]
- 6.Oerke EC. 2006. Crop losses to pests. J Agric Sci 144:31–43. doi: 10.1017/S0021859605005708. [DOI] [Google Scholar]
- 7.Alexander HM. 2010. Disease in natural plant populations, communities, and ecosystems: insights into ecological and evolutionary processes. Plant Dis 94:492–503. doi: 10.1094/PDIS-94-5-0492. [DOI] [PubMed] [Google Scholar]
- 8.Seabloom EW, Borer ET, Gross K, Kendig AE, Lacroix C, Mitchell CE, Mordecai EA, Power AG. 2015. The community ecology of pathogens: coinfection, coexistence and community composition. Ecol Lett 18:401–415. doi: 10.1111/ele.12418. [DOI] [PubMed] [Google Scholar]
- 9.Plowright RK, Sokolow SH, Gorman ME, Daszak P, Foley JE. 2008. Causal inference in disease ecology: investigating ecological drivers of disease emergence. Front Ecol Environ 6:420–429. doi: 10.1890/070086. [DOI] [Google Scholar]
- 10.Woolhouse MEJ, Gowtage-Sequeria S. 2005. Host range and emerging and reemerging pathogens. Emerg Infect Dis 11:1842–1847. doi: 10.3201/eid1112.050997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morris CE, Bardin M, Kinkel LL, Moury B, Nicot PC, Sands DC. 2009. Expanding the paradigms of plant pathogen life history and evolution of parasitic fitness beyond agricultural boundaries. PLoS Pathog 5:e1000693. doi: 10.1371/journal.ppat.1000693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.López MM, Bertolini E, Olmos A, Caruso P, Gorris MT, Llop P, Penyalver R, Cambra M. 2003. Innovative tools for detection of plant pathogenic viruses and bacteria. Int Microbiol 6:233–243. doi: 10.1007/s10123-003-0143-y. [DOI] [PubMed] [Google Scholar]
- 13.Schaad NW, Schuenzel E. 2010. Sensitive molecular diagnostic assays to mitigate the risks of asymptomatic bacterial diseases of plants. Crit Rev Immunol 30:271–275. doi: 10.1615/CritRevImmunol.v30.i3.40. [DOI] [PubMed] [Google Scholar]
- 14.Clark MF, Adams AN. 1977. Characteristics of the microplate method of enzyme-linked immunosorbent assay for the detection of plant viruses. J Gen Virol 34:475–483. doi: 10.1099/0022-1317-34-3-475. [DOI] [PubMed] [Google Scholar]
- 15.Mullis K, Faloona F, Scharf S, Saiki R, Horn G, Erlich H. 1986. Specific enzymatic amplification of DNA in vitro—the polymerase chain reaction. Cold Spring Harb Symp Quant Biol 51:263–273. doi: 10.1101/SQB.1986.051.01.032. [DOI] [PubMed] [Google Scholar]
- 16.Roux KH. 1995. Optimization and troubleshooting in PCR. Genome Res 4:S185–S194. doi: 10.1101/gr.4.5.S185. [DOI] [PubMed] [Google Scholar]
- 17.Vaneechoutte M, VanEldere J. 1997. The possibilities and limitations of nucleic acid amplification technology in diagnostic microbiology. J Med Microbiol 46:188–194. doi: 10.1099/00222615-46-3-188. [DOI] [PubMed] [Google Scholar]
- 18.Champlot S, Berthelot C, Pruvost M, Bennett EA, Grange T, Geigl E-M. 2010. An efficient multistrategy DNA decontamination procedure of PCR reagents for hypersensitive PCR applications. PLoS One 5:e13042. doi: 10.1371/journal.pone.0013042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilson IG. 1997. Inhibition and facilitation of nucleic acid amplification. Appl Environ Microbiol 63:3741–3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roux KH. 2009. Optimization and troubleshooting in PCR. Cold Spring Harb Protoc 2009:pdb.ip66. doi: 10.1101/pdb.ip66. [DOI] [PubMed] [Google Scholar]
