

Summary
Recent studies in humans and in genetic mouse models have identified Slitrks as candidate genes for neuropsychiatric disorders. All Slitrk isotypes are highly expressed in the CNS, where they mediate neurite outgrowth, synaptogenesis and neuronal survival. However, the molecular mechanisms underlying these functions are not known. Here, we report that Slitrk5 modulates BDNF-dependent biological responses through direct interaction with TrkB receptors. Under basal conditions, Slitrk5 interacts primarily with a trans-synaptic binding partner, PTPδ; however, upon BDNF stimulation, Slitrk5 shifts to cis-interactions with TrkB. In the absence of Slitrk5, TrkB has a reduced rate of ligand-dependent recycling and altered responsiveness to BDNF treatment. Structured illumination microscopy revealed that Slitrk5 mediates optimal targeting of TrkB receptors to Rab11-positive recycling endosomes through the recruitment of a Rab11 effector protein, Rab11-FIP3. Thus, Slitrk5 acts as a TrkB co-receptor that mediates its BDNF-dependent trafficking and signaling.
eTOC
Slitrk family proteins are emerging as candidate genes involved in neuropsychiatric disorders. Song et al. show that Slitrk5 modulates BDNF-dependent biological responses by directly regulating TrkB receptor recycling via recruitment of Rab11-FIP3.
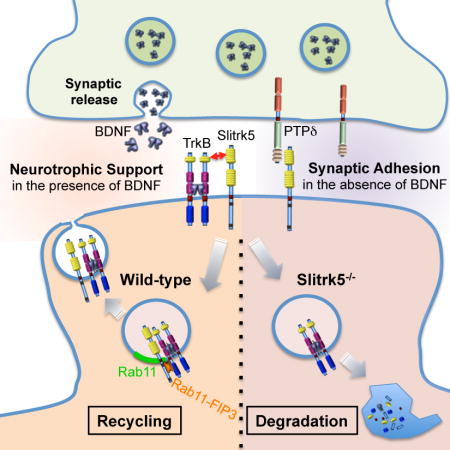
Introduction
Slit- and NTRK-like family (Slitrks) are type 1 transmembrane proteins that localize to and function at central nervous system (CNS) synapses where they mediate synapse formation through trans-synaptic interactions of their ectodomains with presynaptic binding partners (Linhoff et al., 2009; Takahashi et al., 2012; Yim et al., 2013). Their cytoplasmic tails, larger than those of other well-studied synaptic adhesion molecules (SAMs) e.g. Neurexins, Neuroligins, LRRTMs, SynCAMs, N-Cadherins, and L1 adhesion molecules (Aruga and Mikoshiba, 2003; Proenca et al., 2011), suggest functional interactions with other molecules. Slitrks have structural and functional similarity to Trk neurotrophin receptors. They contain N-terminal leucine-rich repeat (LRR) domains and intracellular tyrosine-based motifs similar to those found in TrkA, TrkB, and TrkC. Furthermore, Slitrks mediate biological functions similar to Trk receptors including neurite outgrowth and dendritic elaboration, synapse formation and neuronal survival (Aruga and Mikoshiba, 2003; Ko, 2012; Proenca et al., 2011; Yim et al., 2013). It is well established that Trk receptors mediate this broad range of functions through their selective binding to 3 major ligands: nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), and NT-3 (Chao, 2003).
A key question is how such a limited repertoire of neurotrophins and Trk receptors mediate such a diverse array of biological actions. In the peripheral nervous system (PNS), the basic strategy to generate signaling diversity has been elucidated. In sensory and sympathetic neurons, Trk receptors are expressed at high levels and form combinatorial complexes with their co-receptor, p75NTR at the plasma membrane. These interactions increase their affinity and selectivity for specific neurotrophins (Benedetti et al., 1993; Hempstead et al., 1991; Kuruvilla et al., 2004; Lee et al., 1994). In contrast, in the adult CNS, the strategy underlying signaling diversity remains essentially unknown. Whereas TrkB and TrkC receptors are expressed at high levels in the CNS, p75NTR has a limited distribution, restricted to primarily the basal forebrain (Huang and Reichardt, 2003; Lee et al., 2001). Hence, it is likely that regulation of Trk function in the CNS involves alternative molecules.
Slitrks are excellent candidates to perform such regulatory functions. Of the six Slitrk family members expressed in the CNS (Aruga and Mikoshiba, 2003; Shmelkov et al., 2010), Slitrk5 provides the more compelling links with the neurotrophin system. Slitrk5−/− mice display altered anatomical and neuronal morphological phenotypes in the striatum similar to those observed in the striatum of mice with genetic deficiencies in BDNF or TrkB (Emx-BDNF, Tau-BDNF, Dlx5/6-TrkB, and Bf1-BDNF KO mice) (Baquet et al., 2004; Baydyuk et al., 2011; Li et al., 2012; Rauskolb et al., 2010; Shmelkov et al., 2010). This raises the intriguing question of whether in the CNS Slitrk5 functions through direct interaction and modulation of the neurotrophin system. Here, we present mechanistic evidence demonstrating that Slitrk5 acts as a co-receptor of TrkB that modulates its post-endocytic recycling to facilitate BDNF-dependent signaling responses. Our study identifies a key regulator of the diverse array of BDNF functions in the CNS.
Results
Slitrk5 Interacts with TrkB Receptors
To determine whether Slitrk5 and TrkB receptors are physically associated, we carried out co-immunoprecipitation studies in cell lines and primary neurons. Studies using HEK293T cells transfected with FLAG-tagged TrkB and WT Slitrk5 plasmids (Figure 1A) or HEK293 cells stably expressing TrkB (HEK293-TrkB) transfected with GFP-tagged Slitrk5 (Figure 1B), clearly demonstrated that the two proteins interact. Furthermore, co-immunoprecipitation studies in brain lysates using anti-TrkB antibodies or anti-Slitrk5 antibodies demonstrated that endogenous Slitrk5 and TrkB interact in neurons (Figure 1C, S1E). This interaction is specific, as it was not observed for other Slitrk members (Slitrk1-3) (Figure 1D), or for the other major CNS neurotrophin receptor, TrkC, in primary cultured neurons (Figure 1E, S1E). To map the Slitrk5 and TrkB domains that mediate their interaction, we utilized a chimera-based approach. The extracellular domain of Slitrk5 encodes two LRR domains (Figure 1F). LRR domains often mediate protein-protein interactions (Gay et al., 1991; Mandai et al., 2009). We swapped the extracellular domains of Slitrk5 with the corresponding domains of another Slitrk (Slitrk1) (Figure 1F). After transfection of HEK293-TrkB cells with the chimeric Slitrk5 constructs, their interaction with TrkB was assessed by co-immunoprecipitation and immunoblot analyses. These studies demonstrated that the interaction of Skitrk5 with TrkB was mediated by its first LRR (LRR1) domain (Figure 1F). Complementary studies with chimeras between TrkB and TrkC demonstrated that binding of TrkB with Slitrk5 is mediated by its single LRR domain (Figure 1G). Taken together, these studies demonstrate that Slitrk5 and TrkB interact specifically via their extracellular LRR domains.
Figure 1. TrkB receptors interact and co-localize with Slitrk5.
