

Abstract
Objective
The mechanisms underlying bone damage in rheumatoid arthritis (RA) are incompletely understood. We recently identified the shared epitope (SE), an HLA-DRB1-coded 5 amino acid sequence motif carried by the majority of RA patients, as a signal transduction ligand that interacts with cell surface calreticulin (CRT), and accelerates osteoclast (OC)-mediated bone damage in collagen-induced arthritis (CIA). Given the role of the SE-CRT pathway in arthritis-associated bone damage, here we have sought to determine the therapeutic targetability of CRT.
Methods
A library of backbone-cyclized peptidomimetic compounds, all carrying an identical core DKCLA sequence was synthesized. The ability of these compounds to inhibit SE-activated signaling and OC differentiation was tested in vitro. The effect on disease severity and OC-mediated bone damage was studied by weekly intraperitoneal administration of compounds to DBA/1 mice with CIA.
Results
Two members of the peptidomimetics library were found to have in vitro SE-antagonistic and anti-osteoclast differentiation effects at picomolar concentrations. A lead mimetic compound, designated HS(4–4)c Trp, potently ameliorated arthritis and bone damage in vivo when administered in picogram doses to mice with CIA. Another mimetic analog, designated HS(3–4)c Trp, was found to lack activity, both in vitro and in vivo. The differential activity of the two analogs depended on minor differences in their respective ring sizes, and correlated with distinctive geometry when computationally docked onto the SE binding site on CRT.
Conclusion
These findings identify CRT as a novel therapeutic target in erosive arthritis and provide sound rationale and early structure-activity relationships for future drug design.
INTRODUCTION
Despite the advent of biologic agents, preventing bone damage in rheumatoid arthritis (RA) remains a challenging endeavor. Due to insufficient understanding of the mechanisms that trigger RA onset and determine disease severity, most current and emerging drugs are targeted at generic, ‘downstream’ immune pathways or inflammatory cytokines. As a result, drug failure and/or side effects are all too common.
The etiology and pathogenesis of RA are incompletely understood. However, it has been long observed that the majority of RA patients carry HLA-DRB1 alleles that code a five amino acid sequence motif called the ‘shared epitope’ (SE) in the region 70–74 of the DRβ chain (1). The SE not only confers a higher risk for RA, but also increases the likelihood of developing a more severe disease. SE-coding HLA-DRB1 alleles are associated with earlier disease onset and accelerated bone damage (2–5). Furthermore, there is evidence of gene-dose effect, where the extent of bone destruction in RA correlates positively with the number of SE-coding HLA-DRB1 alleles (3–5).
The underlying mechanisms by which the SE affects susceptibility to – or severity of – RA are unknown. We have recently identified the SE as a signal transduction ligand that binds to a well-defined site on cell surface calreticulin (CRT) (6) in a strictly allele-specific manner and activates nitric oxide (NO)-mediated signaling (7–11) with resultant enhanced osteoclast (OC) differentiation and activation, both in vitro and in vivo (12, 13).
OC-mediated bone damage is a common, unfortunate outcome in RA (14, 15). In addition to juxta-articular bone erosion, RA patients also experience periarticular and systemic osteoporosis (16). The common mechanism underlying these bone pathologies is believed to involve dysregulation of the balance between bone formation and resorption due to excessive cellular activity of OCs (17), as a result of intricate crosstalk with other cells in the synovium that produce the receptor activator of nuclear-κB ligand (RANKL) (18–20).
In previous studies, we have demonstrated that the SE ligand has a dual enhancing effect on OC differentiation and activation in vitro: an indirect effect through polarization toward RANKL-expressing Th17 cells, and a direct differentiation effect on OC precursors. When administered in vivo to mice with collagen-induced arthritis (CIA), the SE ligand increased joint swelling, synovial tissue abundance of active OCs and erosive bone damage (12, 13).
Given the emerging evidence that the SE acts as a signal transduction ligand that directly contributes to bone damage in arthritis, we have undertaken to explore ways to specifically inhibit this pathway. Here we describe a peptidomimetic SE-antagonistic ligand (SEAL) with highly potent anti-osteoclastogenic and anti-arthritic effects. These findings suggest that targeting the SE-activated pathway might be a useful therapeutic strategy.
MATERIALS AND METHODS
Reagents, peptidomimetics, cells and mice
Ficoll-Paque™, 4,5-Diaminofluorescein Diacetate (DAF-2 DA), macrophage colony-stimulating factor (M-CSF), RANKL, chicken collagen type II (CII), and complete Freund’s Adjuvant (CFA) were purchased from previously listed sources (13). All other commercial reagents were purchased from Sigma (St Louis, MO). Linear 5-mer peptides DKCLA, QKCLA and DERAA, as well as 15-mer peptides 65–79*0401 (KDLLEQKRAAVDTYC) and 65–79*0404 (KDLLEQRRAAVDTYC) were all synthesized and purified (> 90%) as we previously described (9, 10). The urea backbone cyclic peptidomimetics, designated generically HS(m-n)c Trp, were synthesized according to a previously described procedure (21, 22) using various Alloc-protected glycine building units, where ‘m’ stands for the number of methylene groups in the N-alkyl chain on the glycine at position 2, and ‘n’ stands for the number of methylene groups in the N-alkyl chain on the glycine building unit at position 6. A tryptophan residue in position 1 was used for tracing and quantitation. The isolation of human peripheral blood mononuclear cells (PBMCs), mouse primary bone marrow cells (BMCs) and the culture of M1 fibroblasts were previously described (13). DBA/1 mice, 6 to 10 weeks old, were purchased from the Jackson Laboratory (Bar Harbor, Maine). Mice were maintained and housed at the University of Michigan-Unit for Laboratory Animal Medicine facility, and all experiments were performed in accordance with protocols approved by University of Michigan Committee on Use and Care of Animals.
