

Abstract
The production of the proinflammatory cytokines TNF-α and IL-6 is regulated by various mRNA-binding proteins, influencing stability and translation of the respective transcripts. Research in macrophages has shown the importance of the p38-MK2-tristetraprolin (TTP) axis for regulation of TNF-α mRNA stability and translation. In the current study we examined a possible involvement of p38 and TTP in LPS-induced cytokine production in bone marrow-derived mast cells (BMMCs). Using pharmacological inhibitors we initially found a strong dependence of LPS-induced TNF-α and IL-6 production on p38 activation, whereas activation of the Erk pathway appeared dispensable. LPS treatment also induced p38-dependent expression of the TTP gene. This prompted us to analyze the proinflammatory cytokine response in BMMCs generated from TTP-deficient mice. Unexpectedly, there were no significant differences in cytokine production between TTP-deficient and WT BMMCs in response to LPS. Gene expression and cytokine production of TNF-α and IL-6 as well as stability of the TNF-α transcript were comparable between TTP-deficient and WT BMMCs. In contrast to TTP mRNA expression, TTP protein expression could not be detected in BMMCs. While we successfully precipitated and detected TTP from lysates of LPS-stimulated RAW 264.7 macrophages, this was not accomplished from BMMC lysates. In contrast, we found mRNA and protein expression of the other TIS11 family members connected to regulation of mRNA stability, BRF1 and BRF2, and detected their interaction with 14-3-3 proteins. These data suggest that control of cytokine mRNA stability and translation in MCs is exerted by proteins different from TTP.
1. Introduction
Placed at the contact points between host and external environment, mast cells (MCs) are strategically positioned to respond to invading microorganisms [1]. For recognition of microbial pathogens, MCs express various Toll-like receptors (TLRs) [2]. These germline-encoded pattern recognition receptors (PRRs) are able to detect a broad spectrum of structures shared by pathogens, also known as pathogen-associated molecular patterns (PAMPs). Signaling of TLR4, the receptor for lipopolysaccharides (LPS) of Gram-negative bacteria, has been thoroughly investigated in MCs. In general, LPS bound by the LPS-binding protein (LBP) is thought to be transferred by either secreted or membrane bound (m) CD14 onto the TLR4/MD-2 complex, leading to receptor activation and downstream signaling. MCs have been demonstrated to lack expression of mCD14 [3]. Thus, they are able to recognize R-chemotypes of LPS by TLR4/MD-2 only, however, cannot be activated by S-chemotypes of LPS, which requires the expression of mCD14 [3]. A comparable response profile was observed in CD14-deficient macrophages [4].
Stimulation of TLR4 on MCs and macrophages leads to NF-kB-dependent production of proinflammatory cytokines, such as TNF-α and IL-6 [5-7]. Apart from NF-kB, TLR4 signaling can also stimulate MAP kinase pathways, resulting in the activation of Erk and p38 [2]. The influence of p38 on LPS-induced TNF-α and IL-6 production in macrophages via the p38-MAPKAP kinase 2 (MK2)-tristetraprolin (TTP) axis has been shown in numerous studies [5, 8]. Briefly, activation of p38 leads to phosphorylation of its downstream target MK2, which in turn phosphorylates serines 52 and 178 of TTP, a mRNA destabilizing protein [9]. TTP, also known as ZFP36, belongs to the TIS11 family of proteins that bind to mRNAs containing AU-rich elements (AREs) in their 3' UTRs. This leads to deadenylation and subsequent destabilization of ARE-containing transcripts [10]. Among these mRNAs are cytokines such as TNF-α and IL-6, but also growth factors like GM-CSF and VEGF [11]. Besides TTP, two other TIS11 family members BRF1 and BRF2, also known as ZFP36L1 and ZFP36L2, respectively, show the ability to destabilize ARE-containing mRNAs [11]. Apart from destabilization, proteins binding AREs can also lead to stabilization of the respective mRNAs. Exemplarily, the ubiquitous nuclear protein HuR has been shown to bind to ARE-containing transcripts, thereby increasing their half-life [12]. HuR and the recently discovered ARE-binding protein Zfand5 bind to the AREs in the TNF-α mRNA, competitively counteracting the effect of TTP binding and thus leading to stabilization and enhanced translation of the TNF-α transcript [13, 14]. The balance between mRNA stabilizing and destabilizing proteins competing for the same ARE sites adds another layer of regulation to the overall translation of proteins.
The vast amount of research on the functions of TTP was performed in macrophage cell systems. To our knowledge there has only been one study published about TTP activity in bone marrow-derived MCs (BMMCs). In this study, a role of TTP in IL-4 mediated downregulation of antigen-induced TNF-α production was described [15].
In a previous study we have shown that pharmacological inhibition of p38 activity leads to a complete shutdown of LPS-induced IL-6 production in BMMCs [16]. Based on the fact that IL-4-induced TTP protein expression was shown in BMMCs in the aforementioned study, this prompted us to investigate a possible role of TTP in LPS-induced cytokine production in BMMCs. In the present study we show that inhibition of p38 leads to a strong decrease of both IL-6 and TNF-α production. Inhibition of MEK1/2, on the other hand, only moderately reduced cytokine production. Using the well-established TTP knockout mice [17], we have generated TTP-deficient BMMCs. Differentiation of these cells as well as degranulation response to antigen was comparable to wildtype BMMCs. Surprisingly, we could not find any differences in response to LPS stimulation between wildtype and TTP-deficient BMMCs, neither in IL-6 and TNF-α expression nor in production of IL-6 and TNF-α mRNA. Stability of TNF-α mRNA was also similar in wildtype and TTP-deficient BMMCs. Even though we can show considerable TTP mRNA expression in wildtype BMMCs in response to LPS, our data suggest that, in contrast to macrophages, TTP is not involved in the regulation of LPS-induced TNF-α and IL-6 production in MCs.
2. Materials and Methods
2.1 Cell Culture
BMMCs were generated according to procedures established by Razin et al. [18]. Bone marrow cells (1 × 106/ml) from 6 to 8 week old mice (129/Sv × C57BL/6) were cultured (37 °C, 5% CO2) as single cell suspensions in RPMI 1640 medium containing 12% FBS, 1% X63Ag8-653-conditioned medium, as a source of IL-3 [19], 2 mM L-glutamine, 1 × 10−5 M 2-mercaptoethanol, 50 units/ml penicillin, and 50 mg/ml streptomycin. At weekly intervals, the non-adherent cells were reseeded at 5 × 105 cells/ml in fresh medium. By 4–6 weeks in culture, greater than 99% of the cells were c-kit and FcεR1 positive as assessed by phycoerythrin-labeled anti-c-kit antibodies (Pharmingen, Mississauga, Canada) and FITC-labeled anti-FcεRIα antibodies (eBioscience, San Diego, CA, USA), respectively. Wildtype (TTP-expressing) and TTP-deficient BMMCs were in vitro differentiated using the same protocol but starting from bone marrow cells of 6-8 week old wild-type and TTP-deficient littermates (129/Sv × C57BL/6) [17]. RAW 264.7 cells were cultured (37 °C, 5% CO2) as adherent cell culture in RPMI 1640 medium containing 10% FBS, 2 mM L-glutamine, 1 × 10−5 M 2-mercaptoethanol, 50 units/ml penicillin, and 50 mg/ml streptomycin.