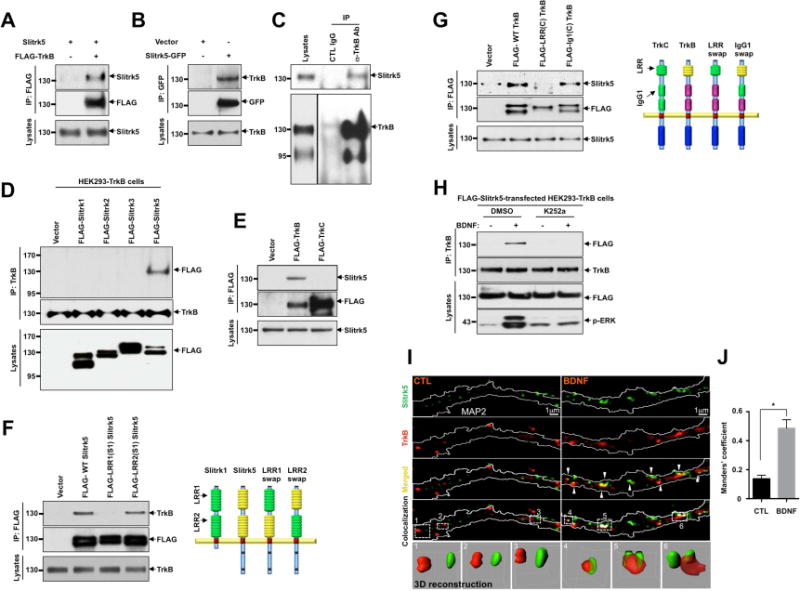
(A) Interaction between TrkB receptors and Slitrk5 was assessed in HEK293T overexpressing cDNAs encoding FLAG-TrkB, Slitrk5, and empty vector. Cell lysates were immunoprecipitated with anti-FLAG antibodies and immunoblotted with anti-Slitrk5 antibodies. (B) Interaction between TrkB receptors and Slitrk5 was assessed in HEK293-TrkB cells. Lysates from Slitrk5-GFP or empty vector transfected HEK293-TrkB cells were immunoprecipitated with anti-GFP antibodies and immunoblotted with anti-TrkB antibody. (C) Endogenous association of Slitrk5 and TrkB. Mouse whole-brain lysates (2 months old) were subjected to immunoprecipitation with anti-TrkB antibody (Millipore) or control IgG. The immune protein complex was then eluted, and TrkB and Slitrk5 were detected by immunoblotting. (D) and (E) Interaction between TrkB and Slitrk5 is specific. (D) Interaction between TrkB and Slitrk family members. FLAG-tagged Slitrk family isotypes were expressed in HEK293-TrkB cells. Cell lysates were immunoprecipitated with anti-TrkB (Millipore) antibodies and immunoblotted with anti-FLAG (M2) antibodies (E) Dissociated E16 mouse cortical neurons were electroporated (Amaxa) with FLAG-TrkB or FLAG-TrkC. At DIV6, cell lysates were precipitated with anti-FLAG (M2) antibodies and immunoblotted with anti-Slitrk5 antibodies. (F) First LRR domain of Slitrk5 mediates TrkB binding. HEK293-TrkB cells were transfected with FLAG-tagged WT Slitrk5, chimeric FLAG-LRR1-domain-swapped (FLAG-LRR1(S1) Slitrk5) and LRR2-domain-swapped Slitrk5 (FLAG-LRR2(S1) Slitrk5) or empty vector. Cell lysates were precipitated with anti-FLAG (M2) antibodies and immunoblotted with anti-TrkB (Millipore) antibodies. Schematic representation of chimeric Slitrk5 mutants shown on the right. (G) LRR domain of TrkB mediates Slitrk5 binding. Dissociated E16 mouse cortical neurons were transfected with FLAG-tagged WT TrkB, LRR domain-swapped TrkB (FLAG-LRR(C) TrkB), and IgG1 domain-swapped TrkB (FLAG-IgG1(C) TrkB) or empty vector. At DIV6, cell lysates were immunoprecipitated with anti-FLAG antibodies and immunoblotted with anti-Slitrk5. Schematic representation of chimeric TrkB mutants shown on the right. (H) BDNF-dependent and TrkB kinase activity-dependent binding of TrkB and Slitrk5. After transfection of FLAG-tagged Slitrk5 into HEK293-TrkB cells, cells were pretreated with DMSO or K252a for 30 min to inhibit kinase activity of TrkB after overnight serum starvation. Cells were treated with or without BDNF, and their binding was examined by immunoprecipitation with anti-TrkB antibodies followed by immunoblotting with anti-FLAG antibodies. (I) TrkB receptors co-localize with Slitrk5 in a BDNF-dependent manner. DIV6 striatal neurons were treated with or without BDNF (25ng/ml) for 30 min after incubating with anti-TrkB antibody to specifically label surface TrkB. Endogenous Slitrk5 and MAP2 were visualized with specific antibodies after fixation and permeabilization. Super resolution images were acquired using a Nikon N-SIM structured illumination microscope. The co-localization of TrkB and Slitrk5 was presented using co-localization highlighter (ImageJ) and 3D reconstruction (IMARIS) to show more convincing co-localization signals. (J) Histogram showing mean Manders’ coefficients for co-localization of TrkB and Slitrk5 (n=20 for each condition). High Manders’ coefficients indicate better co-localization of TrkB with Slitrk5. Results are means ± SEM from 3 independent experiments. 20–30 neurons were analyzed per condition per experiment. *P<0.05 significantly different from control condition (Student’s t test).
Next, we carried out co-immunoprecipitation experiments to examine whether the interaction between Slitrk5 and TrkB is modulated by BDNF-dependent TrkB activation. These experiments showed that, compared to the control condition (10% FBS, Figure 1A, B), serum starvation (0% FBS) significantly reduced the basal interaction between FLAG-tagged Slitrk5 and TrkB, (Figure 1H). In contrast, upon BDNF stimulation, the interaction between Slitrk5 and TrkB was significantly increased, and blocked by pretreatment with K252a, an inhibitor of Trk kinases (Figure 1H). These studies suggest that Slitrk5 optimally interacts with a TrkB receptor complex that is activated by BDNF.
We next considered whether Slitrk5 and TrkB receptors co-localized within neurons. To test this hypothesis, we examined the subcellular localization of endogenous Slitrk5 and TrkB receptors in cultured striatal neurons using Structured Illumination Microscopy (SIM), a form of high-resolution microscopy that uses high frequency striped pattern of light to illuminate the sample in multiple angles to enhance image resolution up to 85 nm (Gustafsson, 2000). In untreated striatal neurons, TrkB and Slitrk5 localized separately in dendrites, visualized with MAP2 staining (Figure 1I, left panels, quantified in Figure 1J). In contrast, after BDNF treatment, TrkB receptors significantly co-localized with Slitrk5 in enlarged punctate structures, presumably reflecting its presence in endosomes (Figure 1I, right panels, quantified in Figure 1J, Figure S1F). The co-localization of TrkB and Slitrk5 was visualized with co-localization highlighter (ImageJ), as well as with 3D reconstruction (IMARIS). Together these interaction and immunocytochemical studies suggest that Slitrk5 and TrkB receptors interact in a BDNF-dependent manner.
BDNF Shifts Slitrk5 binding from PTPδ to TrkB Receptors
Recently, one of the Slitrk isotypes, Slitrk3, was reported to mediate inhibitory synapse formation through trans-synaptic interactions with presynaptic receptor-type protein tyrosine phosphatases δ (PTPδ) (Takahashi et al., 2012). In the same study, the authors also showed that Slitrk5 was capable of interacting with PTPδ (Takahashi et al., 2012). Therefore, we investigated whether the BDNF-induced cis-interaction of Slitrk5 with TrkB disrupts the trans-interaction of Slitrk5 with PTPδ (Figure 2). First, utilizing a binding assay in which we exposed HEK293T cells expressing full-length or mutant Slitrk5 to soluble purified PTPδ ectodomain fused to the Fc region of human immunoglobulin (PTPδ-Fc), we showed that Slitrk5 binds PTPδ through its LRR1 domain (Figure 2A, B). Next, using a heterophilic cell adhesion assay (Fogel et al., 2007), we tested whether the presence of TrkB receptors in cells expressing Slitrk5 affected the trans-interaction of Slitrk5 with PTPδ. Briefly, to mimic cis- and trans-interactions of Slitrk5 with TrkB and PTPδ, respectively, in the neuronal synapse, we co-cultured HEK293 cells co-expressing FLAG-Slitrk5 and TrkB with HEK293T cells expressing HA-PTPδ. Surface proteins were labeled with respective antibodies at the beginning of the experiments, and their localization was analyzed by fluorescence microscopy for cis- and trans-interactions in the presence or absence of BDNF treatment. In the absence of BDNF, all FLAG-Slitrk5 was recruited to regions of heterophilic interaction with HA-PTPδ, where the interacting proteins formed stretched zipper-like structures, with minimal co-localization with TrkB (Figure 2C–E). In contrast, upon addition of BDNF, FLAG-Slitrk5 co-localized with TrkB in punctate endosomal structures. Quantification of their co-localization showed that the binding preference of FLAG-Slitrk5 shifted from PTPδ to TrkB upon BDNF stimulation (Figure 2D, E). To further support this notion of a ligand–dependent shift in interactions, we employed additional quantitative approaches. We examined PTPδ-Fc binding to Slitrk5 with increasing amounts of PTPδ-Fc in the presence or absence of BDNF (25 ng/ml). Results indicate that the maximal binding capacity (Bmax) of PTPδ-Fc, but not dissociation constant (Kd), was decreased in the presence of BDNF (Figure S2A). This result indicates that available PTPδ-Fc binding sites were reduced by BDNF treatment without changing affinity of PTPδ-Fc to Slitrk5. The apparent dissociation constant (Kd) value of PTPδ-Fc binding to Slitrk5 was ~100 nM regardless of BDNF treatment. Next, we measured dissociation of pre-bound PTPδ-Fc to surface-expressed HA-Slitrk5 with increasing BDNF concentrations. In accordance with previous competition data (Figure 2C–E), BDNF potently induced dissociation of pre-bound PTPδ-Fc from HA-Slitrk5-expressing HEK293-TrkB cells (Figure 2F, S2B). Based on the PTPδ-Fc dissociation curve, the Kd value of BDNF to HA-Slitrk5 was ~ 0.9 nM. These results suggest that both PTPδ and TrkB compete for binding to Slitrk5 and that, whereas under basal conditions, the interaction with PTPδ predominates, BDNF stimulation directs Slitrk5 to cis-interactions with TrkB receptors.
Figure 2. PTPδ and TrkB compete for binding to Slitrk5.