Surface plasmon resonance
A Biacore2000 Biosensor System (Pharmacia/LKB Biotechnology) was used to assay the interaction between soluble ligands and recombinant mouse CRT (6,11). A surface plasmon resonance (SPR) assay is based on a biosensor chip with a dextran-coated gold surface that is coated with a covalently immobilized protein. Binding interactions between an injected ligand (the “analyte”) and the immobilized protein result in SPR signals that are directly proportional to the amount and molecular mass of the ligand. Results are read in real time as resonance units (RU).
Before use, biosensor chips CM5 (Biacore) were preconditioned in water at 100 μl/min by applying two consecutive 20-μl pulses of 50 mM NaOH, followed by 10 mM HCl, and finally 0.1% SDS. CM5 surface was activated by a 7-min injection of 200 mM 1-ethyl-3-(−3-dimethylaminopropyl) carbodiimide hydrochloride in 50 mM N-hydroxysuccinimide. Purified CRT was immobilized by a standard primary amine coupling at 25°C in HBS-EP buffer (10 mM HEPES (pH 7.4), 150 mM NaCl, 1 mM EDTA, and 0.005% surfactant P-20), at a flow rate of 10 μl/min. The protein was injected manually at a concentration of 100 μg/ml until approaching 5500 RU. The remaining activated groups were blocked by ethanolamine (1 M).
Binding assays were performed at 25°C in a binding buffer (10 mM HEPES (pH 7.4), 50 mM KCl, 0.5 mM CaCl2, 100 μM ZnCl2, and 0.005% surfactant P-20) at a flow rate of 10 μl/min. Ligands were applied in the analyte at various concentrations. The RU signal intensity at a given molar concentration is proportional to the molecular mass of the analyte. Accordingly, when a competitive small ligand is mixed together with a larger ligand, a drop in the signal relative to the RU produced by the latter ligand alone is observed. This characteristic was used here to determine competitive inhibition of the 15-mer peptides 65–79*0401 (MW: 1751 Da) or 15-mer peptide 65–79*0404 (MW: 1751 Da) binding to CRT by the 5-mer linear peptide DKCLA (MW: 590 Da), or the cDKCLA mimetic (MW: 902 Da). All SPR data were calculated using the BIAevaluation version 3.0.1 program (Biacore).
In vitro assays
Nitric oxide (NO) signal transduction assays were performed using the NO probe DAF-2 DA. The fluorescence level was recorded every 5 min over a period of 500 min using a Fusion αHT system (PerkinElmer Life Sciences) at an excitation wavelength of 488 nm and emission wavelength of 515 nm. In vitro assays for OC differentiation were performed using primary mouse BMCs isolated from femurs and tibias, or human PBMCs isolated from healthy blood donors. BMCs were cultured in 48-well plates (2 × 105/well) in MEM medium supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin in the presence of 10 ng/ml M-CSF alone during the first 2 d, followed by 4 additional d in the presence of 10 ng/ml of M-CSF, plus 20 ng/ml RANKL. Human OCs were differentiated from PBMCs derived from random blood donors, irrespective of their HLA-DRB1 genotype. Cells (2 × 105/well) were cultured in 48-well plates for 7 d in 100 ng/ml M-CSF and 100 ng/ml RANKL-supplemented in 10% FCS DMEM. To quantify the number of OCs, cultures were fixed and stained for tartrate-resistant acid phosphatase (TRAP) activity using an acid phosphatase kit (Kamiya Biomedical Company, Seattle, WA) according to the manufacturer’s instructions. TRAP-positive multinucleated OCs (more than three nuclei) were counted using a tissue-culture inverted microscope.
CIA induction, in vivo compound administration, joint tissue studies and imaging
DBA/1 mice were immunized with chicken CII in CFA. In brief, 50 μl of an emulsion containing 100 μg of CII in 25 μl of 0.05 M acetic acid and 25 μl of CFA was injected intradermally at the base of the tail. Mice were injected once per week intraperitoneally with 50 μl of either PBS, (HS4-4)c Trp, or (HS-3-4)c Trp in PBS. Arthritis severity was determined using a visual scoring system on a 4-point scale for each paw: 0 = no arthritis, 1 = swelling and redness confined to digits, 2 = minor swelling and redness spreading from the digits to the distal paw, and 3 = major swelling and redness extending proximally from the paw.
Limbs were dissected and decalcified in 10% EDTA. After decalcification, the specimens were processed for paraffin embedding and serial sectioned. The histological sections were deparaffinized, rehydrated and stained with tartrate-resistant acid phosphatase (TRAP) activity using a commercial kit (Kamiya Biomedical Company, Seattle, WA). To determine OC abundance, TRAP-positive multinucleated cells were counted. Data represent mean ± SEM of the total number of OCs in front and rear paws + knees.
For micro-computerized tomography (CT), front and hind limbs were dissected, fixed in 10% formalin, and stored in 70% ethanol. Limbs were scanned ex vivo by a micro-CT system (eXplore Locus SP; GE Healthcare Pre-Clinical Imaging, London, ON, Canada) in distilled water. The protocol included a source powered at 80 kV and 80 μA. In addition to a 0.508-mm Al filter, an acrylic beam flattener was used to reduce beam-hardening artifact. Exposure time was defined at 1600 ms/frame with 400 views taken at increments of 0.5°. With four frames averaged and binning at 2 × 2, the images were reconstructed with an 18-μm isotropic voxel size.
Radiographs were taken using a microradiography system (Faxitron X-ray Corporation, Wheeling, IL, USA) with the following operating settings: 27 peak voltage, 2.5 mA anode current, and an exposure time of 4.5 seconds. Coded radiographs were evaluated by an experienced rheumatologist who was blinded to the treatment. Bone erosions were scored on a scale of 0–5 for each of the four paws.