2.2 Reagents
R-form LPS from S. Minnesota mutant R595 was extracted and purified as described [20-22] and was a gift from M. Freudenberg and C. Galanos (MPI for Immunobiology, Freiburg, Germany). PD0325901 was purchased from axon Medchem (Groningen, Netherlands) and BIRB0796 from the Division of Signal Transduction Therapy, College of Life Sciences, University of Dundee, Dundee, Scotland, U.K. DNP-HSA containing 30–40 moles DNP per mole albumin and monoclonal IgE with specificity for DNP (SPE-7) were obtained from SIGMA (Deisenhofen, Germany). Protein A-HRP conjugate was purchased from Merck (Darmstadt, Germany). Monoclonal anti-GAPDH (6C5, sc-32233) and monoclonal anti-pan-14-3-3 (H8, sc-1657) antibodies were obtained from Santa Cruz Biotechnology (Heidelberg, Germany). Polyclonal anti-BRF1/2 (#2119) and anti-phospho-MAPKAPK-2 (phospho-MK2, Thr334, #3007) antibodies were purchased from Cell Signaling Technology (Frankfurt a.M., Germany). Polyclonal rabbit anti-TTP antisera were a gift from A. Clark (SAK21A, described here [23]) and P. Kovarik (K2, described here [24]).
2.3 Cytokine ELISAs
Mouse IL-6 ELISAs (BD Pharmingen, Heidelberg, Germany) and mouse TNF-α ELISAs (R&D Systems, Wiesbaden-Nordenstadt, Germany) were performed according to the manufacturer's instructions. Absolute levels of cytokines in culture supernatants varied between experiments and BMMC cultures. Qualitative differences or similarities between WT and mutant cells, however, were consistent throughout the study.
2.4 Degranulation assay
For degranulation studies, cells were preloaded with 0.15 μg/ml IgE anti-DNP overnight at 37 °C. The cells were then washed and resuspended in Tyrode's buffer (130 mM NaCl, 5 mM KCl, 1.4 mM CaCl2, 1 mM MgCl2, 5.6 mM glucose, and 0.1% bovine serum albumin (BSA) in 10 mM Hepes, pH 7.4). The cells were adapted to 37 °C for 20 min and then treated for 20 min at 37 °C as mentioned. The degree of degranulation was determined by measuring the release of β-hexosaminidase [25].
2.5 RNA preparation and quantitative RT-PCR
RNA from 3 million BMMCs was extracted using the RNeasy Mini Kit (Qiagen) according to the manufacturer's instructions. For measuring RNA stability, 5 μg/ml Actinomycin D (AcD) was added to stimulated samples and after the respective time-points the reaction was stopped by peletting, lysing and freezing the cells in liquid nitrogen. Total RNA (1 μg) was reverse transcribed using random hexamers (Roche) and the Omniscript RT Kit (Qiagen) according to the manufacturer's instructions. qPCR was performed on a Rotorgene (Qiagen) by using SYBR green reaction mix (Bioline #QT650-02). Expression was normalized to the housekeeper genes gusb or gapdh. The relative expression ratio including primer efficiencies was calculated by the Pfaffl method [26]. Primer sequences and efficiency data are specified in Table 1. Experiments were performed with cells from independent BMMC cultures. SD and respective statistics were calculated and indicated in cases of qualitatively and quantitatively comparable results between the different cultures. In cases of qualitatively comparable, but quantitatively different results due to the use of independent cell cultures, relative expression ratios including primer efficiencies according to Pfaffl [26] were shown without SD (see exemplary Fig. 2A and respective Suppl. Fig. 1). Values are depicted relative to the values obtained in untreated WT cells.
Table 1.
Primer sequences and efficiencies
| Murine gene | Forward primer | Reverse primer | Efficiency |
|---|---|---|---|
| Gapdh | ACT CAA GAT TGT CAG CAA TGC A | TGG TCA TGA GCC CTT CCA CAA | 1.9483 |
| Gusb | QuantiTect Primer Assay (Qiagen #QT00176715) | 2.01478 | |
| IL-6 | TCC AGT TGC CTT CTT GGG AC | GTG TAA TTA AGC CTC CGA CTT G | 2.00934 |
| TNF-α | AGC ACA GAA AGC ATG ATC CGC | TGC CAC AAG CAG GAA TGA GAA G | 2.1854 |
| TTP | GAG GGC CGA AGC TGC GGC TGG GT | GGT GGC GAT TGG CTT GGC GAA G | 1.92195 |
| BRF1 | GAC CTT CAC GAC ACA CCA GAT | CGC TGG GAG TGC TGT AGT TG | 2.02463 |
| BRF2 | CAC AAC TTT CCG TCC CTC CTT | TTC TGG GTC CTG TAA TGG TCG | 1.99879 |
| Zfand5 | AGC CAG TTG TCA CTC AGC CCA | CCA CAT CGG CAG TCA AAC CCT GT | 2.05232 |
Primer efficiencies were determined as described by Pfaffl [26].
Fig. 2.
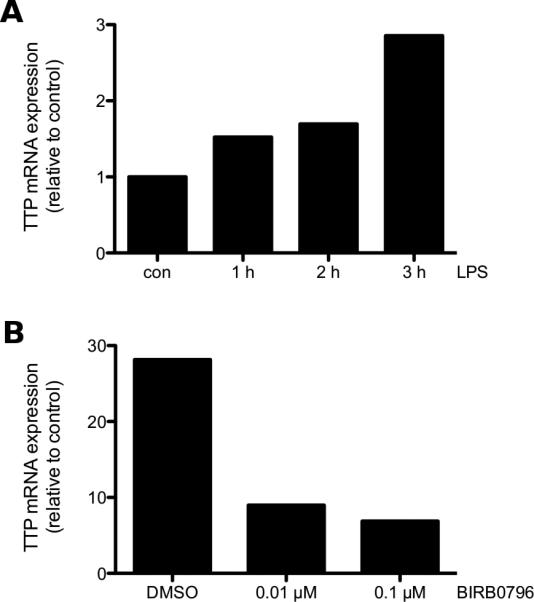
LPS induces TTP gene expression in BMMCs in a p38-dependent manner. (A) WT BMMCs were left untreated (con) or stimulated with 3 μg/ml LPS for the indicated times. TTP mRNA expression was analyzed by RT-qPCR. Data is representative of three independent experiments using seperate cell cultures (see Suppl. Fig. 1). (B) WT BMMCs were incubated with either DMSO or the indicated concentrations of BIRB0796 for 20 min and subsequently stimulated with 3 μg/ml LPS for 2 h. TTP mRNA was analyzed by RT-qPCR. Data is representative of three independent experiments using separate cell cultures (see Suppl. Fig. 2).