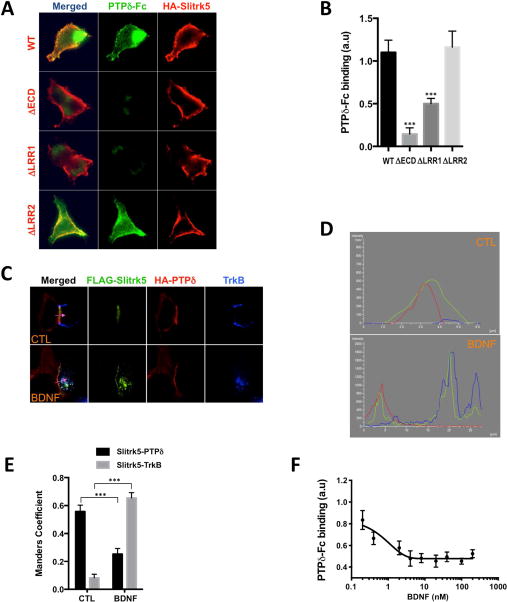
(A) Slitrk5 binds PTPδ through LRR1. Soluble purified PTPδ-Fc chimeras were added to HEK293T cells expressing WT and indicated deletion mutants of Slitrk5 and the binding analyzed by immunofluorescence microscopy. Note that the observed binding to WT Slitrk5 is abolished by deletion of Slitrk5’s extracellular domain (ECD) or LRR1 but not LRR2. (B) Quantitative analysis of these results. Results are means ± SEM from 3 independent experiments. 20–30 cells were analyzed per condition per experiment. ***P< 0.0001 significantly different from WT condition. (One-way ANOVA and Dunnett’s multiple comparisons test). (C) BDNF displaces Slitrk5 binding from PTPδ to TrkB. Heterophilic cell adhesion assay in which HEK293-TrkB cells expressing FLAG-Slitrk5 are co-cultured with HEK293T cells expressing HA-PTPδ. Surface proteins were visualized by fluorescence microscopy in the presence or absence of BDNF treatment (as described in Supplemental Experimental Procedures). (D) Fluorescence intensity trace of FLAG-Slitrk5 (green), HA-PTPδ (red), and TrkB (blue) scanning across corresponding purple arrow as indicated in (C). (E) Quantitative colocalization analysis of FLAG-Slitrk5-HA-PTPδ and FLAG-Slitrk5-TrkB in the presence or absence of BDNF treatment in (C). Results are means ± SEM from 3 independent experiments. 10–15 cells showing heterophilic adhesion were analyzed per condition per experiment. ***P< 0.0001 significantly different from control condition. (2-way ANOVA and Sidak’s multiple comparisons test). (F) BDNF-induced dissociation of pre-bound PTPδ-Fc from HA-Slitrk5-expressing HEK293-TrkB cells. HA-Slitrk5-expressing HEK293-TrkB cells were pre-incubated with saturating condition of PTPδ-Fc (400 nM) for 1hr. After washing, cells were incubated with indicated dose of BDNF for 30 min. Remaining PTPδ-Fc binding was analyzed by immunofluorescence microscopy. Results are means ± SEM from 3 independent experiments. 20–30 cells were analyzed per condition per experiment.
TrkB Receptor Signaling Is Impaired in the Striatum of Slitrk5−/− Mice
Next, we investigated whether Slitrk5 affects BDNF-mediated TrkB signaling. First, we examined the impact of Slitrk5 on steady-state BDNF-TrkB signaling in vivo. Western blot analyses of striatal lysates obtained from adult WT and Slitrk5−/− mice showed that TrkB receptor activation was significantly reduced in Slitrk5−/− mice relative to WT mice (Figure 3A, B). This effect on TrkB receptor activation was also reflected in a reduction in the activation of its downstream targets, Akt and ERK/MAPK, in the striatum of Slitrk5−/− mice (Figure 3A, B). Control experiments showed that the reduction in TrkB receptor activation and downstream signaling in Slitrk5−/− mice was not due to alterations in BDNF protein levels in the striatum (Figure S2A).
Figure 3. Altered TrkB receptor activation and signaling in the striatum of Slitrk5−/− mice.
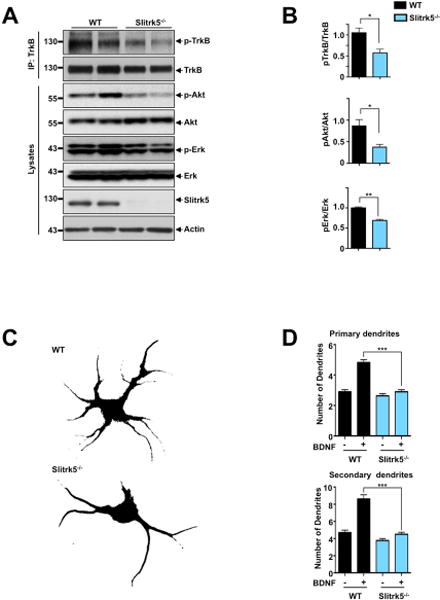
(A) Representative blots showing effect of loss of Slitrk5 on TrkB receptor activation and its downstream signaling. Striatal lysates from WT and Slitrk5−/− mice (3 months old, n=5 for each genotype) were used for immunoblot analysis for phospho-TrkB, phospho-Akt and phospho-Erk, with respective loading controls. (B) Densitometric quantification of the results shown on the right. Results are means ± SEM from 3 independent experiments. (C) and (D) Effects of BDNF on the growth of WT and Slitrk5−/− striatal neurons. Cultured striatal neurons from WT and Slitrk5−/− mice were treated with or without BDNF (40 ng/ml) at DIV2. After 5 days exposure of BDNF, the cultures were fixed and stained with anti-GAD65/67 antibody. Neuronal processes were counted with fluorescent microscopy. Representative images of BDNF-treated WT and Slitrk5−/− striatal neuron were shown in (C). Quantitation of the number of primary and secondary dendrites in WT and Slitrk5−/− was shown in (D). Results are presented as means ± SEM from 3 independent experiments determined from analysis of 40 neurons per condition per experiment (***P < 0.0001, Student’s t test).
Second, we carried out experiments with cultured striatal neurons to determine whether the decreased steady-state levels of TrkB signaling in the striatum of Slitrk5−/− mice might be linked to altered biological responsiveness to BDNF. A previous study has demonstrated that prolonged BDNF treatment (5 days) of cultured striatal neurons leads to a significant increase in the number and length of dendrites (Rauskolb et al., 2010). After prolonged (5 days) exposure to BDNF (40 ng/ml), WT striatal neurons displayed 4.82 ± 0.19 primary dendrites and 8.62 ± 0.48 secondary dendrites, a two-fold increase comparing with untreated neurons. In contrast, after 5 days of BDNF exposure, Slitrk5−/− neurons developed only 2.91 ± 0.14 primary dendrites and 4.5 ± 0.24 secondary dendrites on average, which was not significantly different from untreated neurons (Figure 3C, D). These in vitro findings indicate that the absence of Slitrk5 causes no impairment in BDNF-independent morphogenesis of striatal neurons but near completely abrogate their response to chronic BDNF. Taken together, these in vivo and in vitro results demonstrate that Slitrk5 is necessary for optimal long-term BDNF-dependent TrkB signaling in striatal neurons.
Slitrk5 Plays Pivotal Role in the Endocytic Recycling of TrkB Receptors
To obtain additional insights on the mechanisms underlying the role of Slitrk5 in BDNF-mediated TrkB signaling, we conducted a series of biochemical and fluorescence-based immunocytochemical assays. First, we investigated whether Slitrk5 acts, like p75NTR, by enhancing Trk receptor responsiveness to its ligand. To this end, we tested in WT vs Slitrk5−/− striatal neurons whether a low dose of BDNF (1.0 ng/ml; 15 min) can acutely activate TrkB receptors more potently in the presence of Slitrk5. We chose an early time point at which stage minimal TrkB receptor recycling or degradation occurs (Chen et al., 2005). Our studies demonstrated no change in short term TrkB activation or its downstream signaling pathways (Akt, ERK/MAPK) in response to low dose BDNF (Figure S4A). Next, we hypothesized that the decreased steady-state TrkB signaling observed in Slitrk5−/− striatum (Figure 3A) may be due to alterations in endocytic TrkB receptor trafficking at a particular stage after ligand binding. It has been established that upon binding to neurotrophins, Trk receptors are rapidly internalized in a clathrin-dependent manner that engages endocytic adaptors (Grimes et al., 1997; Zheng et al., 2008; Zhou et al., 2011). Trk receptor endocytosis is also required for certain ligand-mediated downstream signaling (Riccio et al., 1997; Zheng et al., 2008; Zhou et al., 2011). To determine whether Slitrk5 functions to regulate endocytosis, we utilized a fluorescent ratiometric internalization assay we developed to study Trk receptors (Chen et al., 2005), based on similar assays to study other signaling receptors such as EGF receptors and G protein coupled receptors (GPCR) (Gage et al., 2001; Tanowitz and von Zastrow, 2003; Vargas and Von Zastrow, 2004). Quantification of these results confirmed that there was no significant difference in TrkB endocytosis between WT and Slitrk5−/− striatal neurons after BDNF treatment (Figure S4B), in agreement with our observation of similar levels of BDNF-induced TrkB activation at this time point. An alternative internalization assay using RFP-tagged BDNF confirmed that there was no alteration in BDNF-dependent TrkB internalization in the absence of Slitrk5 (Figure S4C–D).
The fate of Trk receptors after ligand binding and receptor internalization has significant impact on the physiological responses to neurotrophins, as it determines the strength and duration of signaling cascades initiated by activated Trk receptors. One of the established postendocytic pathways of Trk receptors is their retrograde trafficking from axons to cell bodies, which is reportedly required for trophic responses in the PNS, but not in the CNS (Ginty and Segal, 2002; Riccio et al., 1997). Alternatively, Trk receptors can undergo (1) trafficking to lysosomes, which is reflected in decreased number of surface Trk receptors and decreased responsiveness to ligand (Sommerfeld et al., 2000), or (2) recycling to the plasma membrane, which can lead to functional resensitization of cell surface-specific signaling events (Chen et al., 2005; Huang et al., 2009). The mechanisms regulating the sorting of endocytosed Trk receptors into these diverse pathways, a complex and highly regulated process, remain unclear.