Docking models
The BioMedCAChe 6.1 software (Fujitsu, Sunnyvale, CA) was used for modeling of the interaction between ligands and the CRT P-domain. Cyclic peptidomimetics, loaded as flexible molecules, or the physiologically folded SE (residues 70–74 in the β-chain third allelic hypervariable region of HLADR4 (PDB ID: 2SEB) as a rigid conformation, were used as ligands. Rat CRT P-domain (PDB ID: 1HHN) in its rigid conformation, was used as receptor. Docking scores were calculated using the manufacturer’s software. In all cases, the minimum potential energy and chemical bonds were determined for the most stable geometry.
Statistical analysis
Data are expressed as mean ± SEM. Unless otherwise stated, statistical analyses were performed using a Student’s T-test (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
RESULTS
DKCLA - a short linear synthetic peptide with SE-antagonistic ligand (SEAL) properties
We identified a 5mer peptide expressing the sequence DKCLA that interacted with CRT in a SPR-based assay (Fig. 1A), and specifically inhibited interaction between CRT and 15mer peptide SE ligands corresponding to the 65–79 region coded by HLA-DRB1*04:01 (Fig. 1B), or DRB1*04:04 (Fig. 1C) at IC50 = 67 μM (Fig. 1D) and IC50 = 289 μM (Fig. 1E), respectively. Additionally, linear DKCLA specifically blocked SE ligand-activated NO signaling (Fig. 1F). Thus, the linear peptide DKCLA is a low-potency competitive inhibitor of the SE ligand, a functional characteristic referred hereto as ‘SE antagonistic ligand’ (SEAL).
Figure 1. SEAL activity in vitro.

A. Interaction between linear DKCLA and recombinant CRT; B. Linear DKCLA (508 μM) specifically inhibits the interaction between SE ligand 65–79*0401 (171 μM) and CRT; C. Linear DKCLA specifically inhibits the interaction between SE ligand 65–79*0404; D. Dose-response inhibition of the interaction between SE ligand 65–79*0401 (171 μM) and CRT by Linear DKCLA; E. Dose-response inhibition of the interaction between SE ligand 65–79*0404 (169 μM) and CRT by Linear DKCLA; F. Linear DKCLA (170 μM) specifically inhibits SE ligand 65–79*0401(57μM)-activated NO signaling; G. Structural formula of the cDKCLA compound library. H. Dose-response curve of the inhibitory effect of HS(4–4)c Trp on SE ligand 65–79*0401 (57 μM)-activated signaling. I. Competitive inhibition of SE ligand 65–79*0401 (229 μM) binding to CRT by HS(4–4)c Trp; J. HS(4–4)c Trp inhibits unstimulated (left) and SE ligand 65–79*0401 (29 μM)-activated (right) osteoclastogenesis in primary mouse BMCs. Asterisks denote statistical significance compared to the respective control culture: PBS, or 65–79*0401; K. Compound HS(3–4)c Trp does not inhibit osteoclastogenesis in mouse BMC; L. HS(4–4)c Trp and HS(3–3)c Trp inhibit SE ligand-activated osteoclastogenesis in human PBMC. #, p < 0.05 compared to PBS cultures; * p < 0.05 compared to cultures with 65–79*0401.
DKCLA peptidomimetics
Linear peptides are short-lived, and their biologic activity is further compromised by their random conformation in solution. Accordingly, as a peptide-stabilization strategy, we have synthesized, according to a previously described procedure (21,22) a library of backbone cyclic DKCLA (cDKCLA) analogs, all carrying an identical primary sequence, but differing in the sizes of their ring (Fig. 1G). Members of this library were screened in SE ligand-activated NO signaling assays to determine the relative SEAL potency of individual compounds in vitro. Of the 16 analogs tested, several were found to be exceptionally potent (e.g., the compounds HS(4–4)c Trp, HS(3–3)c Trp and HS(6–4)c Trp, listed in Table I), with IC50 values as low as 1.6 × 10−12 M (Fig. 1H). The lead compound HS(4–4)c Trp was tested in SPR assays, and was found to be a potent competitive inhibitor of SE-CRT interaction, with an IC50 = 2.4 × 10−12 M (Fig. 1I). Thus, cDKCLA compound HS(4–4)c Trp is a 1,000,000 fold more potent SEAL than the linear DKCLA peptide.
Table I.
Differential inhibition of SE-activated NO signaling by cDKCLA compounds
| cDKCLA compound
|
IC50 (M)
|
|---|---|
| HS(4–4)c Trp | 1.6 × 10−12 |
| HS(3–3)c Trp | 1.8 × 10−12 |
| HS(6–4)c Trp | 6.3 × 10−9 |
| HS(4–6)c Trp | 5.6 × 10−7 |
| HS(6–2)c Trp | 7.7 × 10−7 |
| HS(3–4)c Trp | 4.3 × 10−6 |
cDKCLA compounds inhibit OC differentiation in vitro
Since OC play a key role in bone erosion-associated conditions, an attempt to inhibit these cells is a desirable therapeutic goal. Given the high SEAL potency of compounds HS(4–4)c Trp and HS(3–3)c Trp in signal transduction (Table I), we have carried out experiments to determine their effectiveness as OC inhibitors.
