

Abstract
We report on a target-based approach to identify possible Mycobacterium tuberculosis DXS inhibitors from the structure of a known transketolase inhibitor. A small focused library of analogs was assembled in order to begin elucidating some meaningful structure–activity relationships of 3-(4-chloro-phenyl)-5-benzyl-4H-pyrazolo[1,5-a]pyrimidin-7-one. Ultimately we found that 2-methyl-3-(4-fluorophenyl)-5-(4-meth-oxy-phenyl)-4H-pyrazolo[1,5-a]pyrimidin-7-one, although still weak, was able to inhibit M. tuberculosis DXS with an IC50 of 10.6 μM.
Keywords: Drug design, DXS, Enzyme, SAR, Tuberculosis
Despite the availability of effective antituberculosis drugs, tuberculosis (TB) is still a major cause of disability and death globally. Improved TB medications need to be developed to shorten the duration of the treatment period, to reduce the amount of drugs required while making costs affordable, and to provide a more effective treatment against persistent TB infection.1 To develop new treatments for TB, many drug discovery programs target enzymes considered to be essential for Mycobacterium tuberculosis survival. It is hoped that this target-based approach will identify specific inhibitors of bacterial enzymes, thus blocking the bacterial growth while showing little if any toxicity toward the host organism. Potential lead compounds against these targets can be identified from high-throughput screening (HTS) campaigns or through ligand- and structure-based design methods.2
Within this context, early steps in the isoprenoid biosynthetic pathway are being investigated, as isoprenoids are known to be required for M. tuberculosis survival. Examples of these approaches encompass molecules such as decaprenyl phosphate3 which is involved in the biosynthesis of major cell wall components of M. tuberculosis (peptidoglycan,4 lipoarabinomannan,5 and arabinomannan6), and menaquinone which is the only lipoquinone involved in the mycobacterial electron transport chain.7 The universal precursors of isoprenoids, isopentenyl diphosphate (IPP), and dimethylallyl diphosphate (DMAPP) are synthesized solely through the 2C-methyl-D-erythritol 4-phosphate (MEP) pathway in M. tuberculosis. This biosynthetic pathway does not exist in mammalian biochemistry, and therefore it opens alternative approaches to developing novel antituberculosis drugs.8–10 In the initial rate-limiting step11 in the MEP pathway (Scheme 1), 1-deoxy-D-xylulose 5-phosphate (DXP) is formed from the condensation of pyruvate and D-glyceraldehyde-3-phosphate (GAP) catalyzed by 1-deoxy-D-xylulose 5-phosphate synthase (DXS) in the presence of thiamine pyrophosphate (TPP). The formation of DXP is not a committed step in isoprenoid biosynthesis as this molecule is also a precursor of pyridoxol (vitamin B6)12 or thiamine (vitamin B1).13,14
Scheme 1.
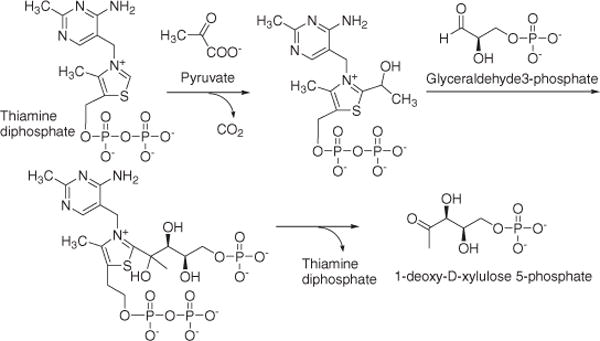
Mechanism of DXS.
Himar1-based transposon mutagenesis predicted that M. tuberculosis DXS is required for M. tuberculosis survival.15 Although there are no structural data available, M. tuberculosis DXS shows 38% amino acid sequence identity with Escherichia coli DXS, for which a crystal structure has been reported.16 These proteins have well-conserved regions predicted to be involved in substrate binding and catalytic activity.8 The overall structure is similar to the members of the mammalian transketolase (TK) superfamily, including pyruvate dehydrogenase E1 subunit17 and 2-oxoisovalerate dehydrogenase.18 These enzymes are all dimers, composed of three different domains, and require TPP as a cofactor.16
In this study, a previously established in vitro DXS assay8 was utilized to study the structure–activity relationships (SARs) of a small, focused compound library based on the structure of a known TK inhibitor,22 3-(4-chloro-phenyl)-5-benzyl-4H-pyrazolo[1,5-a]pyrimidin-7-one (1). Although DXS was previously characterized in several organisms including E. coli23 and M. tuberculosis,8 this is the first study describing the SAR of inhibitors against the M. tuberculosis DXS.