To investigate whether interactions with Slitrk5 control the postendocytic fates of TrkB, we examined the fate of TrkB receptors after BDNF treatment in WT and Slitrk5−/− striatal neurons, using standard biotinylation experiments in which cell surface TrkB receptors are labeled and their degradation measured after ligand treatment by immunoblot analysis (Arevalo et al., 2006; Chen et al., 2005). In the WT striatal neurons, around 30% of TrkB was degraded within 90 min after BDNF treatment (25 ng/ml). Interestingly, we observed increased TrkB degradation (~70% in 90 min) in Slitrk5−/− striatal neurons (Figure 4A, B). The rate of TrkB degradation was reduced to control levels after lentiviral transduction of WT Slitrk5 into Slitrk5−/− neurons, but not after transduction of a chimeric Slitrk5 lacking its endogenous LRR1 domain (LRR1(S1)-Slitrk5) that has been shown not to interact with TrkB receptors.
Figure 4. Slitrk5 plays pivotal role in ligand dependent TrkB receptor recycling.
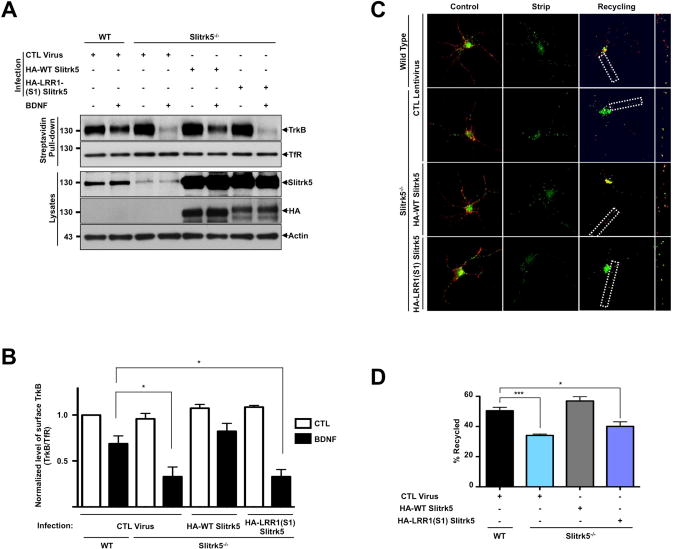
(A) Representative blot showing enhanced BDNF-induced TrkB degradation in Slitrk5−/− striatal neurons. WT and Slitrk5−/− striatal neurons were transduced with HA-tagged WT Slitrk5, or chimeric HA-tagged-LRR1-domain-swapped (HA-LRR1(S1) Slitrk5)-expressing, or empty vector lentivirus at DIV2. Neurons were surface biotinylated and incubated at 37°C for 90 min in the absence or presence of 25 ng/ml BDNF at DIV6. Surface-labeled receptors were detected by streptavidin pull-down followed by anti-TrkB immunoblotting. (B) Densitometric quantification of the results from 3 independent experiments shown in (A) (*P < 0.05, Student’s t test). (C) and (D) TrkB recycling was impaired in Slitrk5−/− striatal neurons. TrkB recycling was examined in WT and Slitrk5−/− neurons with live-cell fluorescence ratiometric recycling assay. WT and Slitrk5−/− striatal neurons were co-transfected with FLAG-tagged TrkB lentivirus, HA-tagged WT Slitrk5 or chimeric HA-tagged-LRR1-domain-swapped (HA-LRR1(S1) Slitrk5)-expressing, or empty vector lentivirus at DIV2. Internalization of TrkB receptor was induced by BDNF treatment for 30 min after labeling surface FLAG-tagged TrkB with Alexa-488 dye-conjugated anti-FLAG (M1) antibody. Remaining surface anti-FLAG (M1) antibodies were removed with EDTA-containing Ca2+/Mg2+ free PBS, and then recycling of FLAG-tagged TrkB were monitored in the presence of Cy3-conjugated anti-mouse secondary antibody in culture medium. (C) Representative images from FLAG-tagged TrkB recycling experiment in striatal neurons. Control refers to the 100% surface TrkB receptor control, Strip refers to the 0% recycled control. The right panels of each images showed enlarged images of framed regions. (D) Receptor recycling was quantitated as described in Experimental Procedures. Results are presented as means ± SEM from 3 independent experiments determined from analysis of 30 neurons per condition per experiment (*** P <0.001, *P < 0.05, Student’s t test).
How does the absence of Slitrk5 in cultured striatal neurons promote accelerated ligand-dependent degradation of TrkB? We hypothesized that Slitrk5 might promote TrkB recycling and its absence might allow targeting of endocytosed TrkB receptors to the degradative pathway. To test this hypothesis, we used a live cell assay that was previously used to study recycling to the plasma membrane for TrkB and TrkA receptors (Chen et al., 2005; Huang et al., 2013; Huang et al., 2009). This assay is based on the FLAG-epitope system that allows rapid removal of fluoresceinated FLAG antibodies that bind to extracellular FLAG epitopes on signaling receptors in a calcium-dependent manner (Guan et al., 1992; Tanowitz and von Zastrow, 2003; Vargas and Von Zastrow, 2004). In WT striatal neurons, the level of TrkB recycling was 51 ± 1.4% (Figure 4C, D). In contrast, in the Slitrk5−/− striatal neurons, the level of ligand-dependent TrkB recycling was significantly reduced (33 ± 0.5%). This reduction in TrkB recycling was specifically rescued after lentiviral transduction of WT Slitrk5 (56 ± 1.8%), but not after transduction of TrkB binding-deficient Slitrk5 (39 ± 1.8%) (Figure 4C, D). In accordance with the binding experiments (Figure 1E, S1E), TrkC recycling was not affected in the absence of Slitrk5 (Figure S4E, F). Together, these results suggest that Slitrk5 is required for efficient TrkB receptor recycling after ligand treatment in striatal neurons.
Slitrk5 Facilitates TrkB Receptor Recruitment into Rab11 Endosomes
The experiments described above indicate that the persistent down-regulation of TrkB receptors in Slitrk5−/− striatal neurons may be due to its reduced recycling rate; possibly explaining our findings of decreased steady-state BDNF-TrkB signaling in the striatum of Slitrk5−/− mice (Figure 3A), and altered striatal neuronal morphology (Shmelkov et al., 2010). To elucidate how Slitrk5 regulates TrkB receptor recycling, we hypothesized that Slitrk5 might act as a specialized sorting protein that enhances recycling of endocytosed TrkB receptors and prevents their incorporation into a degradative route. Recent studies have emphasized the importance of targeting TrkB receptors to a Rab11-positive recycling endosomes as an essential step for the physiological function of TrkB signaling (Huang et al., 2013; Lazo et al., 2013). Furthermore, it has been shown that TrkB can form a complex with Rab11 that may modulate synaptic plasticity (Huang et al., 2013), as well as BDNF-dependent dendritic branching (Lazo et al., 2013). To test our hypothesis, we initially examined whether Slitrk5 mediates recycling of TrkB into Rab11-positive endosomes in cultured striatal neurons. After transduction with lentivirus encoding HA-tagged Slitrk5 construct, DIV6 WT striatal neurons were labeled using a “live-feeding” method to specifically visualize cell surface HA-tagged Slitrk5 and endogenous TrkB receptors with their respective antibodies (Chen et al., 2005; Huang et al., 2013; Huang et al., 2009). Neurons were stained for Rab11 after fixation and permeabilization. In the absence of BDNF treatment, only minimal co-localization of TrkB, HA-tagged Slitrk5 and Rab11 was detected in WT striatal neurons. In contrast, after 30 min of BDNF treatment, there was a significant overlap of Slitrk5 and TrkB in Rab11 positive compartments (Figure 5A). This result suggests that Slitrk5 and TrkB receptors are sorted to Rab11-positive compartments after ligand treatment. We next investigated whether Slitrk5 is required for TrkB localization to the Rab11-positive compartments. In WT striatal neurons, there was 3-fold increase in TrkB co-localization with Rab11 at 30 min after BDNF treatment (Figure 5B, C). This colocalization was significantly reduced after similar treatment of BDNF in Slitrk5−/− striatal neurons (Figure 5B, C). Together, these results suggest that Slitrk5 plays an important role in TrkB recycling by facilitating TrkB sorting into Rab11-positive compartments after BDNF treatment.
Figure 5. Slitrk5 facilitates TrkB receptor recruitment into Rab11-positive compartments.
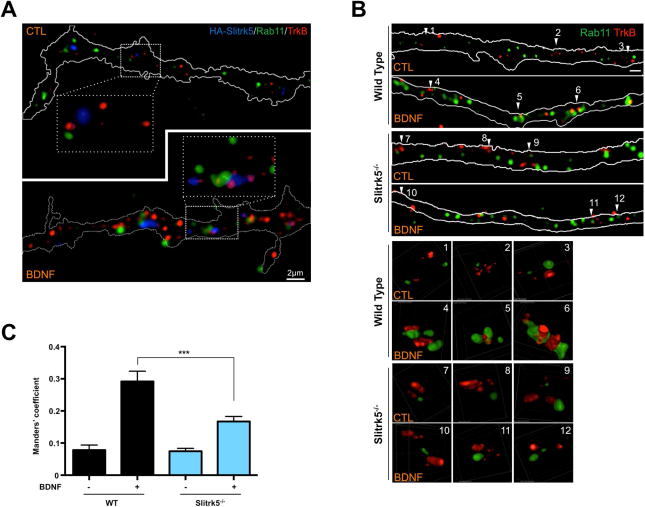
(A) Representative images showing co-localization of TrkB receptors and Slitrk5 in Rab11-positive compartments. WT striatal neurons were transduced with HA-tagged Slitrk5 lentivirus at DIV2. Live neurons were incubated with anti-TrkB and anti-HA antibody to specifically label cell surface proteins, and then stimulated with or without BDNF for 30 min at DIV6. Rab11 was stained after fixation and permeablization. Super resolution images were acquired using a Nikon N-SIM structured illumination microscope. (B) Representative images showing requirement of Slitrk5 for TrkB localization into Rab11-positive compartments after BDNF treatment. Co-localization of TrkB receptors and Rab11 was examined with WT and Slitrk5−/− striatal neurons in the presence or absence of BDNF. Lower panels showed enlarged images of numbered regions in upper panels. (C) Co-localization of TrkB and Rab11 was quantitated as described in Experimental Procedure. Results are presented as means ± SEM from 3 independent experiments determined from analysis of n ≥ 30 neurons per condition per experiment (*** P <0.001, Student’s t test).