- 21.Stukenbrock EH, McDonald BA. 2008. The origins of plant pathogens in agro-ecosystems. Annu Rev Phytopathol 46:75–100. doi: 10.1146/annurev.phyto.010708.154114. [DOI] [PubMed] [Google Scholar]
- 22.Harlan JR. 1973. Comparative evolution of cereals. Evolution 27:311–325. doi: 10.2307/2406971. [DOI] [PubMed] [Google Scholar]
- 23.Sánchez-Navarro JA, Cañizares MC, Cano EA, Pallás V. 2007. Plant tissue distribution and chemical inactivation of six carnation viruses. Crop Prot 26:1049–1054. doi: 10.1016/j.cropro.2006.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pereira AM, Lister RM. 1989. Variations in virus content among individual leaves of cereal plants infected with Barley yellow dwarf virus. Phytopathology 79:1348–1353. doi: 10.1094/Phyto-79-1348. [DOI] [Google Scholar]
- 25.Reich PB, Walters MB, Ellsworth DS, Vose JM, Volin JC, Gresham C, Bowman WD. 1998. Relationships of leaf dark respiration to leaf nitrogen, specific leaf area and leaf life-span: a test across biomes and functional groups. Oecologia 114:471–482. doi: 10.1007/s004420050471. [DOI] [PubMed] [Google Scholar]
- 26.Liu JX, Zhang DQ, Zhou GY, Duan HL. 2012. Changes in leaf nutrient traits and photosynthesis of four tree species: effects of elevated [CO2], N fertilization and canopy positions. J Plant Ecol 5:376–390. doi: 10.1093/jpe/rts006. [DOI] [Google Scholar]
- 27.Rädstrom P, Lofstrom C, Lovenklev M, Knutsson R, Wolffs P. 2008. Strategies for overcoming PCR inhibition. CSH Protoc 2008:pdb.top20. doi: 10.1101/pdb.top20. [DOI] [PubMed] [Google Scholar]
- 28.Alaeddini R. 2012. Forensic implications of PCR inhibition—a review. Forensic Sci Int Genet 6:297–305. doi: 10.1016/j.fsigen.2011.08.006. [DOI] [PubMed] [Google Scholar]
- 29.Opel KL, Chung D, McCord BR. 2010. A study of PCR inhibition mechanisms using real time PCR. J Forensic Sci 55:25–33. doi: 10.1111/j.1556-4029.2009.01245.x. [DOI] [PubMed] [Google Scholar]
- 30.Samanta JN, Mandal K. 2013. In planta detection of Xanthomonas axonopodis pv. commiphorae using fyuA and rpoD genes. Indian J Exp Biol 51:470–476. [PubMed] [Google Scholar]
- 31.De Boer SH, Ward LJ, Li X, Chittaranjan S. 1995. Attenuation of PCR inhibition in the presence of plant compounds by addition of BLOTTO. Nucleic Acids Res 23:2567–2568. doi: 10.1093/nar/23.13.2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wiedbrauk DL, Werner JC, Drevon AM. 1995. Inhibition of pcr by aqueous and vitreous fluids. J Clin Microbiol 33:2643–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chomczynski P, Sacchi N. 2006. The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: twenty-something years on. Nat Protoc 1:581–585. doi: 10.1038/nprot.2006.83. [DOI] [PubMed] [Google Scholar]
- 34.Jordon-Thaden IE, Chanderbali AS, Gitzendanner MA, Soltis DE. 2015. Modified CTAB and TRIzol protocols imporve RNA extraction from chemically complex Embryophyta. Appl Plant Sci 3:apps.1400105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gambino G, Perrone I, Gribaudo I. 2008. A rapid and effective method for RNA extraction from different tissues of grapevine and other woody plants. Phytochem Anal 19:520–525. doi: 10.1002/pca.1078. [DOI] [PubMed] [Google Scholar]
- 36.Rubio-Pina JA, Zapata-Perez O. 2011. Isolation of total RNA from tissues rich in polyphenols and polysaccharides of mangrive plants. Electron J Biotechnol 14:1–2. [Google Scholar]