2.6 Mast cell stimulation, immunoprecipitation and Western blotting
IgE-preloaded (0.15 μg/ml overnight) cells were washed with PBS and resuspended in RPMI/0.1% BSA. Cells were adapted to 37 °C for 20 min and stimulated with the indicated concentrations of the stimuli. After stimulation for different lengths of time, cells were pelleted and solubilized with 0.5% NP-40 and 0.1% deoxycholate in phosphorylation solubilization buffer (PSB) at 4 °C [27]. After normalizing for protein content, the postnuclear supernatants were subjected directly to SDS-PAGE and Western blot analysis. For immunoprecipitation the respective antibody was added to cell lysates normalized for protein content according to manufacturer's instructions. Lysates were then incubated on a rotator overnight at 4 °C. The next day protein G sepharose beads were added to the lysates and incubated for 1 h on a rotator at 4 °C. Precipitates were washed three times with PSB containing one third of detergent concentration used for lysis and analyzed by Western blotting.
2.7 Satistical analysis
P values were calculated by the paired two-tailed Student's t test. P values of * < 0.05, ** < 0.005, and *** < 0.0005 were considered statistically significant. Values higher than a p value of 0.05 were regarded as not significant (n.s.).
3. Results
3.1 p38 is essential for LPS-induced cytokine production in mast cells
Previously we have shown that the PI3K pathway differentially regulates cytokine production in response to LPS in BMMCs [28]. As activation of the p38 and Erk MAPK pathways has also been shown downstream of TLR signaling [2], we wanted to assess the effect of specific inhibition of the respective pathways on cytokine production. For this purpose, we used the p38 inhibitor BIRB0796 and the MEK1/2 inhibitor PD0325901. WT BMMCs were preincubated with the respective inhibitors or vehicle (DMSO) and subsequently stimulated with LPS for 4 h. We found that treatment with PD0325901 only moderately decreased LPS-induced TNF-α and IL-6 production, while incubation with BIRB0796 drastically diminished both production of TNF-α and IL-6 (Fig. 1). The concentration of PD0325901 used was previously shown to be sufficient to inhibit Erk activation in BMMCs [29]. These data demonstrate the importance of the p38 MAPK pathway in regulation of LPS-induced cytokine production in BMMCs.
Fig. 1.
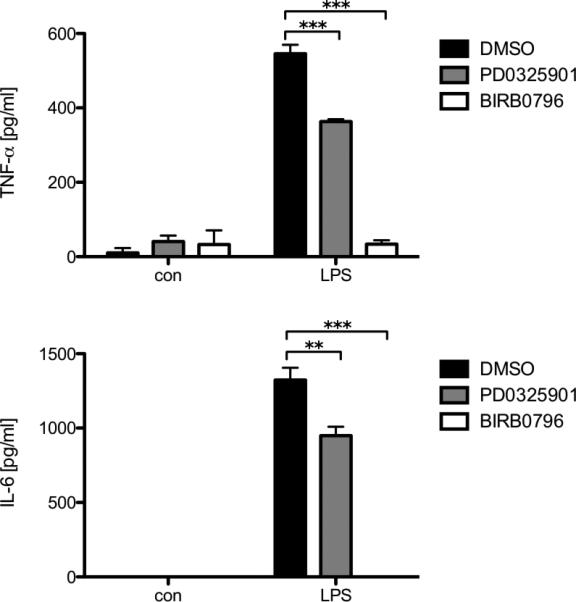
Inhibition of p38 shuts down LPS-induced cytokine production while inhibition of the Erk pathway only moderately decreases it. WT BMMCs were pretreated for 20 min with either DMSO, 1 μM PD0325901 or 0.1 μM BIRB0796, then left untreated (con) or stimulated with 3 μg/ml LPS for 4 h. Subsequently, the amounts of TNF-α (upper panel) and IL-6 (lower panel) were determined by ELISA. Each bar represents the mean of triplicates ± SD. Comparable results were obtained in several experiments using different BMMC cultures (n ≥ 3).
3.2 LPS induces p38-dependent TTP-mRNA expression in BMMCs
For both cytokines extensive research has been performed regarding regulation of mRNA stability and translation via TTP [5, 8, 11]. As the activity of TTP is regulated via the p38 MAPK pathway, we checked if TTP mRNA expression is induced after LPS treatment. WT BMMCs were either left untreated or stimulated with LPS for either 1, 2 or 3 h. The TTP gene transcript was already detectable after 1 h and increased until 3 h of LPS treatment (Fig. 2A and Suppl. Fig. 1). A previous study in macrophages showed that inhibition of the p38 MAPK pathway leads to reduction of TTP gene expression [30]. As we had seen strong influence of p38 on cytokine production in MCs, we investigated the potential p38-dependent regulation of TTP mRNA expression. Indeed, we found strong reduction of TTP mRNA expression already with a low concentration of inhibitor in comparison to the control (Fig. 2B and Suppl. Fig. 2). These data suggest the presence of TTP and its potential regulation via transcription or mRNA stability by p38 in BMMCs.
3.3 TTP is dispensable for development of BMMCs
To check for the function of TTP in MCs we generated BMMCs from TTP KO mice [17]. After 5 weeks in culture we found surface expression of the markers FcεRI and c-Kit to be comparable between TTP-deficient and WT BMMC cultures (Fig. 3A). To test for general responsiveness of the cells, we performed a degranulation assay comparing TTP-deficient and WT BMMCs. As we have shown in previous publications, degranulation is a fast reaction of the MCs to antigen stimulation, occurring in a matter of minutes. [31] As the described functions of TTP are all mRNA related, TTP deficiency should have no influence on the process of degranulation. Indeed measuring β-hexosaminidase activity as readout for degranulation we found no significant differences in antigen-triggered degranulation between TTP-deficient and WT BMMCs (Fig. 3B). These data show that BMMCs lacking expression of TTP develop normally and show no impairment in the degranulation response.
Fig. 3.

TTP-deficient BMMCs develop and degranulate comparable to WT BMMCs. (A) Differentiation of WT and TTP-deficient BMMCs after 5 weeks in culture was determined by FACS analysis measuring surface expression of FcεRI and c-Kit. Comparable results were obtained in 5 independent WT and TTP-deficient BMMC cultures. (B) WT (■) and TTP-deficient (KO) BMMCs (□) were preloaded with IgE overnight. Cells were left untreated (con) or stimulated with 20 ng/ml antigen (DNP-HSA) for 20 min. Degranulation was assessed by relative release of β-hexosaminidase. Each bar represents the mean ± SD of three independent BMMC cultures, each taken from the mean of duplicates.