As can be seen, in mouse cells (Fig. 1J) the lead cDKCLA compound HS(4–4)c Trp was found to inhibit very efficiently RANKL-activated OC differentiation. Moreover, it significantly inhibited the incremental SE-activated OC differentiation. SEAL activity was found to be structure-specific, since the cDKCLA analog HS(3–4)c Trp, which carries an identical core amino acid sequence and only differs in the ring size, failed to inhibit OC differentiation (Fig. 1K), consistent with its low inhibitory effect in signal transduction studies (Table I). The anti-osteoclastogenic effect of SEAL compounds HS(4–4)c and HS(3–3)c tested in human PBMCs as well, and showed potent anti-osteoclastogenic effect in the absence or presence of an exogenously-added SE ligand (Figs. 1J and 1L). The effect was found in both SE-positive and SE-negative PBMCs (data not shown). Additionally, preliminary data suggest that similar to their effect on PBMC-derived OCs, SEAL compounds inhibited signal transduction in human fibroblast-like synoviocytes irrespective of the SE status of the donor as well (not shown).
cDKCLA analog-specific effects in vivo
The effects of SEAL compounds were studied in CIA. As shown in Figure 2, mice injected weekly with picogram-doses of HS(4–4)c Trp intra-peritoneally experienced delayed onset (Fig. 2A), lower incidence (Fig. 2B), and significantly milder (Fig. 2C) arthritis. Their joint histology showed lower OC abundance in synovial tissues (Fig. 2D and 2E). Interestingly, although at the end of the treatment period (day 45 post immunization) joint swelling was seen in all groups, the pannus of PBS-treated mice showed greater lymphocytic infiltration compared to the synovial tissue of the HS(4–4)c Trp-treated mice (Fig. 2E). Micro-CT and radiographic imaging demonstrated lesser extent of bone destruction in the treated groups (Figs. 2F and 2G). In contrast, consistent with its inactivity in vitro, the HS(3–4)c Trp analog had no effect on day of arthritis onset (Fig. 3A), incidence (Fig. 3B), disease severity (Fig. 3C), radiographic bone damage (Fig. 3D), or OC abundance in synovial tissues (Fig. 3E).
Figure 2. HS(4–4)c Trp ameliorates CIA.

A. HS(4–4)c Trp, 36 pg/gm (blue) or 360 pg/gm (red), or PBS (black) was administered ip weekly and day of arthritis onset was recorded. N=10 per group. B. CIA mice were treated as above and arthritis incidence was recorded over time; C. CIA mice were treated as above and joint swelling was recorded over time. P values were calculated using a paired t-test compared to PBS treated mice; D. Synovial OCs were counted in the joints of CIA mice treated as above. N=5 per group. E. Wrist histology of CIA mice treated with PBS (left column), or 360 pg/gr of HS(4–4)c Trp (right column). Sections were stained with either TRAP (upper row, 4x magnification), or H&E (lower row, 100x magnification). The region identified by a white arrow is magnified in the boxed image in the lower right corner of the image. F. Micro-CT images of front (left) and rear (right) paws of CIA mice treated with PBS (upper row), 36 pg/gr (middle row), or 360 pg/gr (bottom row) of HS(4–4)c Trp. G. Bone erosion scores of CIA mice treated as above.
Figure 3. HS(3–4)c Trp is inactive.
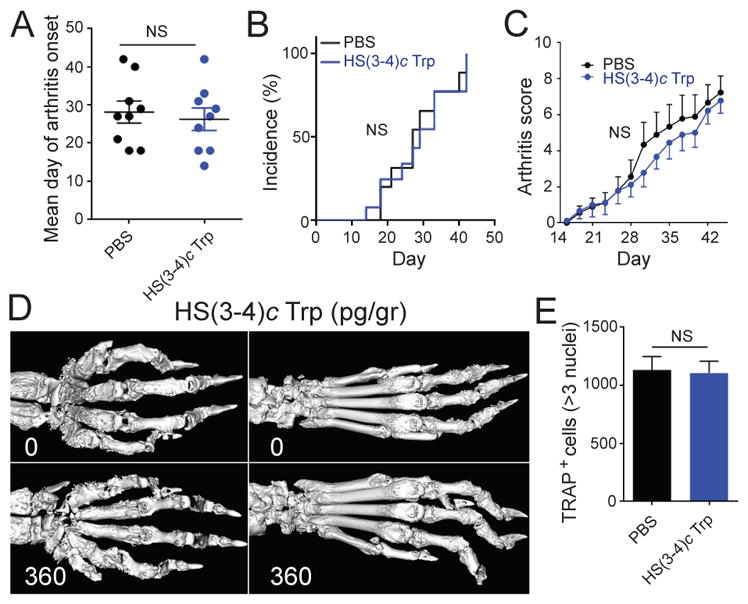
A. Arthritis day of onset in DBA/1 CIA mice treated with HS(3–4)c Trp (administered ip weekly at a dose of 360 pg/gr- blue) or with PBS (black). N=10 per group. B. Arthritis incidence curves of CIA mice treated with 360 pg/gr of HS(3–4)c Trp (blue) or PBS (black). N=10 per group. C. Arthritis scores of CIA mice treated with 360 pg/gr of HS(3–4)c Trp (blue) or with PBS (black). N=10 per group; D. Comparative micro-CT images of paws of CIA mice treated with PBS (upper panel) or HS(3–4)c Trp, 360 pg/gr (lower panel); E. OCs were counted in the joints of CIA mice treated with 360 pg/gr HS(3–4)c Trp (blue) or PBS (black) as above. N=5 per group.
No behavioral, physical, radiological or histological aberrations were noticed in SEAL treated mice, compared to controls. Untreated and treated mice (highest dose 360 pg/gr per week for up to 7 weeks of treatment) showed similar general health parameters (body weight, fur appearance, mobility, and food and water consumption rates).