Slitrk5 Facilitates Rab11-FIP3 Recruitment of TrkB Receptors to Rab11 Compartments
To obtain additional mechanistic insight on how Slitrk5 mediates the sorting of endocytosed TrkB to the Rab11-positive compartments, we screened for TrkB interacting proteins using a yeast two-hybrid assay. An 85 amino acid intracellular juxtamembrane region of the TrkB receptor was selected as bait, since the region was shown to be important for endocytic TrkB recycling (Chen et al., 2005; Huang et al., 2009). Among the positive clones was Rab11-FIP3 a protein that has been also established to interact with ADP-ribosylation factors (ARF5, ARF6), as well as with motor proteins (kinesin I, dynein light intermediate chain) (Horgan et al., 2010; Prekeris, 2003; Simon and Prekeris, 2008) and was previously shown to modulate recycling of various cargoes (Horgan and McCaffrey, 2009; Prekeris, 2003). We confirmed with co-immunoprecipitation studies that Rab11-FIP3 and TrkB receptors interacted (Figure S5A). To test whether there was a physical interaction between Slitrk5 and Rab11-FIP3, we transfected FLAG-tagged Slitrk5 and HA-tagged Rab11-FIP3 plasmids into HEK293T cells, and confirmed their binding with co-immunoprecipitation study (Figure 6A). Additional experiments with a panel of Rab11-FIP’s demonstrated that Slitrk5 bound selectively and directly to Rab11-FIP3 but not to other Rab11-FIP’s (Figure 6B, S6A), and facilitated the interaction between TrkB and Rab11-FIP3 (Figure S5B). Rab11 was not required for Rab11-FIP3 binding to Slitrk5, however Rab11-FIP3 was essential for the recruitment of Rab11 to Slitrk5 (Fig S6B–D). In parallel with this biochemical study, we performed fluorescent microscopy studies to assess co-localization of Slitrk5, TrkB and Rab11-FIP3 in cultured striatal neurons. Endogenous TrkB and transfected HA-tagged Slitrk5 were visualized using the “live feeding” method with respective antibodies to specifically label cell surface TrkB and HA-tagged Slitrk5, and then endogenous Rab11-FIP3 was imaged after fixation and permeabilization. The results showed that, indeed, TrkB and Slitrk5 were co-localized with Rab11-FIP3 after BDNF treatment (Figure 6C). We were also able to see co-localization of TrkB, Slitrk5, Rab11-FIP3 and Rab11 after BDNF treatment (Figure S6E). These findings support a scenario in which Slitrk5 recruits Rab11-FIP3 to TrkB receptors during post-endocytic trafficking to properly target TrkB receptors to the recycling pathway.
Figure 6. Slitrk5 interacts with Rab11-FIP3 to facilitate TrkB receptor trafficking to Rab11-positive recycling endosomes.
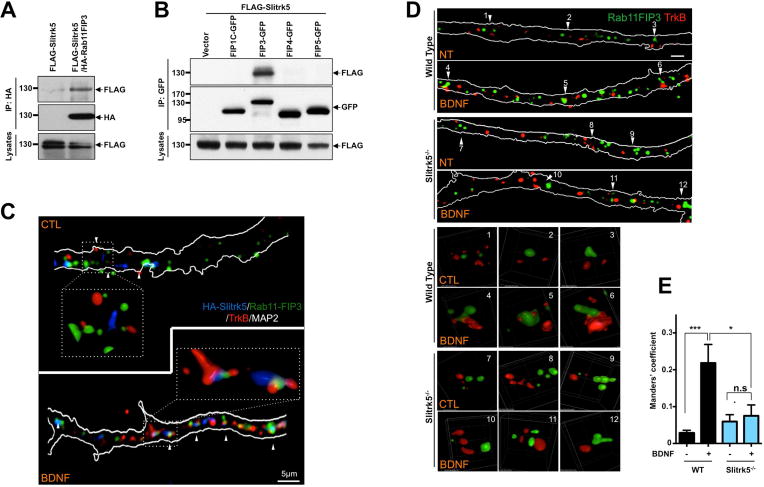
(A) and (B). Representative blots showing specific interaction between Slitrk5 and Rab11-FIP3. (A) HEK293T cells were transfected with cDNAs encoding FLAG-Slitrk5, and HA-Rab11-FIP3. Cell lysates were immunoprecipitated with anti-HA antibodies and immunoblotted with anti-FLAG antibodies. (B) HEK293T cells were transfected with cDNAs encoding FLAG-Slitrk5, and either empty vector, Rab11-FIP3-GFP, FIP1C-GFP, FIP4-GFP or FIP5-GFP. Cell lysates were immunoprecipitated with anti-GFP antibodies and blotted with anti-FLAG antibodies. (C) Representative image showing the co-localization of TrkB and Slitrk5 with Rab11-FIP3 with BDNF-dependent manner. WT striatal neurons expressing HA-Slitrk5 were stimulated with or without BDNF after incubating with anti-TrkB and anti-HA antibody for surface protein labeling. Neurons were stained with anti-Rab11-FIP3 antibody after fixation and permeablization. Super resolution images were acquired using a Nikon N-SIM structured illumination microscope. (D) Representative images showing requirement of Slitrk5 for TrkB receptor localization in Rab11-FIP3 compartments. Co-localization of TrkB and Rab11-FIP3 was examined with WT and Slitrk5−/− striatal neurons in the presence or absence of BDNF. Lower panels showed enlarged images of numbered regions in upper panels. (E) Co-localization of TrkB and Rab11-FIP3 was quantitated as described in Experimental Procedure. Results are presented as means ± SEM from 3 independent experiments determined from analysis of n ≥ 20 neurons per condition per experiment (*P<0.05, ***P<0.001, Student’s t test).
We next investigated whether Slitrk5 is required for TrkB localization to the Rab11-FIP3-positive compartments. We used a “live-feeding” method to specifically visualize cell surface TrkB receptors with anti-TrkB antibodies (Chen et al., 2005; Huang et al., 2013; Huang et al., 2009) and stained for Rab11-FIP3 after fixation and permeabilization. Results showed impaired TrkB localization in Rab11-FIP3-positive compartments in Slitrk5−/− striatal neurons after BDNF treatment (Figure 6D, E). However, there was a significant overlap of TrkB in compartments that were positive for Rab11-FIP3 in WT striatal neurons after BDNF treatment (Figure 6D, E).
Slitrk5-mediated Rab11-FIP3 recruitment is required for TrkB Recycling
To further examine whether Rab11-FIP3 is directly involved in TrkB recycling, we performed live-cell fluorescent ratiometric recycling assays with control siRNA or Rab11-FIP3 siRNA transfected striatal neurons. Knockdown efficiency of siRNA targeting Rab11-FIP3 was validated with Western blot and quantitative PCR analysis (Figure S7A, B). Quantification of these results by ratiometric analysis confirmed that TrkB recycling was impaired in striatal neurons upon Rab11-FIP3 knockdown (36.95 ± 1.75%) compared to control siRNA transfected (48.12 ± 2.48%) (Figure 7A, S7C). We also confirmed that TrkB recycling was impaired in striatal neurons transfected with Rab11-FIP3ΔRBD or ΔERM mutant, presumably due to the impaired Rab11 binding or Slitrk5 binding, respectively (Figure S7D–E).
Figure 7. Slitrk5-mediated Rab11-FIP3 recruitment is required for TrkB recycling.