3.4 TTP-deficient BMMCs show no alterations in LPS-induced cytokine production compared to WT BMMCs
The major readout for loss of function of TTP in macrophages is the increase in both TNF-α transcript and protein expression, which has also been shown for IL-6 [5, 8, 32]. First we checked TTP-deficient and WT BMMCs for differences in gene expression of the cytokines TNF-α and IL-6. TTP-deficient and WT BMMCs were either left untreated or stimulated with LPS for 90 min or 3 h. Induction of TNF-α mRNA expression did not further increase from 90 min to 3 h (data not shown) and there was no significant difference between TTP-deficient and WT BMMCs (Fig. 4A, left panel). For expression of IL-6 mRNA we saw an increase from 90 min to 3 h, but also no significant difference between TTP-deficient and WT BMMCs at any time-point (Fig. 4A, right panel). We then looked for differences in cytokine production, as the amount of protein translated from mRNA might differ between TTP-deficient and WT cells. TTP has recently been shown not only to decrease stability but also to block translation of transcripts in two independent studies [14, 33]. Thus the lack of TTP could still lead to increased cytokine production. However, production of both TNF-α and IL-6 did not show significant differences between TTP-deficient and WT cells after LPS treatment (Fig. 4B). Taken together these results imply a function of TTP in BMMCs that is different from its published role in macrophages. This suggests that there should not be any difference regarding stability of TNF-α mRNA between TTP-deficient and WT BMMCs. To investigate this, we treated LPS stimulated TTP-deficient and WT BMMCs with the transcription inhibitor Actinomycin D (AcD) and harvested AcD-treated cells at several time-points. In agreement with the previous data we found no significant difference between TTP-deficient and WT BMMCs regarding stability of TNF-α mRNA (Fig. 4C). All together the data presented show that TTP-deficiency in BMMCs causes no evident phenotype regarding LPS-induced production of proinflammatory cytokines (TNF-α and IL-6).
Fig. 4.

Gene expression and protein production of TNF-α and IL-6 is comparable between WT and TTP-deficient BMMCs. (A) WT (■) and TTP-deficient (KO) BMMCs (□) were left untreated (con) or stimulated with 3 μg/ml LPS for 90 min or 3 h. TNF-α (left panel) and IL-6 mRNA (right panel) expression was analyzed by RT-qPCR. Each bar represents the mean ± SD of three independent BMMC cultures. Comparable results were obtained in several experiments (n ≥ 3). (B) WT (■) and TTP-deficient (KO) BMMCs (□) were left untreated (con) or stimulated with 3 μg/ml LPS for 4 h. Subsequently, the amounts of TNF-α (left panel) and IL-6 (right panel) were determined by ELISA. Each bar represents the mean ± SD of three independent BMMC cultures, each taken from the mean of triplicates. Comparable results were obtained in several experiments (n ≥ 5). (C) WT (■) and TTP-deficient BMMCs (□) were treated with 3 μg/ml LPS for 2 h. Subsequently, 5 μg/ml Actinomycin D (AcD) were added. After 0 min, 30 min, 1 h and 2 h cells were harvested and TNF-α mRNA was analyzed by RT-qPCR. All samples were normalized to the respective control. Each bar represents the mean ± SD of three independent BMMC cultures. Statistical significance refers to the differences between WT and TTP-deficient BMMCs.
3.5 Lack of phenotype of TTP-deficient cells may be due to undetectable protein expression of TTP in wildtype BMMCs
Since all of the previous results oppose the established function of TTP in LPS signal transduction, we had to make sure that there is a significant difference in TTP expression between TTP-deficient and WT BMMCs. Therefore, TTP-deficient and WT BMMCs were either left untreated or stimulated with LPS for 1, 2 or 3 h. While we could confirm TTP mRNA expression in WT BMMCs, peaking at 2 h of stimulation, we could not detect TTP transcripts in the TTP-deficient BMMCs (Fig. 5A and Suppl. Fig. 3). Several TTP antisera have been developed for detection of TTP by Western blotting. We obtained two antisera raised against the C-terminus of TTP, SAK21A [23], and K2 [24], to check protein expression. TTP-deficient, WT BMMCs and RAW 264.7 macrophages, as a positive control, were either left untreated or stimulated with LPS for 4 h. The BMMCs had to be treated with a higher LPS concentration than the RAW macrophages, as the reaction of BMMCs to LPS is weaker than of macrophages, possibly due to absence of mCD14 and the TRIF signaling pathway [3, 34]. Detection with both antisera showed strong expression of TTP in RAW 264.7 macrophages after LPS treatment, which was absent in LPS-stimulated WT BMMCs (Fig. 5B). The basal level of TTP in unstimulated RAW 264.7 cells already appeared stronger than TTP expression in LPS-stimulated BMMCs (Fig. 5B). To be sure that neither our method of lysis nor our blotting procedure is the reason for not being able to detect TTP in BMMCs, we tested hot lysis of cell pellets as well as wet blotting in contrast to the previously used semi-dry blotting. None of these changes led to detection of TTP protein after LPS treatment in WT BMMCs (data not shown). Since the levels of TTP protein may simply be too low for detection via Western blotting, we looked at TTP protein upon immunoprecipitation using an N-terminal antiserum, SAK20A [23]. RAW 264.7 macrophages were used as positive control. Postnuclear supernatants (PS) again showed induction of TTP protein expression only in LPS treated RAW 264.7 cells but not in WT BMMCs (Fig. 5C, first panel from top). As well, TTP protein could only be precipitated from unstimulated and LPS-stimulated RAW 264.7 macrophages. As the signal of the heavy chain (HC) of the precipitating antibody can superimpose the signal of proteins around 50 kDa size, as in the case for TTP, we used protein A conjugated with HRP for detection as described [35]. While we still saw a strong signal of the HC, we could readily detect TTP in LPS treated RAW 264.7 cells and weakly in untreated RAW cells, but not in WT and TTP-deficient BMMCs (Fig. 5C, third panel from top). As the signal detected from the HC after immunoprecipitation may still mask modified TTP protein running at a larger size, we also checked for interaction of TTP with 14-3-3 proteins. It has been shown before that LPS-induced TTP expression also increases binding of 14-3-3 proteins to TTP [9]. Confirming our previous results we only found precipitated 14-3-3 proteins in untreated and LPS-stimulated RAW 264.7 macrophages with the expected increase in interaction in response to LPS stimulation. In both TTP-deficient and WT BMMCs we were not able to detect 14-3-3 proteins in the anti-TTP precipitates (Fig. 5C, fourth panel from top). These data suggest a lack of TTP protein expression in LPS-stimulated BMMCs.
Fig. 5.