- 37.Rädström P, Knutsson R, Wolffs P, Lövenklev M, Löfström C. 2004. Pre-PCR processing: strategies to generate PCR-compatible samples. Mol Biotechnol 26:133–146. doi: 10.1385/MB:26:2:133. [DOI] [PubMed] [Google Scholar]
- 38.Omrani M, Ansari MHK, Agaverdizadae D. 2009. PCR and Elisa methods (IgG and IgM): their comparison with conventional techniques for diagnosis of Mycobacterium tuberculosis. Pak J Biol Sci 12:373–377. doi: 10.3923/pjbs.2009.373.377. [DOI] [PubMed] [Google Scholar]
- 39.Kumar M, Nandi S, Chidri S. 2010. Development of a polyclonal antibody-based AC-ELISA and its comparison with PCR for diagnosis of canine parvovirus infection. Virol Sin 25:352–360. doi: 10.1007/s12250-010-3132-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Demeke T, Jenkins GR. 2010. Influence of DNA extraction methods, PCR inhibitors and quantification methods on real-time PCR assay of biotechnology-derived traits. Anal Bioanal Chem 396:1977–1990. doi: 10.1007/s00216-009-3150-9. [DOI] [PubMed] [Google Scholar]
- 41.Miller WA, Rasochova L. 1997. Barley yellow dwarf viruses. Annu Rev Phytopathol 35:167–190. doi: 10.1146/annurev.phyto.35.1.167. [DOI] [PubMed] [Google Scholar]
- 42.Gray S, Gildow FE. 2003. Luteovirus-aphid interactions. Annu Rev Phytopathol 41:539–566. doi: 10.1146/annurev.phyto.41.012203.105815. [DOI] [PubMed] [Google Scholar]
- 43.D'arcy CJ, Burnett PA (ed). 1995. Barley yellow dwarf, 40 years of progress. The American Phytopathological Society, St. Paul, MN. [Google Scholar]
- 44.Perry KL, Kolb FL, Sammons B, Lawson C, Cisar G, Ohm H. 2000. Yield effects of Barley yellow dwarf virus in soft red winter wheat. Phytopathology 90:1043–1048. doi: 10.1094/PHYTO.2000.90.9.1043. [DOI] [PubMed] [Google Scholar]
- 45.Malmstrom CM, McCullough AJ, Johnson HA, Newton LA, Borer ET. 2005. Invasive annual grasses indirectly increase virus incidence in California native perennial bunchgrasses. Oecologia 145:153–164. doi: 10.1007/s00442-005-0099-z. [DOI] [PubMed] [Google Scholar]
- 46.Borer ET, Hosseini PR, Seabloom EW, Dobson AP. 2007. Pathogen-induced reversal of native dominance in a grassland community. Proc Natl Acad Sci U S A 104:5473–5478. doi: 10.1073/pnas.0608573104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schrotenboer AC, Allen MS, Malmstrom CM. 2011. Modification of native grasses for biofuel production may increase virus susceptibility. Glob Change Biol Bioenergy 3:360–374. doi: 10.1111/j.1757-1707.2011.01093.x. [DOI] [Google Scholar]
- 48.Power AG, Gray SM. 1995. Aphid Transmission of barley yellow dwarf viruses: interactions between viruses, vectors and host plants, p 259–289. In D'arcy CJ, Burnett PA (ed), Barley yellow dwarf: 40 years of progress. The American Phytopathological Society, St. Paul, MN. [Google Scholar]
- 49.Leclercq-Le Quillec F, Plantegenest M, Riault G, Dedryver CA. 2000. Analyzing and modeling temporal disease progress of Barley yellow dwarf virus serotypes in barley fields. Phytopathology 90:860–866. doi: 10.1094/PHYTO.2000.90.8.860. [DOI] [PubMed] [Google Scholar]
- 50.Seabloom EW, Borer ET, Mitchell CE, Power AG. 2010. Viral diversity and prevalence gradients in North American Pacific Coast grasslands. Ecology 91:721–732. doi: 10.1890/08-2170.1. [DOI] [PubMed] [Google Scholar]