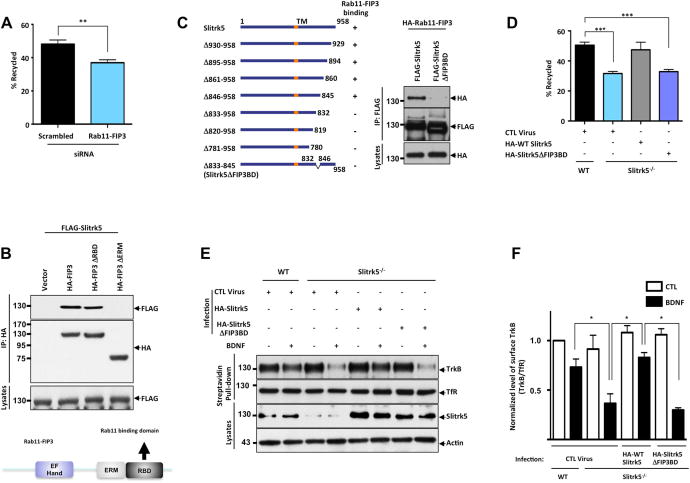
(A) TrkB recycling was quantified with live-cell fluorescence ratiometric recycling assay in the control siRNA (Scrb) or Rab11-FIP3 siRNA transfected striatal neuron. Knockdown efficiency of siRNA targeting Rab11-FIP3 and representative images of live-cell fluorescence ratiometric recycling assay were shown in supplementary information (Figure S4A–C). The error bars represent the SEM of three independent experiments (n ≥ 30 cells for each condition per experiment; ** P <0.001, Student’s t test). (B) ERM domain of Rab11-FIP3 mediates Slitrk5 binding. HEK293T cells were co-transfected with cDNAs encoding FLAG-Slitrk5, and either empty vector, HA-Rab11-FIP3, HA-Rab11-FIP3ΔRBD, or HA-Rab11-FIP3ΔERM. Cell lysates were immunoprecipitated with anti-HA antibodies and immunoblotted with anti-FLAG antibodies. Schematic of Rab-FIP3 denoting established domains (EF Hand, ERM (ezrin/radixin/moesin domain), RBD (Rab11 binding domain). (C) Mapping domain in Slitrk5 that mediates association with Rab11-FIP3. Schematic representation shows a series of Slitrk5 deletion mutants that were tested for the capacity to interact with Rab11-FIP3 (Left). HEK293T cells were co-transfected with cDNAs encoding HA-Rab11-FIP3 and either FLAG-Slitrk5 or FLAG-Slitrk5ΔFIP3BD. Cell lysates were immunoprecipitated with anti-FLAG antibodies and immunoblotted with anti-HA antibodies (Right). (D) Rab11-FIP3 binding is required for Slitrk5 to rescue reduced recycling of TrkB in Slitrk5−/− striatal neurons. WT and Slitrk5−/− striatal neurons were co-transfected at DIV2 with FLAG-tagged TrkB lentivirus, and either empty vector, HA-Slitrk5 or HA-tagged Rab11-FIP3 binding-deficient Slitrk5 (HA-Slitrk5ΔFIP3BD). BDNF-induced TrkB recycling was measured with live-cell fluorescence ratiometric recycling assay at DIV6, as described in Experimental Procedure. The error bars represent the SEM of three independent experiments (n ≥ 30 cells for each condition per experiment; *** P <0.0001, Student’s t test). (E) A cell surface biotinylation assay shows that RAB11-FIP3 binding-deficient Slitrk5 is not able to rescue enhanced degradation of TrkB. WT and Slitrk5−/− striatal neurons were transduced with empty vector, HA-tagged WT Slitrk5, or HA-Slitrk5ΔFIP3BD-expressing lentivirus at DIV2. At DIV6 neurons were surface biotinylated and incubated in the presence or absence of BDNF (25ng/ml; 90 min). Cell lysates were subjected to avidin pull-down, and TrkB levels assessed by immnunoblotting with anti-TrkB antibodies. (F) Densitometric quantification of the results was shown. The error bars represent the SEM of three independent experiments (*P < 0.05, Student’s t test).
Rab11-FIP3 contains several conserved domains that may be involved in protein-protein interactions. The central ERM domain is known to interact with actin cytoskeleton while Rab11 binding domain is localized in the C-terminus. To determine the Rab11-FIP3 domain responsible for binding to Slitrk5, we constructed a series of deletion mutants and tested their ability to interact with Slitrk5. Co-immunoprecipitation experiments showed that the ERM domain of Rab11-FIP3 is essential for the Slitrk5 binding (Figure 7B).
We next mapped the Rab11-FIP3 binding region on Slitrk5 using a series of C-terminal deletion mutants of Slitrk5. We found that deletion of 13 amino acids in the intracellular region of Slitrk5 abolished Rab11-FIP3 binding (Figure 7C). Next, we took advantage of this Rab11-FIP3 binding-deficient mutant (Slitrk5ΔFIP3BD) to determine whether Rab11-FIP3 is necessary for ligand-dependent TrkB recycling. Using fluorescent ratiometric recycling assays, we demonstrated that Slitrk5ΔFIP3BD was not able to rescue the decreased recycling of TrkB in Slitrk5−/− striatal neurons (Figure 7D) in contrast to WT Slitrk5. In accordance with this result, a TrkB degradation experiment showed that Slitrk5ΔFIP3BD was not able to reverse the enhanced rate of TrkB degradation in Slitrk5−/− striatal neurons (Figure 7E–F). These complementary experiments highlight the importance of Rab11-FIP3 binding to Slitrk5 in mediating efficient TrkB recycling in striatal neurons and suggest that Slitrk5 plays a pivotal role in the Rab11-mediated TrkB recycling by facilitating the recruitment of a Rab11 interacting protein, Rab11-FIP3, into the TrkB receptor complex for efficient targeting to the recycling endosomes.
Discussion
LRR domain-containing proteins play pivotal roles in the regulation of various neuronal functions, such as neurite outgrowth, synapse formation, and dendritic morphogenesis. Two basic mechanisms are employed by LRR proteins to execute such functions. First, LRR proteins function in trans as cell-cell adhesion molecules that mediate axon-dendrite adhesion (Gur et al., 2004; Laederich et al., 2004; Lin et al., 2003; Shattuck et al., 2007; Zhao et al., 2008). Second, LRR proteins act in cis to regulate cell surface receptor function (Gur et al., 2004; Laederich et al., 2004; Shattuck et al., 2007; Zhao et al., 2008). Here, we have shown that Slitrk5, a postsynaptic plasma membrane protein containing extracellular LRR domains, interacts under basal conditions with the presynaptic adhesion molecule PTPδ in trans, but in the presence of BDNF, shifts to a cis-interaction with TrkB receptor that mediates its postendocytic recycling, leading to functional resensitization of neurotrophic signaling. Of note, the Slitrk5 interaction with TrkB receptors represents the first reported interaction amongst LRR proteins and RTK’s that is mediated by the respective LRR domains. This likely contributes to the high specificity of this interaction and to the exclusion of other Slitrk and Trk family members (Figure 1). In contrast to other LRR containing proteins, such as Lrigs, which negatively regulate EGFR and Met, Slitrk5 positively regulates TrkB receptor activity. In addition, a recent study shows that another LRR containing protein, Linx, is required for NGF–TrkA and GDNF-GFRα1/Ret mediated sensory and motor axonal projections. Linx forms physical complex with Trk receptors (TrkA and TrkC, but not with TrkB) and Ret receptors, however, it was not determined how Linx modulates RTK complexes in that study (Mandai et al., 2009). Linx has a relatively short cytoplasmic domain (134 aa) compared to Slitrk5 (271 aa) and does not have features required for downstream signaling in the cytoplasmic domain, e.g. a tyrosine phosphorylation site or a SH2 domain binding site, both observed in the intracellular domain of Slitrk5. Interestingly, we found that Linx is required for the recycling of TrkA (Figure S7F–H). Together with suggested role of Lrigs on EGFR ubiquitination and downregulation (Gur et al., 2004; Laederich et al., 2004), these results suggest that LRR protein-mediated regulation of trafficking fate could be a universally employed mechanism for modulating RTK signaling.
In addition to the originally established function of Slitrk family members in regulating neuronal processes outgrowth (Abelson et al., 2005; Aruga and Mikoshiba, 2003), recent studies have focused on Slitrks’ involvement in synapse formation. All Slitrk family members have been shown to induce presynaptic neuronal differentiation in a cellular co-culture system (Takahashi et al., 2012). Slitrks have been shown to interact with presynaptic receptor-type protein tyrosine phosphatase, PTPδ. Thus, Slitrks were only considered to mediate a trans interaction with a presynaptic receptor-type protein tyrosine phosphatase. Our current studies elucidate a new aspect of Slitrk5 function in which Slitrk5 has a cis interaction with activated TrkB receptors on the surface of postsynaptic sites via extracellular interactions. Intriguingly, the cis interactions of Slitrk5 with TrkB receptors compete with trans interactions with presynaptic partners PTPδ, and the competition was modulated by BDNF stimulation (Figure 2). It will be important to study further the significance of the interplay between these three synaptic molecules during steady-state and activity-dependent synaptic remodeling. It is interesting to note that significant co-localization of Slitrk5 and TrkB receptors occurs only after BDNF treatment, suggesting that the interaction is enhanced after TrkB dimerization, involving the 2 LRR domains in the TrkB dimer, leading to optimal interaction with the LRR1 domain of Slitrk5. The interaction between the extracellular domains of Slitrk5 and TrkB allows their intracellular interaction with Rab11-FIP3, which mediates the recycling of TrkB receptors to the cell surface via Rab11 recycling endosomes, while not trafficking the other TrkB isoform, truncated TrkB receptor, to the same compartments. Truncated TrkB receptors have been shown to act in a dominant negative manner, sequestering BDNF from full-length TrkB (Eide et al., 1996). In striatal neurons, with limiting BDNF supplies, Slitrk5 could thus act to efficiently recycle activated TrkB receptors, facilitating BDNF-dependent signaling pathways.
In summary, the present studies identify an unanticipated role of a cell surface transmembrane protein in regulating TrkB receptor endocytic trafficking to recycling endosomes leading to facilitation of neurotrophic signaling in neurons. Engagement of TrkB receptors with Slitrk5, as a co-receptor, represents a new mechanism of how neurons within a particular brain region with limiting BDNF levels can expand the strength and duration of neurotrophic factor signaling. The striatum is the largest component of the basal ganglia and loss of striatal function has been implicated in neurodegenerative disorders such as Huntington and Parkinson disease. While neurotrophins and neurotrophin receptors have been thought of as potential therapeutic targets for these disorders, there has been limited success in these lines of investigation due to pharmacokinetic and delivery issues. Slitrk5 represents a new potential target for therapeutics for these neurodegenerative disorders in which selective facilitation of BDNF-dependent signaling could be achieved in a region-specific manner by enhancing interaction of TrkB receptors with Slitrk5 in the absence of exogenous neurotrophic factors.