Despite TTP gene expression no TTP protein is detectable in WT BMMCs. (A) WT (■) and TTP-deficient (KO) BMMCs (□) were left untreated (con) or stimulated with 3 μg/ml LPS for the indicated times. TTP mRNA was analyzed by RT-qPCR. Data is representative of three independent experiments using seperate cell cultures (see Suppl. Fig. 3). (B) WT BMMCs, TTP-deficient BMMCs, and RAW 264.7 macrophages were left untreated (con) or stimulated with LPS for 4 h (3 μg/ml for BMMCs, 0.1 μg/ml for RAW macrophages). Postnuclear supernatants (PS) (40 μg protein for BMMCs, 20 μg protein for RAW macrophages) were analyzed by immunoblotting with antisera raised against the C-terminus of TTP (SAK21A [23] top panel, K2 [24] middle panel) and an antibody against GAPDH (bottom panel). (C) WT, TTP-deficient BMMCs and RAW 264.7 macrophages were left untreated (con) or stimulated with LPS for 3 h (3 μg/ml for BMMCs, 0.1 μg/ml for RAW macrophages). Pellets were lysed and PS were subjected to anti-TTP immunoprecipitation. Precipitates (IP, third and fourth panel from top) as well as PS (first and second panel from top) were analyzed by anti-TTP (first and third panel from top) and anti-14-3-3 (second and fourth panel from top) immunoblotting. HC, heavy chain
3.6 BMMCs and RAW macrophages show differential mRNA expression of TIS11 family members
As the previous results showed great differences of protein expression of TTP between BMMCs and RAW macrophages, we were interested if mRNA expression of TTP would show the same trends. We found higher TTP mRNA expression in RAW 264.7 macrophages in comparison to WT BMMCs already in unstimulated cells. The discrepancy in TTP mRNA levels between the two cell types increased further with LPS treatment. The greatest disparity was found after 1 h, with the RAW macrophages showing a 6 times higher mRNA expression of TTP than WT BMMCs (Fig. 6A). TTP is part of the TIS11 protein family and two other members of this family, BRF1 and BRF2, have been shown to be comparable in their function and regulation [11]. We investigated if mRNA expression of the other TIS11 family proteins was regulated in the same way as TTP mRNA expression. In contrast to TTP, mRNA expression of both BRF1/2 in WT BMMCs was higher than in RAW macrophages at all times. While LPS stimulation upregulated mRNA expression of BRF1/2 over time in BMMCs, only a minor regulation occurred in RAW macrophages (Fig. 6B/C). Apart from TIS11 family proteins we also investigated mRNA expression of the recently discovered Zfand5, a protein stabilizing ARE-containing transcripts [13]. While we found a higher expression of Zfand5 transcript in RAW macrophages than in WT BMMCs that were either untreated or stimulated with LPS for 1 h, after 3 h of LPS treatment mRNA expression of Zfand5 was comparable between the different cell types (Fig. 6D). This data shows that the transcripts for TIS11 family proteins and Zfand5 are differentially expressed in WT BMMCs and RAW 264.7 macrophages.
Fig. 6.

Differential expression of TTP and BRF1/2 transcripts in BMMCs and RAW 264.7 macrophages. RAW 264.7 macrophages (□) and WT BMMCs (■) were left untreated (con) or stimulated with LPS (3 μg/ml for BMMCs, 0.1 μg/ml for RAW macrophages) for 1 h or 3 h. TTP (A), BRF1 (B), BRF2 (C) and Zfand5 mRNA (D) was analyzed by RT-qPCR. Each bar represents the mean ± SD of three independent RAW 264.7 or BMMC cultures.
3.7 The TIS11 family proteins BRF1 and BRF2 are expressed in BMMCs and may compensate for the lack of TTP
Next, we were interested whether TTP-deficient BMMCs show differences in mRNA expression of BRF1 and BRF2 in comparison to WT BMMCs. Deficiency of TTP did not lead to a significant difference in mRNA expression of either BRF1 or BRF2 when compared to wildtype cells (Fig. 7A, top and middle panels). As well expression of the transcript for Zfand5 showed no significant difference over the course of LPS stimulation between TTP-deficient and WT BMMCs (Fig. 7A, bottom panel).
Fig. 7.

Other TIS11 family proteins as well as the ARE-mRNA stabilizing protein Zfand5 are expressed in BMMCs. (A) WT (■) and TTP-deficient BMMCs (□) were left untreated (con) or stimulated with 3 μg/ml LPS for 90 min or 3 h. BRF1 (top panel), BRF2 (middle panel) and Zfand5 mRNA (bottom panel) was analyzed by RT-qPCR. Each bar represents the mean ± SD of three independent BMMC cultures. Comparable results were obtained in several experiments (n ≥ 3). (B) WT BMMCs were left untreated (con) or stimulated with 3 μg/ml LPS for 2 h. Pellets were lysed and PS were subjected to anti-14-3-3 immunoprecipitation. Precipitates (IP, left panels) as well as PS (right panels) were analyzed by anti-BRF1/2 (top panels) and anti-14-3-3 (bottom panels) immunoblotting. Densitometry was performed and relative expression levels (normalization on precipitated 14-3-3) are indicated.
As we have seen TTP transcript but could not detect TTP protein, we wanted to investigate protein expression of BRF1 and BRF2. In PS of untreated and LPS stimulated BMMCs we could not detect protein at the predicted sizes of BRF1 and BRF2 (Fig. 7B, right panel). It has been shown before that both BRF1 and TTP bind to 14-3-3 proteins when phosphorylated due to activation of PKB or p38 MAPK signaling pathways [9, 36]. In 14-3-3 precipitates we could detect protein at the predicted sizes of both BRF1 and BRF2, although, according to densitometry normalizing on precipitated 14-3-3, interaction did not change between untreated and LPS-stimulated WT BMMCs (Fig. 7B, left panel). As the used antibody detects both BRF1 and BRF2 and their predicted sizes are similar, we were not able to determine if only one of the two or both are binding to 14-3-3. We also checked for precipitation of TTP with anti-14-3-3 antibodies, but as implied by previous data, no interaction could be detected (data not shown). Taken together these data indicate that while we found no protein expression of TTP, the TIS11 family proteins BRF1 and BRF2 are both expressed at mRNA and protein level in BMMCs and may compensate for the lack of TTP expression.
4. Discussion
Correct regulation of cytokine production is vital for an adequate immune response, limiting damage to the host and preventing the development of chronic diseases. Overexpression of both TNF-α and IL-6 was reported, amongst other syndromes and diseases, to cause cachexia, rheumatoid arthritis, and atherosclerosis [37-40]. Especially for production of TNF-α, intensive research has shown that stability and rate of translation of TNF-α mRNA is tightly regulated by various proteins [5, 12-14, 33]. Some of these pathways were also confirmed to play roles for mRNA stability in regulation of IL-6 production [8]. Central to regulation of TNF-α production in macrophages is the mRNA-binding protein TTP. So far, most of the research on TTP was based on macrophage cell systems, in particular in the context of LPS stimulation. To our knowledge only one study has been published investigating TTP in BMMCs, but in contrast to our study the authors concentrated on signaling induced by antigen stimulation and induction of TTP by IL-4 [15]. This prompted us to investigate the involvement of TTP in LPS-induced cytokine production in BMMCs.
The principal finding of our study was that in response to LPS stimulation only the TTP transcript was produced but no TTP protein in BMMCs was detectable. Instead we found mRNA and protein expression of two other TIS11 family proteins BRF1/2 and mRNA expression of a recently described mRNA-stabilization promoting protein, named Zfand5. These results imply that proteins other than TTP may partake in balancing the response of BMMCs to LPS treatment.