Expression and purification of DXS
M. tuberculosis DXS was cloned, expressed, and purified in E. coli as a fusion protein bearing a His6-tag. The purified M. tuberculosis DXS was estimated to be at least 95% pure by SDS–PAGE analysis on 12% gels. A clear band was observed corresponding to a molecular weight (67.8 kDa) consistent with the expected size. Western blot analysis with anti-His antibody confirmed that the single band corresponds to DXS. Purified DXS (59 pmol) were used for the in vitro enzyme assay to screen the compounds.
Determination of the IC50 of active compounds against M. tuberculosis DXS
As has been reported, the catalytic function of M. tuberculosis DXS combines the mechanism of the pyruvate decarboxylase and TK.8 Therefore, the known TK inhibitor 1 (Table 1), inhibiting the TK superfamily with an IC50 of 3.9 μM,22 was expected to inhibit M. tuberculosis DXS as well. However, the IC50 value of the compound for M. tuberculosis DXS (114.1 μM) was much higher than that found for the TK superfamily, suggesting that there may be sufficient variation between the binding pockets of TK and M. tuberculosis DXS. Indeed, the domain arrangements of DXS differ from those of TK, resulting in differences in the formation of the active site.19–21 In DXS, three subunits of one domain form the active site. However, in the TK superfamily, the largest subunit of one domain contacts smaller subunits of the other domains forming the active site.17 These differences are possibly caused by a longer linker (95 amino acids) between domains in the TK superfamily,16 whereas DXS contains only 20 amino acid residues in this linker.16 Although the catalytic mechanism of M. tuberculosis DXS appears to be similar to that of TK superfamily, inhibitory specificity may be achieved through these structural differences. Moreover, the M. tuberculosis DXS active site can accommodate a relatively broad spectrum of substrates, such as D- or L-glyceraldehyde and D-erythrose 4-phosphate,8 indicating that there is flexibility in the types of molecules that can be accommodated in the active site. Based on this information together with the difference in IC50 values for inhibitor 1 between M. tuberculosis DXS and TK, we were encouraged to select some modifications to this molecule in order to gain possibly better selectivity. Based on the compound drug likeness as defined by the guidelines of Lipinski’s Rule of Five,24 we purchased compounds from different commercial vendors in order to build a preliminary body of SAR information (Fig. 1). In order to explore both steric and electronic effects at the active site, various modifications were included: different substitutions were introduced at the 2-position; the chloro group on the phenyl ring located at the 3-position was replaced by an electron-withdrawing or -donating group; the carbon linker length was varied and electron-withdrawing/donating groups were introduced at different positions of the 5-position benzyl side chain; alkyl or aryl substituents were introduced at the 6 and 7-positions.
Table 1.
Inhibition activity of different scaffold compounds against M. tuberculosis DXS
| Compounda |
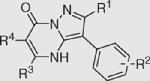
|
DXS IC50 (μM) | |||
|---|---|---|---|---|---|
| R1 | R2 | R3 | R4 | ||
| 1 | H | 4-Cl | PhCH2 | H | 114.1 ±0.6 |
| 2 | H | 4-Cl | CH3OCH2 | H | >200 |
| 3 | H | 4-Cl | CH3 | H | >200 |
| 4 | H | H | CH3 | CH2COOEt | >200 |
| 5 | H | H | ClCH2 | H | >200 |
| 6 | CF3 | H | PhCH2 | H | 71.1 ± 5.1 |
| 7 | CF3 | H | Ph | H | 23.0 ± 4.1 |
| 8 | CF3 | 4-Cl | PhCH2 | H | 14.0 ± 1.1 |
| 9 | CF3 | 4-CH3O | Ph | H | 20.2 ± 0.6 |
| 10 | CF3 | 4-Br | CH3OCOCH2 | H | 73.0 ± 6.4 |
| 11 | CF3 | 4-Br | CH3 | H | >200 |
| 12 | CF3 | 4-Cl | CH3 | H | 98.4 ± 13.8 |
| 13 | CF3 | 4-Cl | CH3 | PhCH2 | >200 |
| 14 | CH3 | 4-CH3O | Ph | H | >200 |
| 15 | CH3 | 4-CH3O | Ph | H | 90.7 ± 4.6 |
| 16 | CH3 | 4-CH3O | 4-CH3OC6H4 | H | 42.4 ± 3.8 |
| 17 | CH3 | 4-Cl | Ph | H | 34.2 ± 5.8 |
| 18 | CH3 | 4-Cl | 4-CH3OC6H4 | H | 10.9 ±1.5 |