- 51.French R. 1995. Barley yellow dwarf: diagnostic procedures and reagents, p 293–305. In D'arcy CJ, Burnett PA (ed), Barley yellow dwarf: 40 years of progress. The American Phytopathological Society, St., Paul, MN. [Google Scholar]
- 52.Savenkov EI, Valkonen JPT. 2001. Potyviral helper-component proteinase expressed in transgenic plants enhances titers of Potato leaf roll virus but does not alleviate its phloem limitation. Virology 283:285–293. doi: 10.1006/viro.2000.0838. [DOI] [PubMed] [Google Scholar]
- 53.McCullagh P, Nelder J. 1989. Generalized linear models. Chapman and Hall, New York, NY. [Google Scholar]
- 54.Pinheiro JC, Bates DM. 2000. Mixed effects models in S and S-Plus. Springer-Verlag, New York, NY. [Google Scholar]
- 55.Malmstrom CM, Shu R. 2004. Multiplexed RT-PCR for streamlined detection and separation of barley and cereal yellow dwarf viruses. J Virol Methods 120:69–78. doi: 10.1016/j.jviromet.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 56.Lacroix C, Seabloom EW, Borer ET. 2014. Environmental nutrient supply alters prevalence and weakens competitive interactions among coinfecting viruses. New Phytol 204:424–433. doi: 10.1111/nph.12909. [DOI] [PubMed] [Google Scholar]
- 57.Deb M, Anderson JM. 2008. Development of a multiplexed PCR detection method for Barley and Cereal yellow dwarf viruses, Wheat spindle streak virus, Wheat streak mosaic virus and Soil-borne wheat mosaic virus. J Virol Methods 148:17–24. doi: 10.1016/j.jviromet.2007.10.015. [DOI] [PubMed] [Google Scholar]
- 58.Kreader CA. 1996. Relief of amplification inhibition in PCR with bovine serum albumin or T4 gene 32 protein. Appl Environ Microbiol 62:1102–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abu Al-Soud W, Radstrom P. 2000. Effects of amplification facilitators on diagnostic PCR in the presence of blood, feces, and meat. J Clin Microbiol 38:4463–4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu JR, Yeh YC. 1973. Requirement of a functional gene 32 product of bacteriophage T4 in UV repair. J Virol 12:758–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Robertson NL, French R, Gray SM. 1991. Use of group-specific primers and the polymerase chain-reaction for the detection and identification of luteoviruses. J Gen Virol 72:1473–1477. doi: 10.1099/0022-1317-72-6-1473. [DOI] [PubMed] [Google Scholar]
- 62.Chomic A, Pearson MN, Clover GRG, Farreyrol K, Saul D, Hampton JG, Armstrong KF. 2010. A generic RT-PCR assay for the detection of Luteoviridae. Plant Pathol 59:429–442. doi: 10.1111/j.1365-3059.2010.02282.x. [DOI] [Google Scholar]
- 63.Kunta M, da Graca JV, Malik NSA, Louzada ES, Setamou M. 2014. Quantitative distribution of “Candidatus Liberibacter asiaticus” in the aerial parts of the Huanglongbing-infected citrus trees in Texas. HortScience 49:65–68. [Google Scholar]
- 64.Rashed A, Workneh F, Paetzold L, Gray J, Rush CM. 2014. Zebra chip disease development in relation to plant age and time of “Candidatus Liberibacter solanacearum” infection. Plant Dis 98:24–31. doi: 10.1094/PDIS-04-13-0366-RE. [DOI] [PubMed] [Google Scholar]