Initially, we checked if LPS-induced cytokine production in BMMCs is indeed regulated via p38 MAPK activity, as this is the main pathway involved in regulation of TTP [5, 14, 23, 41]. We could show that both LPS-induced TNF-α and IL-6 gene expression and protein production are suppressed when p38 activity is inhibited (Fig. 1 and data not shown). Together with the results showing upregulation of TTP mRNA expression after LPS treatment and decrease of LPS-induced gene expression of TTP in the presence of the p38 inhibitor BIRB0796 (Fig. 2 and Suppl. Figs. 1 and 2), we have confirmed basic results about function and regulation of TTP in macrophages [23, 41] in the model system of BMMCs. In contrast to p38 inhibition, we only found weak effects on LPS-induced cytokine production when inhibiting the Erk pathway (Fig. 1). This was in line with results published about the substances imperatorin and roxatidine, which inhibit LPS-induced p38 but not Erk activity in RAW 264.7 macrophages. These substances were shown to downregulate TNF-α and IL-6 production in LPS-stimulated macrophages, implying dispensability for the Erk pathway in this respect [42, 43]. Interestingly, TACE-mediated processing of pre-TNF was demonstrated to be dependent on Erk signaling in LPS-stimulated macrophages [44], indicating cell type-specific regulation of TNF-α production.
In an attempt to more precisely investigate the role of TTP in LPS-induced signaling in BMMCs, we generated BMMCs from TTP-deficient and WT mice. TTP-deficient and WT BMMCs proliferated and developed comparably in terms of surface expression of FcεRI and c-Kit, and they degranulated in a similar fashion in response to antigen (Fig. 3). While compared to WT BMMCs TTP-deficient cells showed a slightly lower surface expression of FcεRI, we have not found any differences in degranulation in thorough antigen dose-response studies (data not shown).
Surprisingly we found no significant differences between TTP-deficient and WT BMMCs in LPS-induced TNF-α and IL-6 gene and protein expression or stability of TNF-α mRNA (Fig. 4). As well TTP protein was not detectable after LPS treatment up to 8 h in WT BMMCs while we could readily detect TTP in RAW 264.7 macrophages stimulated with LPS (Fig. 5B and data not shown). Concurrently, immunoprecipitation with an N-terminal anti-TTP antibody only precipitated TTP in the RAW 264.7 macrophages, but not in any of the BMMCs (Fig. 5C). TTP protein expression has been shown to be severely diminished in cells devoid of MK2/3 activity [5]. We found this not to be the reason for the absence of TTP protein in BMMCs, as we could detect LPS-induced phosphorylation of MK2 in BMMCs (Suppl. Fig. 5). Also, inhibition of the proteasome has been shown to stabilize TTP protein expression in RAW 264.7 macrophages and in several melanoma cell lines [45, 46]. In contrast to these reports, we could not detect TTP protein in LPS-stimulated WT BMMCs pretreated with the proteasomal inhibitor MG-132 (data not shown). Thus, we conclude while we can reproducibly confirm expression of transcripts for TTP in WT BMMCs, there is no detectable expression of protein occurring. This finding may explain why we could not find any significant differences between TTP-deficient and WT BMMCs in response to LPS.
As we had to assume from our results that LPS does not induce TTP protein expression in BMMCs, we set out to analyze expression of other TIS11 family members, BRF1 and BRF2, in BMMCs, which might compensate for the lack of TTP. The fourth member of the TIS11 family of proteins, Zfp36l3, is restricted in expression to placental and extraembryonal mouse tissue [47]. Indeed we found enhanced mRNA expression of BRF1 and BRF2 after LPS treatment that was comparable between TTP-deficient and WT BMMCs. (Fig. 6 B/C, Fig. 7A, top and middle panel). Similar to TTP, we found no protein expression in total cell lysates but could confirm presence of BRF1/2 protein via coimmunoprecipitation with 14-3-3 (Fig. 7C), a known interaction partner of phosphorylated TTP and BRF1 [9, 48]. For the stimulation time-point chosen we were not able to see an increase in interaction of BRF1/2 and 14-3-3. As the used antibody does not distinguish between BRF1 and BRF2 and both of them are similar in predicted size, 36 and 37 kDa for the murine proteins respectively, we cannot say if either one exclusively or both of them together interact with the 14-3-3 proteins. To our knowledge published data only confirmed interaction of TTP and BRF1 with 14-3-3 [9, 48]. So far no study specifically investigated BRF2 for that matter. Given that TTP and BRF1/2 share over 70 % amino acid identity and TTP and BRF1 have been shown to be regulated in a similar fashion, it is well possible that BRF2 may also interact with 14-3-3 proteins [11]. It seems that BRF1/2 are only produced at a very low level in BMMCs, suggesting weak influence on LPS-induced cytokine production. Apart from mRNA-destabilizing proteins, we also checked for expression of a recently discovered ARE-containing mRNA-stabilizing protein, Zfand5 [13]. Similar to the results of that study we found expression of Zfand5 mRNA after LPS treatment of BMMCs (Fig. 6D, Fig. 7A, bottom panel). This lead to the conclusion that apart from TTP, other mRNA-stabilizing and -destabilizing proteins are expressed in BMMCs in response to LPS treatment,
This difference in LPS-signaling between macrophages that strongly express TTP, and MCs that according to our results do not express detectable amounts of TTP protein, may lie in the disparity of TNF-α production in response to LPS treatment between these two cell types. Macrophages are frequently cited to be the major source of TNF-α during inflammatory processes [17, 49, 50]. In a previous study we have shown in a direct comparison that bone marrow-derived macrophages produced more than hundred-fold greater amounts of TNF-α than BMMCs relatively to the Re-LPS concentration used for stimulation [34]. For such a massive cytokine production, strong negative regulatory mechanisms are needed to keep the response in check. Heavy upregulation of TTP ensures fast control over stability and translation of TNF-α transcripts, as TTP accumulates when inactivated by phosphorylation via the TLR4-induced p38-MK2 axis [30]. As soon as the response to LPS diminishes, activity of p38 and MK2 is reduced and the phosphatase PP2A can dephosphorylate the accumulated TTP protein, which should then be able to rapidly destabilize and inhibit further translation of the TNF-α mRNA [51]. As MCs produce much lower amounts of TNF-α, low expression of the BRF proteins may be enough to control the amount of TNF-α transcripts in these cells without the help of TTP. This is supported by our results showing that RAW macrophages express more than 6 times higher amounts of TTP transcript than WT BMMCs (Fig. 6A). The opposite situation was found for BRF1/2 mRNA expression. While WT BMMCs already show a higher expression of BRF1/2 mRNA in control cells, almost no regulation occurs in RAW macrophages (Fig. 6B/C). While we were not able to detect TTP protein, we have frequently found destabilization of TTP transcripts in experiments with Actinomycin D-treated cells (Suppl. Fig. 4). One possibility could be that the BRF proteins influence the stability of the TTP transcript. It has been shown before that TTP can destabilize its own mRNA and several studies pointed out overlapping binding targets between the TIS11 family members [11, 30]. But so far no study has shown direct binding of BRFs to TTP transcript and thus we can only hypothesize that this might be the reason for destabilization of TTP mRNA in BMMCs. While it seems almost wasteful that TTP mRNA expression occurs without apparent protein expression, comparable examples have been described for other proteins. In K562 cells undergoing erythroid maturation, mRNAs for reticulocyte 15-lipoxygenase (r15-LOX) and c-Src are present at all stages of differentiation, maintaining the same amount of transcripts during the process. However, both proteins can only be detected after 6 to 8 days of maturation as their translation is repressed by heterogeneous nuclear ribonucleoproteins (hnRNPs) K and E1 [52]. Interestingly, an additional mechanism for silencing of translation has been proposed for both TTP and BRF1. It was shown that both proteins are able to localize ARE-containing transcripts into processing bodies (PBs), promoting translational silencing. While proteins involved in mRNA decay are also associated with the PBs, it is not entirely clear whether they are already active in the PBs, or kept inactive until mRNA and enzymes are released from the PBs [53]. This suggests the possibility of transcriptional silencing of TTP mRNA in MCs, as the TTP transcript itself contains AREs.