| 19 | CH3 | 4-F | 4-CH3OC6H4 | H | 10.6 ± 3.1 |
| 20 | CH3 | 4-Br | 2-CH3OC6H4 | H | 53.5 ± 0.5 |
| 21 | CH3 | H | Ph | H | 30.6 ± 1.1 |
| 22 | CH3 | 4-Cl | (1-Phenyl-1H-tetrazol-5-ylsulfanyl)-methylene- | H | 55.1 ± 4.4 |
| 23 | CH3 | 4-Cl | 4-ClC6H4SCH2 | H | 41.1 ± 0.4 |
| 24 | CH3 | H | 4-ClC6H4SCH2 | H | 63.7 ± 1.0 |
| 25 | CH3 | 4-Cl | CH3OCH2 | H | >200 |
| 26 | CH3 | H | Naphthalene-2-yl | H | 19.5 ± 2.9 |
| 27 | CH3 | H | Ph | CH3CH2CH2 | 71.9 ± 10.1 |
| 28 | CH3CH2 | 4-Cl | 4-CH3OC6H4 | H | 19.7 ± 3.4 |
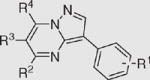
|
|||||
| R1 | R2 | R3 | R4 | ||
|
|
|||||
| 29 | 4-Cl | 4-ClPh | H | CF3 | >200 |
| 30 | 3-Br | Ph | H | Ph | >200 |
Compounds in 2-H series: 1–5, 29, and 30; compounds in 2-CF3 series: 6–13; compounds in 2-CH3 series: 14–27.
Figure 1.
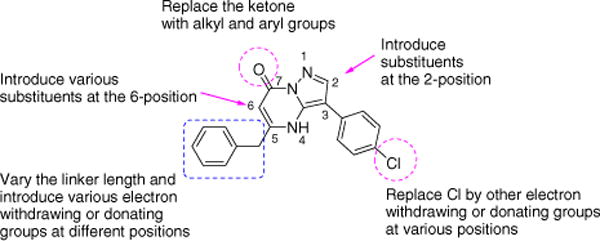
Modifications of reported TK inhibitor.
Modifications of compound 1 are shown in Table 1. When the benzyl group was replaced with a methoxymethyl group as in compound 2 or a methyl group as in 3, the inhibitory activity was abolished, indicating that an aromatic ring at the 5-position is required for the activity. When the chloro substituent was removed at the para-position of phenyl side chain, and a methylene ethyl ester was added at 6-position, compound 4 also lost its activity against M. tuberculosis DXS. In addition, compounds with a trifluoromethyl group (compound 29) and a phenyl group (compound 30) at the 7-position increased the IC50 on M. tuberculosis DXS.
Introduction of an electron-withdrawing trifluoromethyl group at the 2-position reduced the IC50 on M. tuberculosis DXS compared with the 2-H series (compound 8 vs compound 1, Table 1). Addition of a para-methoxy group on the 3-phenyl group did not increase the inhibitory activity (compound 9 vs compound 7). In contrast, introduction of a para-chloro substituent on this phenyl ring decreased the IC50 value (compound 8 vs compound 6). Furthermore, when we compared the impact of different halogens, a para-chloro substituent on the phenyl ring showed better inhibitory activity than bromo (compound 12 vs compound 11). Additionally, compound 7 served as a better inhibitor than compound 6, which indicated that a phenyl substituent is better than a benzyl group at the 5-position. Compound 10 exhibited greater inhibitory activity than that of compound 11, suggesting that there is some space in the binding cavity located in the area surrounding the 5-position of the main core in the active site. However, this is apparently not the case for the 6-position of the main scaffold (cf. compound 13 with compound 12).
The 2-methyl series (compounds 14–27, Table 1) showed less inhibitory activity than the 2-trifluoromethyl series, but the activity was still greater than the 2-H series (compound 17 vs compound 8, compound 17 vs compound 1). A phenyl ring bearing an electron-withdrawing group is preferred at the 3-position as compound 17 served as a better inhibitor than compound 14. Halogen size and/or electronic effects played a role at the 3-phenyl side chain as the smaller fluoro and chloro substituents (compound 19 and compound 18 vs compound 20) showed better inhibition than the bromo substitution. The notion that a phenyl substituent is better than a benzyl group at the 5-position is supported by the comparison of compounds 15 and 14. A para-methoxy group at the 5-position increased the inhibitory activity (cf. compound 16 vs compound 15). As mentioned earlier, the replacement of the benzyl group with a non-aromatic substituent (compound 25) at the 5-position resulted in a loss in activity. However, compounds containing aromatic rings with various linkers (compounds 22 and 23) still maintained some inhibitory activity. The 5-naphthalenyl-substituted compound 26 showed slightly greater inhibitory activity than the 5-benzyl analog 21, indicating that a conjugated aromatic ring system was favored at this position. As indicated earlier, introduction of an appendage at the 6-position led to a loss of activity (27 vs 21).