Experimental Procedures
Reagents and Antibodies
The reagents, antibodies, plasmid construct, siRNA, and primers used in this study are available in Supplemental Experimental Procedures.
Production of soluble PTPδ-Fc protein and binding assays
Based on previously described methods (Takahashi et al., 2012), soluble PTPδ ectodomain fused to Fc (PTPδ-Fc) were generated using HEK-293 cells transfected with the expression vectors and purified from culture media. For testing binding of soluble PTPδ-Fc, HEK293T or HEK293-TrkB cells on coverslips were transfected with the expression vectors for WT and deletion mutants of HA-tagged Slitrk5 and grown for 24 h. The transfected cells were washed with extracellular solution (ECS; containing, in mM: 168 NaCl, 2.4 KCl, 20 HEPES pH 7.4, 10 D-glucose, 2 CaCl2, 1.3 MgCl2) that contained 100 μg/ml BSA (ECS/BSA) and then incubated with ECS/BSA that contained 100 nM purified PTPδ Fc-fusion protein for 1 h at room temperature in the presence or absence of BDNF. The cells were washed in ECS, fixed with 4% paraformaldehyde, incubated with blocking solution, and then with mouse anti-HA antibody. Cells were incubated with subtype-specific fluorescenated secondary antibodies and analyzed by fluorescence microscopy with Alexa488 dye–conjugated anti-human IgG (H+L) antibodies (donkey IgG; 1:400; Jackson ImmunoResearch) for labeling of bound Fc proteins. For PTPδ-Fc dissociation experiment, Slitrk5-expressing HEK293-TrkB cells were incubated with purified PTPδ Fc-fusion protein for 1 h at room temperature. After washing with ECS, PTPδ Fc-bound cells were incubated with indicated dose of BDNF for 30 min at room temperature. For quantification, we measured the average intensity of bound Fc protein per COS cell area, subtracted for off-cell background.
Super-resolution microscopy
For super resolution analysis, imaging was performed on Nikon’s Structured Illumination Microscope (N-SIM) Nikon Eclipse Ti that can bring the resolution to 100-85nm, equipped with ANDOR camera. Images of Alexa fluors 488, 568 and 647 were acquired in 3D-SIM mode using 100x Apo TIRF lens with 1.49 N.A. For every Z-stack, 15 images were generated resulting from three directions and five phases, which were subsequently reconstructed using NIS-Elements software with SIM plugin to generate super resolution data. In order to outline dendritic morphology, MAP2 images were acquired using TIRF 405 laser and combined with SIM images. The background was subtracted and maximum intensity projection collapsed images are portrayed along with cropped segments represented in volume view. Each experiment was repeated at least three times. See the Supplemental Experimental Procedures for details of neuronal culture and imaging sample preparation.
Analysis of Trk Receptor Recycling by Using Fluorescence Ratio Microscopy
To quantify the extent of TrkB recycling in individual neurons, an adapted version of previous receptor recycling methods (Chen et al., 2005; Tanowitz and von Zastrow, 2003; Vargas and Von Zastrow, 2004) was employed. Details are available in Supplemental Experimental Procedures.
Statistical analyses
Statistical analyses were performed using GraphPad Prism V5.0 software. Statistical significance was considered at *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001 between the means of a minimum of three groups was determined using Student’s t test or one-way ANOVA or 2-way ANOVA test as indicated in the figure legends. Results are expressed as the mean value ± SD. All experiments were done with at least three independent biological replicates.
Supplementary Material
HIGHLIGHTS.
Slitrk5, a cell surface LRR protein, is a co-receptor facilitating TrkB signaling
TrkB and Slitrk5 form a physical complex through their leucine-rich repeat domains
TrkB and PTPδ compete for the first LRR domain of Slitrk5
Slitrk5 regulates TrkB receptor recycling by facilitating recruitment of Rab11-FIP3
Acknowledgments
We acknowledge support from US National Institutes of Health grants MH079513 (F.S.L.), 5UL1TR000457 (F.S.L.), and NS052819 (F.S.L.), HL66592 (S.R.), HL097797 (S.R.), AI080309 (S.R.), NS030687 (B.L.H.), and GM804302 (J.B.D.), National Natural Science Foundation of China (No. 31130026; Z.Y.C.), Burroughs Wellcome Foundation (F.S.L.), International Mental Health Research Organization (F.S.L.), the Sackler Institute (F.S.L.), DeWitt-Wallace Fund of the New York Community Trust (F.S.L.), Pritzker Consortium (F.S.L.), Brain and Behavior Research Foundation (F.S.L., D.Q.J., S.V.S.), and National Science Foundation grant NSF960367 (J.B.D.). We acknowledge the resources and staff, as well as Dr. Diana Bratu, of the Bio-Imaging Facility at Hunter College.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Financial Interests
The authors declare no competing financial interests.
Contributions
M.S., C.C.P., Z.Y.C, E.R.B., and F.S.L developed the research. M.S. designed the experiments and conducted biochemical and functional characterization for the research. I.D., D.J. and M.E. conducted biochemical analysis. J.G., J.K. and J.B.D. optimized and performed super-resolution imaging studies. M.S. analyzed, quantified and interpreted the acquired images. S.H.H. and Z.Y.C. conducted Rab11-FIP3 knock-down studies and generated constructs. E.C. generated various TrkB constructs. R.S. and E.R.B supported Rab11 imaging studies. S.V.S. and S.R. generated and analyzed theSlitrk5 knockout mice. R.P. generated Rab11-FIP related reagents. M.V.C. and B.L.H. supported yeast 2 hybrid experiments. M.S. and F.S.L. wrote the manuscript with assistance from B.L.H., Z.Y.C., and E.R.B.
References
- Abelson JF, Kwan KY, O’Roak BJ, Baek DY, Stillman AA, Morgan TM, Mathews CA, Pauls DL, Rasin MR, Gunel M, et al. Sequence variants in SLITRK1 are associated with Tourette’s syndrome. Science. 2005;310:317–320. doi: 10.1126/science.1116502. [DOI] [PubMed] [Google Scholar]
- Arevalo JC, Waite J, Rajagopal R, Beyna M, Chen ZY, Lee FS, Chao MV. Cell survival through Trk neurotrophin receptors is differentially regulated by ubiquitination. Neuron. 2006;50:549–559. doi: 10.1016/j.neuron.2006.03.044. [DOI] [PubMed] [Google Scholar]
- Aruga J, Mikoshiba K. Identification and characterization of Slitrk, a novel neuronal transmembrane protein family controlling neurite outgrowth. Molecular and cellular neurosciences. 2003;24:117–129. doi: 10.1016/s1044-7431(03)00129-5. [DOI] [PubMed] [Google Scholar]
- Baquet ZC, Gorski JA, Jones KR. Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brain-derived neurotrophic factor. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2004;24:4250–4258. doi: 10.1523/JNEUROSCI.3920-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baydyuk M, Russell T, Liao GY, Zang K, An JJ, Reichardt LF, Xu B. TrkB receptor controls striatal formation by regulating the number of newborn striatal neurons. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:1669–1674. doi: 10.1073/pnas.1004744108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti M, Levi A, Chao MV. Differential expression of nerve growth factor receptors leads to altered binding affinity and neurotrophin responsiveness. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:7859–7863. doi: 10.1073/pnas.90.16.7859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao MV. Neurotrophins and their receptors: A convergence point for many signalling pathways. Nat Rev Neurosci. 2003;4:299–309. doi: 10.1038/nrn1078. [DOI] [PubMed] [Google Scholar]
- Chen ZY, Ieraci A, Tanowitz M, Lee FS. A novel endocytic recycling signal distinguishes biological responses of Trk neurotrophin receptors. Molecular biology of the cell. 2005;16:5761–5772. doi: 10.1091/mbc.E05-07-0651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eide FF, Vining ER, Eide BL, Zang K, Wang XY, Reichardt LF. Naturally occurring truncated trkB receptors have dominant inhibitory effects on brain-derived neurotrophic factor signaling. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1996;16:3123–3129. doi: 10.1523/JNEUROSCI.16-10-03123.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogel AI, Akins MR, Krupp AJ, Stagi M, Stein V, Biederer T. SynCAMs organize synapses through heterophilic adhesion. Journal of Neuroscience. 2007;27:12516–12530. doi: 10.1523/JNEUROSCI.2739-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage RM, Kim KA, Cao TT, von Zastrow M. A transplantable sorting signal that is sufficient to mediate rapid recycling of G protein-coupled receptors. The Journal of biological chemistry. 2001;276:44712–44720. doi: 10.1074/jbc.M107417200. [DOI] [PubMed] [Google Scholar]
- Gay NJ, Packman LC, Weldon MA, Barna JC. A leucine-rich repeat peptide derived from the Drosophila Toll receptor forms extended filaments with a beta-sheet structure. FEBS letters. 1991;291:87–91. doi: 10.1016/0014-5793(91)81110-t. [DOI] [PubMed] [Google Scholar]
- Ginty DD, Segal RA. Retrograde neurotrophin signaling: Trk-ing along the axon. Current opinion in neurobiology. 2002;12:268–274. doi: 10.1016/s0959-4388(02)00326-4. [DOI] [PubMed] [Google Scholar]