- 65.Karimi E, Jaafar HZE, Ahmad S. 2011. Phytochemical analysis and antimicrobial activities of methanolic extracts of leaf, stem and root from different varieties of Labisa pumila Benth. Molecules 16:4438–4450. doi: 10.3390/molecules16064438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.El-Bebany AF, Adam LR, Daayf F. 2013. Differential accumulation of phenolic compounds in potato in response to weakly and highly aggressive isolates of Verticillium dahliae. Can J Plant Pathol 35:232–240. doi: 10.1080/07060661.2013.773943. [DOI] [Google Scholar]
- 67.Larbat R, Paris C, Le Bot J, Adamowicz S. 2014. Phenolic characterization and variability in leaves, stems and roots of Micro-Tom and patio tomatoes, in response to nitrogen limitation. Plant Sci 224:62–73. doi: 10.1016/j.plantsci.2014.04.010. [DOI] [PubMed] [Google Scholar]
- 68.Sheldon BC, Verhulst S. 1996. Ecological immunology: costly parasite defences and trade-offs in evolutionary ecology. Trends Ecol Evol 11:317–321. doi: 10.1016/0169-5347(96)10039-2. [DOI] [PubMed] [Google Scholar]
- 69.Ricklefs RE, Wikelski M. 2002. The physiology/life-history nexus. Trends Ecol Evol 17:462–468. doi: 10.1016/S0169-5347(02)02578-8. [DOI] [Google Scholar]
- 70.Miller MR, White A, Boots M. 2007. Host life span and the evolution of resistance characteristics. Evolution 61:2–14. doi: 10.1111/j.1558-5646.2007.00001.x. [DOI] [PubMed] [Google Scholar]
- 71.Lind EM, Borer ET, Seabloom EW, Adler P, Bakker JD, Blumenthal DM, Crawley M, Davies K, Firn J, Gruner DS, Harpole WS, Hautier Y, Hillebrand H, Knops J, Melbourne B, Mortensen B, Risch AC, Schuetz M, Stevens C, Wragg PD. 2013. Life-history constraints in grassland plant species: a growth-defence trade-off is the norm. Ecol Lett 16:513–521. doi: 10.1111/ele.12078. [DOI] [PubMed] [Google Scholar]
- 72.Martin Ii LB, Hasselquist D, Wikelski M. 2006. Investment in immune defense is linked to pace of life in house sparrows. Oecologia 147:565–575. doi: 10.1007/s00442-005-0314-y. [DOI] [PubMed] [Google Scholar]
- 73.Lee KA, Wikelski M, Robinson WD, Robinson TR, Klasing KC. 2008. Constitutive immune defences correlate with life-history variables in tropical birds. J Anim Ecol 77:356–363. doi: 10.1111/j.1365-2656.2007.01347.x. [DOI] [PubMed] [Google Scholar]
- 74.Cronin JP, Welsh ME, Dekkers MG, Abercrombie ST, Mitchell CE. 2010. Host physiological phenotype explains pathogen reservoir potential. Ecol Lett 13:1221–1232. doi: 10.1111/j.1461-0248.2010.01513.x. [DOI] [PubMed] [Google Scholar]
- 75.Rosenthal JP, Dirzo R. 1997. Effects of life history, domestication and agronomic selection on plant defence against insects: evidence from maizes and wild relatives. Evol Ecol 11:337–355. doi: 10.1023/A:1018420504439. [DOI] [Google Scholar]
- 76.Glasa M, Malinowski T, Predajna L, Pupola N, Dekena D, Michalczuk L, Candresse T. 2011. Sequence variability, recombination analysis, and specific detection of the W strain of Plum pox virus. Phytopathology 101:980–985. doi: 10.1094/PHYTO-12-10-0334. [DOI] [PubMed] [Google Scholar]
- 77.Narayanasamy P. 2011. Microbial plant pathogens, vol 3 Detection and disease diagnosis: viral and viroid pathogens. Springer, Dordrecht, The Netherlands. [Google Scholar]
- 78.Smith DS, Maxwell PW, De Boer SH. 2005. Comparison of several methods for the extraction of DNA from potatoes and potato-derived products. J Agric Food Chem 53:9848–9859. doi: 10.1021/jf051201v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.