5. Conclusions
Taken together, while the impact of TTP on cytokine production in macrophages is undeniable, it cannot be concluded that this has to be the case for all other cytokine producing cell types as well. More and more enzymes regulating mRNA stability and translation have been discovered recently, implying that different cell types may use differential expression of these proteins to serve their needs in controlling cytokine production. For treatment of diseases caused by cytokine overproduction it might be worthwhile to look into target proteins other than TTP, depending on which cell type is the cause of the damaging cytokine response and on which proteins are expressed in that particular cell type.
Supplementary Material
Acknowledgements
We thank Drs. A. R. Clark and P. Kovarik for generously providing antibodies against TTP. The expert technical assistance of K. Maschke-Neuß and M. Kauffmann is acknowledged. We thank Dr. M. Kuhny for critical reading of the manuscript. This work was supported by grants from the Deutsche Forschungsgemeinschaft (Hu794/8-1; Priority Programme 1394: “Mast Cells - Promoters of Health and Modulators of Disease”, and Ga 453-13/1).
References
- 1.Marshall JS. Nature reviews in immunology. 2004;4:787–799. doi: 10.1038/nri1460. [DOI] [PubMed] [Google Scholar]
- 2.Sandig H, Bulfone-Paus S. Frontiers in immunology. 2012;3:185. doi: 10.3389/fimmu.2012.00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huber M, Kalis C, Keck S, Jiang Z, Georgel P, Du X, Shamel L, Sovath S, Mudd S, Beutler B, Galanos C, Freudenberg MA. European journal of immunology. 2006;36:701–711. doi: 10.1002/eji.200535593. [DOI] [PubMed] [Google Scholar]
- 4.Jiang Z, Georgel P, Du X, Shamel L, Sovath S, Mudd S, Huber M, Kalis C, Keck S, Galanos C, Freudenberg M, Beutler B. Nature immunology. 2005;6:565–570. doi: 10.1038/ni1207. [DOI] [PubMed] [Google Scholar]
- 5.Ronkina N, Menon MB, Schwermann J, Tiedje C, Hitti E, Kotlyarov A, Gaestel M. Biochemical pharmacology. 2010;80:1915–1920. doi: 10.1016/j.bcp.2010.06.021. [DOI] [PubMed] [Google Scholar]
- 6.Shimura M, Yamamoto M, Fujii G, Takahashi M, Komiya M, Noma N, Tanuma SI, Yanaka A, Mutoh M. Biological & pharmaceutical bulletin. 2012;35:2186–2191. doi: 10.1248/bpb.b12-00575. [DOI] [PubMed] [Google Scholar]
- 7.Zuckerman SH, Evans GF, Guthrie L. Immunology. 1991;73:460–465. [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao W, Liu M, D'Silva NJ, Kirkwood KL. Journal of interferon & cytokine research. 2011;31:629–637. doi: 10.1089/jir.2010.0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chrestensen CA, Schroeder MJ, Shabanowitz J, Hunt DF, Pelo JW, Worthington MT, Sturgill TW. The Journal of biological chemistry. 2004;279:10176–10184. doi: 10.1074/jbc.M310486200. [DOI] [PubMed] [Google Scholar]
- 10.Lai WS, Kennington EA, Blackshear PJ. Molecular and cellular biology. 2003;23:3798–3812. doi: 10.1128/MCB.23.11.3798-3812.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanduja S, Blanco FF, Dixon DA. Wiley interdisciplinary reviews. RNA. 2011;2:42–57. doi: 10.1002/wrna.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fan XC, Steitz JA. The EMBO journal. 1998;17:3448–3460. doi: 10.1093/emboj/17.12.3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He G, Sun D, Ou Z, Ding A. The Journal of biological chemistry. 2012;287:24967–24977. doi: 10.1074/jbc.M112.362020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tiedje C, Ronkina N, Tehrani M, Dhamija S, Laass K, Holtmann H, Kotlyarov A, Gaestel M. PLOS genetics. 2012;8:e1002977. doi: 10.1371/journal.pgen.1002977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suzuki K, Nakajima H, Ikeda K, Maezawa Y, Suto A, Takatori H, Saito Y, Iwamoto I. The Journal of experimental medicine. 2003;198:1717–1727. doi: 10.1084/jem.20031701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zorn CN, Keck S, Hendriks RW, Leitges M, Freudenberg MA, Huber M. Cellular signalling. 2009;21:79–86. doi: 10.1016/j.cellsig.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 17.Carballo E, Gilkeson GS, Blackshear PJ. The Journal of clinical investigation. 1997;100:986–995. doi: 10.1172/JCI119649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Razin E, Cordon-Cardo C, Good RA. Proceedings of the National Academy of Sciences of the United States of America. 1981;78:2559–2561. doi: 10.1073/pnas.78.4.2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karasuyama H, Melchers F. European journal of immunology. 1988;18:97–104. doi: 10.1002/eji.1830180115. [DOI] [PubMed] [Google Scholar]
- 20.Galanos C, Luderitz O. European journal of biochemistry / FEBS. 1975;54:603–610. doi: 10.1111/j.1432-1033.1975.tb04172.x. [DOI] [PubMed] [Google Scholar]
- 21.Galanos C, Luderitz O, Westphal O. European journal of biochemistry / FEBS. 1969;9:245–249. doi: 10.1111/j.1432-1033.1969.tb00601.x. [DOI] [PubMed] [Google Scholar]
- 22.Galanos C, Luderitz O, Westphal O. Zentralblatt fur Bakteriologie, Parasitenkunde, Infektionskrankheiten und Hygiene. Erste Abteilung Originale. Reihe A: Medizinische Mikrobiologie und Parasitologie. 1979;243:226–244. [PubMed] [Google Scholar]