As mentioned above, compounds HS(4–4)c Trp and HS(3–4)c Trp share an identical core sequence, but differ slightly in their ring size (24 versus 23 atoms, respectively). This minor structural dissimilarity, nevertheless, accounted for a major functional disparity between the two compounds. To gain better insights into the structure-function correlates of the two compounds, we docked them virtually onto a physiologically-folded CRT P-domain (6), using the BioMedCAChe 6.1 software. Compounds HS(4–4)c Trp and HS(3–4)c Trp were both predicted to occupy the SE binding pocket (Figure 4); however, their respective chemical interactions within the binding pocket, and their docking energies were distinct (Table II). The lead SEAL compound HS(4–4)c Trp had a higher docking energy compared to the inactive analog HS(3–4)c Trp, and engaged in a dual chemical bond with a single CRT residue, Glu257. The inactive compound HS(3–4)c Trp, on the other hand, interacted weakly with three CRT residues: Asp209, Glu223 and Gly256. For comparison, a similarly docked SE ligand in its native α helical conformation had a single electrostatic bond with CRT residue Glu223, and a dual electrostatic bond with residue Asp209. Thus, the inactive analog HS(3–4)c Trp, similar to the naturally folded SE ligand, was predicted to generate a ‘bridging’ effect across the SE binding pocket, while the SEAL compound HS(4–4)c Trp displayed a strong focal interaction with CRT residue Glu257, at the center of that pocket (Fig. 4). Taken together, the data in Figure 4 and Table II indicate that despite their near-identical chemical structure, the two functionally disparate compounds displayed distinct molecular interactions with the SE binding site on CRT.
Figure 4. Docking models.

Ligands (cyan) were docked onto the CRT surface (light brown). Side chains of interacting CRT residues are highlighted in orange. Dotted lines represent chemical bonds. Red areas represent surface of key CRT residues that interact with ligands. Inserted boxes depict chemical interactions. The CRT residues are identified in a three-letter format. Ligand residues are shown in a single-letter format.
DISCUSSION
The main significance of the findings reported here relates to the fact that they identify the SE-CRT pathway as a novel therapeutic target. To date, treatment modalities in RA have targeted cytokines, their receptors, or other players in the immune-activated final common pathway. Due to their involvement in ‘downstream’ segments of RA pathogenesis, these targets are often redundant and/or non-specific. As a result, current treatment modalities are often ineffective and/or carry high rates of side effects, mainly infection (23). The advantage of a SEAL-based therapeutic approach over current or emerging drugs is that it addresses an unmet need by offering a potent intervention strategy that specifically targets an ‘upstream’ segment in the pathogenic cascade.
The compounds described here were found to be highly potent both in vitro and in vivo. For example, compounds HS(4–4)c Trp and HS(3–3)c Trp competitively blocked SE signaling effects and inhibited OC differentiation at low-pM concentrations, a 1000-fold higher potency than JAK inhibitors (24). The in vivo effect of compound HS(4–4)c Trp, likewise, was much more potent than emerging drugs, reported to exert therapeutic efficacy in CIA at mg/kg-range doses (24–26). By contrast, compound HS(4–4)c Trp described here achieved disease amelioration effects at ng/kg-range doses, i.e., at a 1,000,000-fold higher potency.
Interestingly, despite its seemingly modest anti-osteoclastogenic effect in vitro (Figs 1J and 1L), SEAL compound HS(4–4)c Trp was highly effective in diminishing tissue OCs when administered in vivo to mice with CIA (Fig. 2D). The likely explanation for the apparent discrepancy is that in vitro assays do not accurately replicate the cellular events that take place in vivo. Specifically in this case: in vitro OC differentiation assays were carried out using mouse BMCs (Fig 1J) or human PBMCs (Fig 1L), while in vivo, OC abundance was determined within mouse synovial tissues, which represent a different cellular milieu. Second, in vitro OC differentiation protocols use selected pro-osteoclastogenic factors (M-CSF and RANKL), whereas the in vivo process involves additional pro- and anti-osteoclastogenic factors. Third, the in situ concentrations of RANKL and M-CSF are likely lower than those used to artificially stimulate robust OC differentiation in vitro.
SEAL compound-treated CIA mice showed delayed arthritis onset and incidence; however both the treated and control groups eventually developed joint swelling and reached equivalent disease incidence. This observation is consistent with our prior studies, which demonstrated that the arthritogenic effect of SE agonist ligands affect joint swelling in the early phase of CIA, but less so in the chronic phase (12,13). The mechanistic basis of the differential effect on early versus late joint swelling is unknown, but is likely related to the fact that the early and late stages of CIA are distinct in their pathogenic mechanisms and the degree of their responsiveness to therapeutic interventions (27–31).
Similarly, consistent with our previous findings with SE agonist ligands (12,13), which demonstrated a disproportionate impact on bone erosions relative to their effects on joint swelling, the SEAL compound reversed this trend by affecting primarily bone damage. Such differential effect is not without precedent, as disparate severities of joint inflammation versus bone erosion have been previously reported in experimental arthritis models (32,33), as well as RA (34). Better understanding of the mechanisms governing SEAL bone-selectivity could provide important new insights into the pathogenic mechanisms that specifically underlie inflammation versus bone damage.
It should be pointed out that SEAL compounds inhibited basal osteoclastogenesis in vitro even in the absence of the SE ligand and ameliorated CIA in mice that do not carry SE-coding class II MHC alleles. Therefore, consistent with the SE ligand theory (discussed in 35,36), it is reasonable to propose the existence of a physiologic CRT-binding ligand capable of OC activation during healthy bone remodeling. There are several physiologic ligands that the SE ligand could potentially mimic. For example, circulating levels of adiponectin, a known CRT ligand (37), have been shown to correlate with the extent of radiographic bone damage in RA (38). Noteworthy, adiponectin facilitates Th17 polarization (39), and can activate OCs (reviewed in 40). Thrombospondin-1, likewise, a well-characterized CRT ligand (41) implicated in RA pathogenesis (42, 43), facilitates Th17 polarization (44), and activates OCs (45).