A 2-ethyl substituent showed somewhat less inhibition than the corresponding 2-methyl (compound 28 vs compound 18), therefore, we did not investigate this series further.
Anti-TB activity and toxicity of compounds
The best two inhibitors (the inhibition curve of compound 18 is shown in Fig. S1) were tested for inhibition of growth of M. tuberculosis (Table 2). Both compound 18 and compound 19 demonstrated reasonably good inhibition of bacterial growth. However, they also showed some toxicity against Vero cells,25 with a selectivity index of 2.1 and 4.6, for compounds 18 and 19, respectively. The MICs of compounds 16 and 17 showed some correlation with their IC50s, but their cytotoxicity indicates that these compounds may have off-target activity against mammalian TK or other unknown targets in the Vero cells.
Table 2.
Antituberculosis activity and cytotoxicity of selected inhibitors
| Compound | MABA MIC (μM) | Vero cell IC50 (μM) |
|---|---|---|
| 16 | 61.8 | 80.9 |
| 17 | 14.2 | 39.5 |
| 18 | 7.6 | 16.0 |
| 19 | 7.7 | 35.6 |
In this study, we have successfully utilized a previously established in vitro DXS assay to screen for new inhibitors. Several potential inhibitors of M. tuberculosis DXS, with IC50 values of ~10 μM, have been identified, and preliminary SAR data have been generated. As it is apparent from Figure 2, a trifluoromethyl group is the preferred substituent at the 2-position of the 3-phenyl-4H-pyrazolo[1,5-a]pyrimidin-7-one scaffold; ethyl, methyl, and hydrogen result in increasingly poorer activity. An electron-donating group para-methoxy attached to the phenyl ring is favored, and the 5-naphthenyl analog is even more potent. One of the best inhibitors also exhibited a reasonably good MIC (7.7 μM) against M. tuberculosis H37Rv with a 4.6-fold selectivity index. Active compounds did not inhibit DXS from other organisms or other enzymes from M. tuberculosis (data not shown). Therefore, solution of the X-ray structures of M. tuberculosis DXS in complex with these inhibitors could provide valuable insights into the subtleties of the M. tuberculosis DXS binding site that may allow the design of a new generation of inhibitors specific for M. tuberculosis DXS.
Figure 2.
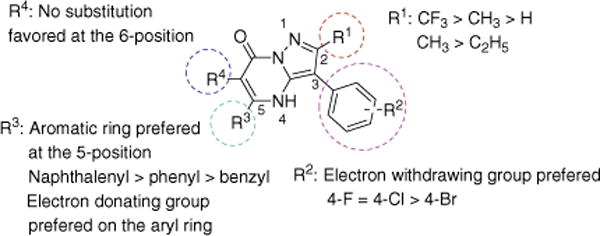
SAR summary of 3-phenyl-4H-pyrazolo[1,5-a]pyrimidin-7-one.
Supplementary Material
Acknowledgments
We thank Hao Xie (Department of Chemistry, Princeton) for his helpful discussions and suggestions for the letter.
Footnotes
Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bmcl.2008.08.034.