- 23.Mahtani KR, Brook M, Dean JL, Sully G, Saklatvala J, Clark AR. Molecular and cellular biology. 2001;21:6461–6469. doi: 10.1128/MCB.21.9.6461-6469.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schaljo B, Kratochvill F, Gratz N, Sadzak I, Sauer I, Hammer M, Vogl C, Strobl B, Muller M, Blackshear PJ, Poli V, Lang R, Murray PJ, Kovarik P. The journal of immunology. 2009;183:1197–1206. doi: 10.4049/jimmunol.0803883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nishizumi H, Yamamoto T. The journal of immunology. 1997;158:2350–2355. [PubMed] [Google Scholar]
- 26.Pfaffl MW. Nucleic acids research. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu L, Damen JE, Cutler RL, Krystal G. Molecular and cellular biology. 1994;14:6926–6935. doi: 10.1128/mcb.14.10.6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hochdorfer T, Kuhny M, Zorn CN, Hendriks RW, Vanhaesebroeck B, Bohnacker T, Krystal G, Huber M. Cellular signalling. 2011;23:866–875. doi: 10.1016/j.cellsig.2011.01.012. [DOI] [PubMed] [Google Scholar]
- 29.Marschall JS, Wilhelm T, Schuh W, Huber M. Cellular signalling. 2012;24:879–888. doi: 10.1016/j.cellsig.2011.12.007. [DOI] [PubMed] [Google Scholar]
- 30.Tchen CR, Brook M, Saklatvala J, Clark AR. The Journal of biological chemistry. 2004;279:32393–32400. doi: 10.1074/jbc.M402059200. [DOI] [PubMed] [Google Scholar]
- 31.Fehrenbach K, Lessmann E, Zorn CN, Kuhny M, Grochowy G, Krystal G, Leitges M, Huber M. The journal of immunology. 2009;182:7897–7905. doi: 10.4049/jimmunol.0801773. [DOI] [PubMed] [Google Scholar]
- 32.Blackshear PJ. Biochemical Society transactions. 2002;30:945–952. doi: 10.1042/bst0300945. [DOI] [PubMed] [Google Scholar]
- 33.Qi MY, Wang ZZ, Zhang Z, Shao Q, Zeng A, Li XQ, Li WQ, Wang C, Tian FJ, Li Q, Zou J, Qin YW, Brewer G, Huang S, Jing Q. Molecular and cellular biology. 2012;32:913–928. doi: 10.1128/MCB.05340-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keck S, Muller I, Fejer G, Savic I, Tchaptchet S, Nielsen PJ, Galanos C, Huber M, Freudenberg MA. The journal of immunology. 2011;186:5478–5488. doi: 10.4049/jimmunol.1000458. [DOI] [PubMed] [Google Scholar]
- 35.Lal A, Haynes SR, Gorospe M. Molecular and cellular probes. 2005;19:385–388. doi: 10.1016/j.mcp.2005.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmidlin M, Lu M, Leuenberger SA, Stoecklin G, Mallaun M, Gross B, Gherzi R, Hess D, Hemmings BA, Moroni C. The EMBO journal. 2004;23:4760–4769. doi: 10.1038/sj.emboj.7600477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Businaro R, Tagliani A, Buttari B, Profumo E, Ippoliti F, Di Cristofano C, Capoano R, Salvati B, Rigano R. Annals of the New York Academy of Sciences. 2012;1262:134–141. doi: 10.1111/j.1749-6632.2012.06600.x. [DOI] [PubMed] [Google Scholar]
- 38.Iwase S, Murakami T, Saito Y, Nakagawa K. European cytokine network. 2004;15:312–316. [PubMed] [Google Scholar]
- 39.Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, Kollias G. The EMBO journal. 1991;10:4025–4031. doi: 10.1002/j.1460-2075.1991.tb04978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Probert L, Keffer J, Corbella P, Cazlaris H, Patsavoudi E, Stephens S, Kaslaris E, Kioussis D, Kollias G. The journal of immunology. 1993;151:1894–1906. [PubMed] [Google Scholar]
- 41.Brook M, Sully G, Clark AR, Saklatvala J. FEBS letters. 2000;483:57–61. doi: 10.1016/s0014-5793(00)02084-6. [DOI] [PubMed] [Google Scholar]
- 42.Cho EJ, An HJ, Shin JS, Choi HE, Ko J, Cho YW, Kim HM, Choi JH, Lee KT. Journal of cellular biochemistry. 2011;112:3648–3659. doi: 10.1002/jcb.23294. [DOI] [PubMed] [Google Scholar]
- 43.Guo W, Sun J, Jiang L, Duan L, Huo M, Chen N, Zhong W, Wassy L, Yang Z, Feng H. Inflammation. 2012;35:1764–1772. doi: 10.1007/s10753-012-9495-9. [DOI] [PubMed] [Google Scholar]
- 44.Rousseau S, Papoutsopoulou M, Symons A, Cook D, Lucocq JM, Prescott AR, O'Garra A, Ley SC, Cohen P. Journal of cell science. 2008;121:149–154. doi: 10.1242/jcs.018671. [DOI] [PubMed] [Google Scholar]
- 45.Bourcier C, Griseri P, Grepin R, Bertolotto C, Mazure N, Pages G. American journal of physiology. Cell physiology. 2011;301:C609–618. doi: 10.1152/ajpcell.00506.2010. [DOI] [PubMed] [Google Scholar]
- 46.Deleault KM, Skinner SJ, Brooks SA. Molecular immunology. 2008;45:13–24. doi: 10.1016/j.molimm.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 47.Blackshear PJ, Phillips RS, Ghosh S, Ramos SB, Richfield EK, Lai WS. Biology of reproduction. 2005;73:297–307. doi: 10.1095/biolreprod.105.040527. [DOI] [PubMed] [Google Scholar]
- 48.Maitra S, Chou CF, Luber CA, Lee KY, Mann M, Chen CY. RNA. 2008;14:950–959. doi: 10.1261/rna.983708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parameswaran N, Patial S. Critical reviews in eukaryotic gene expression. 2010;20:87–103. doi: 10.1615/critreveukargeneexpr.v20.i2.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qiu LQ, Stumpo DJ, Blackshear PJ. The journal of immunology. 2012;188:5150–5159. doi: 10.4049/jimmunol.1103700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun L, Stoecklin G, Van Way S, Hinkovska-Galcheva V, Guo RF, Anderson P, Shanley TP. The Journal of biological chemistry. 2007;282:3766–3777. doi: 10.1074/jbc.M607347200. [DOI] [PubMed] [Google Scholar]
- 52.Naarmann IS, Harnisch C, Flach N, Kremmer E, Kuhn H, Ostareck DH, Ostareck-Lederer A. The Journal of biological chemistry. 2008;283:18461–18472. doi: 10.1074/jbc.M710328200. [DOI] [PubMed] [Google Scholar]
- 53.Franks TM, Lykke-Andersen J. Genes & development. 2007;21:719–735. doi: 10.1101/gad.1494707. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.