Obviously, identification of the specific physiologic CRT ligand that the SE may be mimicking requires further study. However, irrespective of the identity of that physiologic ligand, the model proposed here hypothesizes that the SE may facilitate OC activation and bone damage by mimicking a putative physiologic ligand. Accordingly, SEAL compounds may ameliorate OC activation and arthritis through competitive inhibition of a physiologic ligand, independent of the presence or absence of a SE ligand. This model further posits that individuals with SE-coding HLA-DRB1 alleles are at higher risk of developing erosive arthritis due to the high abundance of cell surface SE-expressing HLA-DR molecules that mimic the physiologic OC-activating ligand. Under certain genetic and environmental circumstances, the SE ligand may over-activate the innate pathway, with resultant excessive activation of OCs. In this context it is worth noting that SE-associated bone damage is not unique to RA, as SE-coding HLA-DRB1 alleles have been shown to associate with accelerated bone erosion in psoriatic arthritis (46), periodontal disease (47) and SLE (48) patients.
Beyond offering new insights into the pathogenesis of bone damage in arthritis, our findings illustrate the therapeutic targetability of the SE-CRT pathway by highly-specific, rationally-designed SEAL compounds. The compounds presented here were highly potent, both in vitro and in vivo, and showed exquisite structure-activity relationships. These facts could facilitate future compound optimization efforts. From the medicinal chemistry perspective it is also worth noting that the cysteine residue does not appear to play a role in the antagonist’s interaction model with CRT (Fig. 4 and Table II). Additionally, since the inactive compound HS(3–4)c Trp contains cysteine in the same position as the active compounds HS(4–4)c Trp and HS(3–3)c Trp, the cysteine residue, including its thiol side chain, is unlikely to play a functional role in the compounds’ biologic effects.
The new strategy presented here may be safer than the use of prevailing drugs, due to the unique role played by this pathway at an ‘upstream’ phase in RA pathogenesis. The SE is the single most significant risk factor for RA. It determines susceptibility, severity and disease penetrance (49). Thus, different from effector cytokines or intracellular enzymes, this pathway is involved in the early segments of disease etiology and pathogenesis. Targeting such an ‘upstream’ pathway might be more effective and would likely produce fewer side effects.
In conclusion, this study suggests that SEAL compounds could be useful tools for studying the mechanisms governing bone erosion in various conditions, including RA. Furthermore, the results of this study provide rationale and early medicinal information that could help developing specific, potent, safe and inexpensive drugs for arthritis and other erosive bone conditions.
Supplementary Material
Acknowledgments
This study was supported by grants from The National Institute of General Medical (5R01GM088560), The National Center for Research Resources (UL 1RR024986), and The National Institute of Arthritis and Musculoskeletal and Skin Diseases (5R01AR059085, 3R01AR059085-03S1 and T32AR07080). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Drs. Ling, Gilon and Holoshitz are named Inventors on US Patents owned by the regents of the University of Michigan, USA and The Hebrew University in Jerusalem, Israel.
References
- 1.Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987;30:1205–13. doi: 10.1002/art.1780301102. [DOI] [PubMed] [Google Scholar]
- 2.Gonzalez-Gay MA, Garcia-Porrua C, Hajeer AH. Influence of human leukocyte antigen-DRB1 on the susceptibility and severity of rheumatoid arthritis. Semin Arthritis Rheum. 2002;31:355–60. doi: 10.1053/sarh.2002.32552. [DOI] [PubMed] [Google Scholar]
- 3.Mattey DL, Hassell AB, Dawes PT, Cheung NT, Poulton KV, Thomson W, et al. Independent association of rheumatoid factor and the HLA-DRB1 shared epitope with radiographic outcome in rheumatoid arthritis. Arthritis Rheum. 2001;44:1529–33. doi: 10.1002/1529-0131(200107)44:7<1529::AID-ART275>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 4.Plant MJ, Jones PW, Saklatvala J, Ollier WE, Dawes PT. Patterns of radiological progression in early rheumatoid arthritis: results of an 8 year prospective study. J Rheumatol. 1998;25:417–26. [PubMed] [Google Scholar]
- 5.Weyand CM, Goronzy JJ. Disease mechanisms in rheumatoid arthritis: gene dosage effect of HLA-DR haplotypes. J Lab Clin Med. 1994;124:335–8. [PubMed] [Google Scholar]
- 6.Ling S, Cheng A, Pumpens P, Michalak M, Holoshitz J. Identification of the rheumatoid arthritis shared epitope binding site on calreticulin. PLoS One. 2010;5:e11703. doi: 10.1371/journal.pone.0011703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Almeida DE, Ling S, Pi X, Hartmann-Scruggs AM, Pumpens P, Holoshitz J. Immune dysregulation by the rheumatoid arthritis shared epitope. J Immunol. 2010;185:1927–34. doi: 10.4049/jimmunol.0904002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ling S, Cline EN, Haug TS, Fox DA, Holoshitz J. Citrullinated calreticulin potentiates rheumatoid arthritis shared epitope signaling. Arthritis Rheum. 2013;65:618–26. doi: 10.1002/art.37814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ling S, Lai A, Borschukova O, Pumpens P, Holoshitz J. Activation of nitric oxide signaling by the rheumatoid arthritis shared epitope. Arthritis Rheum. 2006;54:3423–32. doi: 10.1002/art.22178. [DOI] [PubMed] [Google Scholar]