References and notes
- 1.Tomioka H, Sato K, Shimizu T, Sano C. J Infect. 2002;44:160. doi: 10.1053/jinf.2002.0973. [DOI] [PubMed] [Google Scholar]
- 2.Selzer PM, Brutsche S, Wiesner P, Schmid P, Mullner H. Int J Med Microbiol. 2000;290:191. doi: 10.1016/S1438-4221(00)80090-9. [DOI] [PubMed] [Google Scholar]
- 3.Crick DC, Schulbach MC, Zink EE, Macchia M, Barontini S, Besra GS, Brennan PJ. J Bacteriol. 2000;182:5771. doi: 10.1128/jb.182.20.5771-5778.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mahapatra S, Yagi T, Belisle JT, Espinosa BJ, Hill PJ, McNeil MR, Brennan PJ, Crick DC. J Bacteriol. 2005;187:2747. doi: 10.1128/JB.187.8.2747-2757.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Besra GS, Morehouse CB, Rittner CM, Waechter CJ, Brennan PJ. J Biol Chem. 1997;272:18460. doi: 10.1074/jbc.272.29.18460. [DOI] [PubMed] [Google Scholar]
- 6.Mikusova K, Mikus M, Besra GS, Hancock I, Brennan PJ. J Biol Chem. 1996;271:7820. doi: 10.1074/jbc.271.13.7820. [DOI] [PubMed] [Google Scholar]
- 7.Putra SR, Disch A, Bravo JM, Rohmer M. FEMS Microbiol Lett. 1998;164:169. doi: 10.1111/j.1574-6968.1998.tb13082.x. [DOI] [PubMed] [Google Scholar]
- 8.Bailey AM, Mahapatra S, Brennan PJ, Crick DC. Glycobiology. 2002;12:813. doi: 10.1093/glycob/cwf100. [DOI] [PubMed] [Google Scholar]
- 9.Dhiman RK, Schaeffer ML, Bailey AM, Testa CA, Scherman H, Crick DC. J Bacteriol. 2005;187:8395. doi: 10.1128/JB.187.24.8395-8402.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eoh H, Brown AC, Buetow L, Hunter WN, Parish T, Kaur D, Brennan PJ, Crick DC. J Bacteriol. 2007;189:8922. doi: 10.1128/JB.00925-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kang MJ, Lee YM, Yoon SH, Kim JH, Ock SW, Jung KH, Shin YC, Keasling JD, Kim SW. Biotechnol Bioengin. 2005;91:636. doi: 10.1002/bit.20539. [DOI] [PubMed] [Google Scholar]
- 12.Hill RE, Himmeldirk K, Kennedy IA, Pauloski RM, Sayer BG, Wolf E, Spenser ID. J Biol Chem. 1996;271:30426. doi: 10.1074/jbc.271.48.30426. [DOI] [PubMed] [Google Scholar]
- 13.Therisod M, Fischer JC, Estramareix B. Biochem Biophys Res Commun. 1981;98:374. doi: 10.1016/0006-291x(81)90850-0. [DOI] [PubMed] [Google Scholar]
- 14.White RH. Biochemistry. 1978;17:3833. doi: 10.1021/bi00611a024. [DOI] [PubMed] [Google Scholar]
- 15.Sassetti CM, Boyd DH, Rubin E. J Mol Microbiol. 2003;48:77. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 16.Xiang S, Usunow G, Lange G, Busch M, Tong L. J Biol Chem. 2007;282:2676. doi: 10.1074/jbc.M610235200. [DOI] [PubMed] [Google Scholar]
- 17.Nikkola M, Lindqvist Y, Schneider G. J Mol Biol. 1994;238:387. doi: 10.1006/jmbi.1994.1299. [DOI] [PubMed] [Google Scholar]
- 18.Aevarsson A, Seger K, Turley S, Sokatch JR, Hol WG. Nat Struct Biol. 1999;6:785. doi: 10.1038/11563. [DOI] [PubMed] [Google Scholar]
- 19.Lindqvist Y, Schneider G, Ermler U, Sundstrom M. EMBO J. 1992;11:2373. doi: 10.1002/j.1460-2075.1992.tb05301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gerhardt S, Echt S, Busch M, Freigang J, Auerbach G, Bader G, Martin WF, Bacher A, Huber R, Fischer M. Plant Physiol. 2003;132:1941. doi: 10.1104/pp.103.020982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nilsson U, Lindqvist Y, Kluger R, Schneider G. FEBS Lett. 1993;326:145. doi: 10.1016/0014-5793(93)81779-y. [DOI] [PubMed] [Google Scholar]
- 22.Du MX, Sim J, Fang L, Yin Z, Koh S, Stratton J, Pons J, Wang JJ, Carte B. J Biomol Screening. 2004;9:427. doi: 10.1177/1087057104263913. [DOI] [PubMed] [Google Scholar]
- 23.Sprenger GA, Schorken U, Wiegert T, Grolle S, de Graaf AA, Taylor SV, Begley TP, Bringer-Meyer S, Sahm H. Proc Natl Acad Sci USA. 1997;94:12857. doi: 10.1073/pnas.94.24.12857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Adv Drug Deliv Rev. 2001;46:3. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 25.Falzari K, Zhu Z, Pan D, Liu H, Hongmanee P, Franzblau SG. Antimicrob Agents Chemother. 2005;49:1447. doi: 10.1128/AAC.49.4.1447-1454.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.