- Grimes ML, Beattie E, Mobley WC. A signaling organelle containing the nerve growth factor-activated receptor tyrosine kinase, TrkA. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:9909–9914. doi: 10.1073/pnas.94.18.9909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan XM, Kobilka TS, Kobilka BK. Enhancement of membrane insertion and function in a type IIIb membrane protein following introduction of a cleavable signal peptide. The Journal of biological chemistry. 1992;267:21995–21998. [PubMed] [Google Scholar]
- Gur G, Rubin C, Katz M, Amit I, Citri A, Nilsson J, Amariglio N, Henriksson R, Rechavi G, Hedman H, et al. LRIG1 restricts growth factor signaling by enhancing receptor ubiquitylation and degradation. The EMBO journal. 2004;23:3270–3281. doi: 10.1038/sj.emboj.7600342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson MGL. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J Microsc-Oxford. 2000;198:82–87. doi: 10.1046/j.1365-2818.2000.00710.x. [DOI] [PubMed] [Google Scholar]
- Hempstead BL, Martin-Zanca D, Kaplan DR, Parada LF, Chao MV. High-affinity NGF binding requires coexpression of the trk proto-oncogene and the low-affinity NGF receptor. Nature. 1991;350:678–683. doi: 10.1038/350678a0. [DOI] [PubMed] [Google Scholar]
- Horgan CP, Hanscom SR, Jolly RS, Futter CE, McCaffrey MW. Rab11-FIP3 links the Rab11 GTPase and cytoplasmic dynein to mediate transport to the endosomal-recycling compartment. Journal of cell science. 2010;123:181–191. doi: 10.1242/jcs.052670. [DOI] [PubMed] [Google Scholar]
- Horgan CP, McCaffrey MW. The dynamic Rab11-FIPs. Biochemical Society transactions. 2009;37:1032–1036. doi: 10.1042/BST0371032. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annual review of biochemistry. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Huang SH, Wang J, Sui WH, Chen B, Zhang XY, Yan J, Geng Z, Chen ZY. BDNF-Dependent Recycling Facilitates TrkB Translocation to Postsynaptic Density during LTP via a Rab11-Dependent Pathway. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2013;33:9214–9230. doi: 10.1523/JNEUROSCI.3256-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SH, Zhao L, Sun ZP, Li XZ, Geng Z, Zhang KD, Chao MV, Chen ZY. Essential role of Hrs in endocytic recycling of full-length TrkB receptor but not its isoform TrkB.T1. The Journal of biological chemistry. 2009;284:15126–15136. doi: 10.1074/jbc.M809763200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J. The leucine-rich repeat superfamily of synaptic adhesion molecules: LRRTMs and Slitrks. Molecules and cells. 2012;34:335–340. doi: 10.1007/s10059-012-0113-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuruvilla R, Zweifel LS, Glebova NO, Lonze BE, Valdez G, Ye H, Ginty DD. A neurotrophin signaling cascade coordinates sympathetic neuron development through differential control of TrkA trafficking and retrograde signaling. Cell. 2004;118:243–255. doi: 10.1016/j.cell.2004.06.021. [DOI] [PubMed] [Google Scholar]
- Laederich MB, Funes-Duran M, Yen L, Ingalla E, Wu X, Carraway KL, 3rd, Sweeney C. The leucine-rich repeat protein LRIG1 is a negative regulator of ErbB family receptor tyrosine kinases. The Journal of biological chemistry. 2004;279:47050–47056. doi: 10.1074/jbc.M409703200. [DOI] [PubMed] [Google Scholar]
- Lazo OM, Gonzalez A, Ascano M, Kuruvilla R, Couve A, Bronfman FC. BDNF regulates Rab11-mediated recycling endosome dynamics to induce dendritic branching. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2013;33:6112–6122. doi: 10.1523/JNEUROSCI.4630-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee FS, Kim AH, Khursigara G, Chao MV. The uniqueness of being a neurotrophin receptor. Current opinion in neurobiology. 2001;11:281–286. doi: 10.1016/s0959-4388(00)00209-9. [DOI] [PubMed] [Google Scholar]
- Lee KF, Davies AM, Jaenisch R. p75-deficient embryonic dorsal root sensory and neonatal sympathetic neurons display a decreased sensitivity to NGF. Development. 1994;120:1027–1033. doi: 10.1242/dev.120.4.1027. [DOI] [PubMed] [Google Scholar]
- Li Y, Yui D, Luikart BW, McKay RM, Li Y, Rubenstein JL, Parada LF. Conditional ablation of brain-derived neurotrophic factor-TrkB signaling impairs striatal neuron development. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:15491–15496. doi: 10.1073/pnas.1212899109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JC, Ho WH, Gurney A, Rosenthal A. The netrin-G1 ligand NGL-1 promotes the outgrowth of thalamocortical axons. Nat Neurosci. 2003;6:1270–1276. doi: 10.1038/nn1148. [DOI] [PubMed] [Google Scholar]
- Linhoff MW, Lauren J, Cassidy RM, Dobie FA, Takahashi H, Nygaard HB, Airaksinen MS, Strittmatter SM, Craig AM. An unbiased expression screen for synaptogenic proteins identifies the LRRTM protein family as synaptic organizers. Neuron. 2009;61:734–749. doi: 10.1016/j.neuron.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandai K, Guo T, St Hillaire C, Meabon JS, Kanning KC, Bothwell M, Ginty DD. LIG family receptor tyrosine kinase-associated proteins modulate growth factor signals during neural development. Neuron. 2009;63:614–627. doi: 10.1016/j.neuron.2009.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prekeris R. Rabs, Rips, FIPs, and endocytic membrane traffic. The Scientific World Journal. 2003;3:870–880. doi: 10.1100/tsw.2003.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proenca CC, Gao KP, Shmelkov SV, Rafii S, Lee FS. Slitrks as emerging candidate genes involved in neuropsychiatric disorders. Trends in neurosciences. 2011;34:143–153. doi: 10.1016/j.tins.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauskolb S, Zagrebelsky M, Dreznjak A, Deogracias R, Matsumoto T, Wiese S, Erne B, Sendtner M, Schaeren-Wiemers N, Korte M, et al. Global deprivation of brain-derived neurotrophic factor in the CNS reveals an area-specific requirement for dendritic growth. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2010;30:1739–1749. doi: 10.1523/JNEUROSCI.5100-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccio A, Pierchala BA, Ciarallo CL, Ginty DD. An NGF-TrkA-mediated retrograde signal to transcription factor CREB in sympathetic neurons. Science. 1997;277:1097–1100. doi: 10.1126/science.277.5329.1097. [DOI] [PubMed] [Google Scholar]
- Shattuck DL, Miller JK, Laederich M, Funes M, Petersen H, Carraway KL, 3rd, Sweeney C. LRIG1 is a novel negative regulator of the Met receptor and opposes Met and Her2 synergy. Molecular and cellular biology. 2007;27:1934–1946. doi: 10.1128/MCB.00757-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmelkov SV, Hormigo A, Jing D, Proenca CC, Bath KG, Milde T, Shmelkov E, Kushner JS, Baljevic M, Dincheva I, et al. Slitrk5 deficiency impairs corticostriatal circuitry and leads to obsessive-compulsive-like behaviors in mice. Nature medicine. 2010;16:598–602. doi: 10.1038/nm.2125. 591p following 602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon GC, Prekeris R. Mechanisms regulating targeting of recycling endosomes to the cleavage furrow during cytokinesis. Biochemical Society transactions. 2008;36:391–394. doi: 10.1042/BST0360391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommerfeld MT, Schweigreiter R, Barde YA, Hoppe E. Down-regulation of the neurotrophin receptor TrkB following ligand binding. Evidence for an involvement of the proteasome and differential regulation of TrkA and TrkB. The Journal of biological chemistry. 2000;275:8982–8990. doi: 10.1074/jbc.275.12.8982. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Katayama K, Sohya K, Miyamoto H, Prasad T, Matsumoto Y, Ota M, Yasuda H, Tsumoto T, Aruga J, et al. Selective control of inhibitory synapse development by Slitrk3-PTPdelta trans-synaptic interaction. Nat Neurosci. 2012;15:389–398. S381–382. doi: 10.1038/nn.3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanowitz M, von Zastrow M. A novel endocytic recycling signal that distinguishes the membrane trafficking of naturally occurring opioid receptors. The Journal of biological chemistry. 2003;278:45978–45986. doi: 10.1074/jbc.M304504200. [DOI] [PubMed] [Google Scholar]
- Vargas GA, Von Zastrow M. Identification of a novel endocytic recycling signal in the D1 dopamine receptor. The Journal of biological chemistry. 2004;279:37461–37469. doi: 10.1074/jbc.M401034200. [DOI] [PubMed] [Google Scholar]
- Yim YS, Kwon Y, Nam J, Yoon HI, Lee K, Kim DG, Kim E, Kim CH, Ko J. Slitrks control excitatory and inhibitory synapse formation with LAR receptor protein tyrosine phosphatases. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:4057–4062. doi: 10.1073/pnas.1209881110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Tanegashima K, Ro H, Dawid IB. Lrig3 regulates neural crest formation in Xenopus by modulating Fgf and Wnt signaling pathways. Development. 2008;135:1283–1293. doi: 10.1242/dev.015073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J, Shen WH, Lu TJ, Zhou Y, Chen Q, Wang Z, Xiang T, Zhu YC, Zhang C, Duan SM, et al. Clathrin-dependent endocytosis is required for TrkB-dependent Akt-mediated neuronal protection and dendritic growth. Journal of Biological Chemistry. 2008;283:13280–13288. doi: 10.1074/jbc.M709930200. [DOI] [PubMed] [Google Scholar]
- Zhou P, Alfaro J, Chang EH, Zhao X, Porcionatto M, Segal RA. Numb links extracellular cues to intracellular polarity machinery to promote chemotaxis. Developmental cell. 2011;20:610–622. doi: 10.1016/j.devcel.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.