- 10.Ling S, Li Z, Borschukova O, Xiao L, Pumpens P, Holoshitz J. The rheumatoid arthritis shared epitope increases cellular susceptibility to oxidative stress by antagonizing an adenosine-mediated anti-oxidative pathway. Arthritis Res Ther. 2007;9:R5. doi: 10.1186/ar2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ling S, Pi X, Holoshitz J. The rheumatoid arthritis shared epitope triggers innate immune signaling via cell surface calreticulin. J Immunol. 2007;179:6359–67. doi: 10.4049/jimmunol.179.9.6359. [DOI] [PubMed] [Google Scholar]
- 12.Fu J, Ling S, Liu Y, Yang J, Naveh S, Hannah M, et al. A small shared epitope-mimetic compound potently accelerates osteoclast-mediated bone damage in autoimmune arthritis. J Immunol. 2013;191:2096–103. doi: 10.4049/jimmunol.1203231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holoshitz J, Liu Y, Fu J, Joseph J, Ling S, Colletta A, et al. An HLA-DRB1-coded signal transduction ligand facilitates inflammatory arthritis: a new mechanism of autoimmunity. J Immunol. 2013;190:48–57. doi: 10.4049/jimmunol.1202150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gravallese EM, Manning C, Tsay A, Naito A, Pan C, Amento E, et al. Synovial tissue in rheumatoid arthritis is a source of osteoclast differentiation factor. Arthritis Rheum. 2000;43:250–8. doi: 10.1002/1529-0131(200002)43:2<250::AID-ANR3>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 15.Schett G, Gravallese E. Bone erosion in rheumatoid arthritis: mechanisms, diagnosis and treatment. Nat Rev Rheumatol. 2012;8:656–64. doi: 10.1038/nrrheum.2012.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Punder YM, van Riel PL. Rheumatoid arthritis: understanding joint damage and physical disability in RA. Nat Rev Rheumatol. 2011;7(5):260–1. doi: 10.1038/nrrheum.2011.49. [DOI] [PubMed] [Google Scholar]
- 17.Le Goff B, Berthelot JM, Maugars Y, Heymann D. Osteoclasts in RA: diverse origins and functions. Joint Bone Spine. 2013;80:586–91. doi: 10.1016/j.jbspin.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 18.Kim HR, Kim KW, Kim BM, Jung HG, Cho ML, Lee SH. Reciprocal activation of CD4+ T cells and synovial fibroblasts by stromal cell-derived factor 1 promotes RANKL expression and osteoclastogenesis in rheumatoid arthritis. Arthritis Rheum. 2014;66:538–48. doi: 10.1002/art.38286. [DOI] [PubMed] [Google Scholar]
- 19.Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest. 1999;103:1345–52. doi: 10.1172/JCI5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. 2006;203:2673–82. doi: 10.1084/jem.20061775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hurevich M, Tal-Gan Y, Klein S, Barda Y, Levitzki A, Gilon C. Novel method for the synthesis of urea backbone cyclic peptides using new Alloc-protected glycine building units. J Pept Sci. 2010;16:178–85. doi: 10.1002/psc.1218. [DOI] [PubMed] [Google Scholar]
- 22.Naveh S, Tal-Gan Y, Ling S, Hoffman A, Holoshitz J, Gilon C. Developing potent backbone cyclic peptides bearing the shared epitope sequence as rheumatoid arthritis drug-leads. Bioorg Med Chem Lett. 2012;22:493–6. doi: 10.1016/j.bmcl.2011.10.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Furst DE. The risk of infections with biologic therapies for rheumatoid arthritis. Semin Arthritis Rheum. 2010;39:327–46. doi: 10.1016/j.semarthrit.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 24.Fridman JS, Scherle PA, Collins R, Burn TC, Li Y, Li J, et al. Selective inhibition of JAK1 and JAK2 is efficacious in rodent models of arthritis: preclinical characterization of INCB028050. J Immunol. 2010;184:5298–307. doi: 10.4049/jimmunol.0902819. [DOI] [PubMed] [Google Scholar]
- 25.Stump KL, Lu LD, Dobrzanski P, Serdikoff C, Gingrich DE, Dugan BJ, et al. A highly selective, orally active inhibitor of Janus kinase 2, CEP-33779, ablates disease in two mouse models of rheumatoid arthritis. Arthritis Res Ther. 2011;13:R68. doi: 10.1186/ar3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.William AD, Lee AC, Poulsen A, Goh KC, Madan B, Hart S, et al. Discovery of the macrocycle (9E)-15-(2-(pyrrolidin-1-yl)ethoxy)-7,12,25-trioxa-19,21,24-triaza-tetracyclo[18. 3.1.1(2,5). 1(14,18)]hexacosa-1(24),2,4,9,14(26),15,17,20,22-nonaene (SB1578), a potent inhibitor of janus kinase 2/fms-like tyrosine kinase-3 (JAK2/FLT3) for the treatment of rheumatoid arthritis. J Med Chem. 2012;55:2623–40. doi: 10.1021/jm201454n. [DOI] [PubMed] [Google Scholar]
- 27.Svendsen P, Andersen CB, Willcox N, Coyle AJ, Holmdahl R, Kamradt T, et al. Tracking of proinflammatory collagen-specific T cells in early and late collagen-induced arthritis in humanized mice. J Immunol. 2004;173:7037–45. doi: 10.4049/jimmunol.173.11.7037. [DOI] [PubMed] [Google Scholar]
- 28.Merky P, Batsalova T, Bockermann R, Dzhambazov B, Sehnert B, Burkhardt H, et al. Visualization and phenotyping of proinflammatory antigen-specific T cells during collagen-induced arthritis in a mouse with a fixed collagen type II-specific transgenic T-cell receptor beta-chain. Arthritis Res Ther. 2010;12:R155. doi: 10.1186/ar3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miellot-Gafsou A, Biton J, Bourgeois E, Herbelin A, Boissier MC, Bessis N. Early activation of invariant natural killer T cells in a rheumatoid arthritis model and application to disease treatment. Immunology. 2010;130:296–306. doi: 10.1111/j.1365-2567.2009.03235.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abreu JR, Dontje W, Krausz S, de Launay D, van Hennik PB, van Stalborch AM, et al. A Rac1 inhibitory peptide suppresses antibody production and paw swelling in the murine collagen-induced arthritis model of rheumatoid arthritis. Arthritis Res Ther. 2010;12:R2. doi: 10.1186/ar2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bruhl H, Cihak J, Niedermeier M, Denzel A, Rodriguez Gomez M, Talke Y, et al. Important role of interleukin-3 in the early phase of collagen-induced arthritis. Arthritis Rheum. 2009;60:1352–61. doi: 10.1002/art.24441. [DOI] [PubMed] [Google Scholar]
- 32.Joosten LA, Helsen MM, Saxne T, van De Loo FA, Heinegard D, van Den Berg WB. IL-1 alpha beta blockade prevents cartilage and bone destruction in murine type II collagen-induced arthritis, whereas TNF-alpha blockade only ameliorates joint inflammation. J Immunol. 1999;163:5049–55. [PubMed] [Google Scholar]
- 33.Binder NB, Puchner A, Niederreiter B, Hayer S, Leiss H, Bluml S, et al. Tumor necrosis factor-inhibiting therapy preferentially targets bone destruction but not synovial inflammation in a tumor necrosis factor-driven model of rheumatoid arthritis. Arthritis Rheum. 2013;65:608–17. doi: 10.1002/art.37797. [DOI] [PubMed] [Google Scholar]
- 34.Backhaus M, Burmester GR, Sandrock D, Loreck D, Hess D, Scholz A, et al. Prospective two year follow up study comparing novel and conventional imaging procedures in patients with arthritic finger joints. Ann Rheum Dis. 2002;6:895–904. doi: 10.1136/ard.61.10.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Almeida DE, Holoshitz J. MHC molecules in health and disease: At the cusp of a paradigm shift. Self Nonself. 2011;2:43–8. doi: 10.4161/self.2.1.15757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holoshitz J. The quest for better understanding of HLA-disease association: scenes from a road less travelled by. Discov Med. 2013;16:93–101. [PMC free article] [PubMed] [Google Scholar]
- 37.Takemura Y, Ouchi N, Shibata R, Aprahamian T, Kirber MT, Summer RS, et al. Adiponectin modulates inflammatory reactions via calreticulin receptor-dependent clearance of early apoptotic bodies. J Clin Invest. 2007;117:375–86. doi: 10.1172/JCI29709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Giles JT, van der Heijde DM, Bathon JM. Association of circulating adiponectin levels with progression of radiographic joint destruction in rheumatoid arthritis. Ann Rheum Dis. 2011;70:1562–8. doi: 10.1136/ard.2011.150813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jung MY, Kim H-S, Hong H-J, Youn B-S, Kim TS. Adiponectin induces dendritic cell activation via PLCγ/JNK/NF-κB pathways, leading to Th1 and Th17 polarization. J Immunol. 2012;188:2592–601. doi: 10.4049/jimmunol.1102588. [DOI] [PubMed] [Google Scholar]
- 40.Kanazawa I. Adiponectin in metabolic bone disease. Curr Med Chem. 2012;19:5481–92. doi: 10.2174/092986712803833146. [DOI] [PubMed] [Google Scholar]
- 41.Goicoechea S, Orr AW, Pallero MA, Eggleton P, Murphy-Ullrich JE. Thrombospondin mediates focal adhesion disassembly through interactions with cell surface calreticulin. J Biol Chem. 2000;275:36358–68. doi: 10.1074/jbc.M005951200. [DOI] [PubMed] [Google Scholar]
- 42.Vallejo AN, Mugge LO, Klimiuk PA, Weyand CM, Goronzy JJ. Central role of thrombospondin-1 in the activation and clonal expansion of inflammatory T cells. J Immunol. 2000;164:2947–54. doi: 10.4049/jimmunol.164.6.2947. [DOI] [PubMed] [Google Scholar]
- 43.Rico MC, Rough JJ, Del Carpio-Cano FE, Kunapuli SP, DeLa Cadena RA. The axis of thrombospondin-1, transforming growth factor beta and connective tissue growth factor: an emerging therapeutic target in rheumatoid arthritis. Curr Vasc Pharmacol. 2010;8:338–43. doi: 10.2174/157016110791112296. [DOI] [PubMed] [Google Scholar]
- 44.Yang K, Vega JL, Hadzipasic M, Schatzmann Peron JP, Zhu B, Carrier Y, et al. Deficiency of thrombospondin-1 reduces Th17 differentiation and attenuates experimental autoimmune encephalomyelitis. J Autoimmun. 2009;32:94–103. doi: 10.1016/j.jaut.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carron JA, Walsh CA, Fraser WD, Gallagher JA. Thrombospondin promotes resorption by osteoclasts in vitro. Biochem Biophys Res Commun. 1995;213:1017–25. doi: 10.1006/bbrc.1995.2230. [DOI] [PubMed] [Google Scholar]
- 46.Korendowych E, Dixey J, Cox B, Jones S, McHugh N. The Influence of the HLA-DRB1 rheumatoid arthritis shared epitope on the clinical characteristics and radiological outcome of psoriatic arthritis. J Rheumatol. 2003;30:96–101. [PubMed] [Google Scholar]
- 47.Marotte H, Farge P, Gaudin P, Alexandre C, Mougin B, Miossec P. The association between periodontal disease and joint destruction in rheumatoid arthritis extends the link between the HLA-DR shared epitope and severity of bone destruction. Ann Rheum Dis. 2006;65:905–9. doi: 10.1136/ard.2005.036913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chan MT, Owen P, Dunphy J, Cox B, Carmichael C, Korendowych E, et al. Associations of erosive arthritis with anti-cyclic citrullinated peptide antibodies and MHC Class II alleles in systemic lupus erythematosus. J Rheumatol. 2008;35:77–83. [PubMed] [Google Scholar]
- 49.Jawaheer D, Thomson W, MacGregor AJ, Carthy D, Davidson J, Dyer PA, et al. “Homozygosity” for the HLA-DR shared epitope contributes the highest risk for rheumatoid arthritis concordance in identical twins. Arthritis Rheum. 1994;37:681–6. doi: 10.1002/art.1780370511. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.