

Abstract
The resistance of Plasmodium falciparum to sulfadoxine-pyrimethamine (SP) is an emerging public health threat. Resistance to these drugs is associated with point mutations in the genes encoding dihydropteroate synthase (DHPS) and dihydrofolate reductase (DHFR). We describe here an assay using real-time PCR and sequence-specific probes that detects these mutations. Using DNA from plasmids, cultured strains, and clinical samples, real-time PCR could distinguish four DHPS polymorphisms (codons 437, 540, 581, and 613) and three DHFR polymorphisms (codons 51, 59, and 108). This assay is rapid and sensitive, with a detection limit of 10 copies in most cases. This assay is amenable to large-scale studies of drug resistance.
Malaria is a major public health threat, causing millions of deaths per year. One of the greatest challenges in the control of malaria is drug resistance, which has contributed to its reemergence and spread (23). At present, chloroquine resistance in Plasmodium falciparum is nearly universal; because of chloroquine resistance, five African countries have switched to sulfadoxine-pyrimethamine (SP) as their first-line antimalarial agent (23). However, resistance to SP is spreading.
Sulfadoxine and pyrimethamine inhibit dihydropteroate synthase (DHPS) and dihydrofolate reductase (DHFR), respectively, which are two enzymes involved in folate biosynthesis. SP resistance is conferred by single nucleotide changes in either enzyme, and increases as mutations accumulate (reference 18 and references therein). For DHFR, the mutations Arg-50, Ile-51, Arg-59, and Asn-108 have been strongly associated with in vitro and in vivo resistance and are correlated with SP usage (9, 11, 14, 19). Mutations in DHPS (Ala-436, Phe-436, Gly-437, Lys-540, Gly-581, Thr-613, and Ser-613) have also been associated with in vitro and in vivo resistance, although they have a weaker relationship with SP usage than DHFR mutations (2, 9, 11, 15, 19, 21). Other DHFR polymorphisms are more closely associated with cycloguanil resistance (Val-16 and Thr-108) (16).
Surveillance for antimalarial drug resistance is usually done by using in vitro or in vivo methods. In vitro methods involve the culturing of malaria parasites, which is difficult and requires skilled technicians and tissue culture facilities. In vivo methods require patients to be monitored for at least 14 days, which is often challenging in field conditions. Thus, new surveillance tools are needed. Numerous studies, by using standard PCR methods, have shown that DHPS and DHFR mutations are closely associated with SP resistance. However, the usefulness of these assays is restricted due to difficulty, cost, high risk for contamination, and the inability to pick up low-prevalence genotypes in a mixed sample (4). Real-time PCR, on the other hand, is simpler and less prone to contamination. The purpose of the present study was to develop a real-time PCR assay for DHPS and DHFR mutations.
Real-time PCR minor groove binding (MGB) probes provide a relatively inexpensive and sensitive way to detect single-nucleotide polymorphisms (SNPs) in a large number of samples (1, 13). MGB probes are designed to hybridize to an internal region of the PCR amplicon, thereby providing another level of specificity beyond the site-specific primers. When the Taq DNA polymerase cleaves the probe from the 5′ end, the fluorophore is released from the quencher, allowing it to fluoresce. The increase in fluorescence over time can be measured by using a real-time PCR thermocycler. Single base differences between MGB probes and DNA cause the melting temperature to decrease significantly, which decreases the efficiency of probe hybridization (12, 13). Allelic discrimination is achieved by putting in competition two probes complementary to wild-type and mutant alleles, which are attached to different fluorophores (13). The risk for contamination is much lower in this technique because there is no transfer of material between the addition of the DNA and the acquisition of the results (20). This technique has been used for allelic discrimination in humans, and a similar technique has been used to identify alleles that confer drug resistance in bacteria (8, 10, 13).
The impending loss of SP effectiveness in Africa, due to drug resistance, could have calamitous consequences. Partnering SP with another antimalarial agent, such as artesunate, might preserve SP effectiveness by delaying the onset of SP resistance. In order to measure the effects of combination chemotherapy on the development of SP resistance in Africa, we attempted to develop real-time PCR assays for the three DHFR polymorphisms (Ile-51, Arg-59, and Asn-108) and five DHPS polymorphisms (Ala-436, Gly-437, Lys-540, Gly-581, and Ser-613) reported from Africa.
MATERIALS AND METHODS
DNA.
Six DHPS plasmids (MRA-189 through MRA-194) and five DHFR plasmids (MRA-195 through MRA-199) contained in Escherichia coli were obtained from the Malaria Reagent Repository Resource (http://www.malaria.mr4.org/and references therein), containing the wild-type sequences and the most important mutations for each gene. The E. coli clones were grown on Luria-Bertani medium (1% tryptone, 0.5% yeast extract, 1.5% agarose in deionized H2O) with 50 μg of ampicillin/ml. The plasmids were extracted and purified by using the Promega Wizard Plus Minipreps DNA purification system (Promega, Madison, Wis.). P. falciparum strain 3D7 parasites were kindly provided by Jesse Kwiek, Duke University. DNA was extracted by using the QIAamp DNA minikit (Qiagen, Hilden, Germany). DNA from six strains of P. falciparum (Dd2, HB3, W2, FCR3, K1, and VI/S) was obtained from the Malaria Reagent Repository Resource.
Clinical samples of malaria parasites were obtained from a subgroup of pregnant women enrolled in a study investigating the effect of maternal malarial infection on mother-to-child transmission of human immunodeficiency virus (HIV). Details of the study, which commenced in December 2000, have been reported elsewhere (14a). Briefly, consent was sought from women in their late third trimester of pregnancy before the onset of active labor. Peripheral blood samples were drawn to screen for malaria by using thick-blood-smear microscopy and to detect HIV infection by using rapid antibody tests. All HIV-infected women and a subset of HIV-uninfected were enrolled in that study. For the present study, 24 clinical samples were genotyped: 21 from HIV-infected women with malaria, one from a parasitemic HIV-uninfected woman, and two control samples from HIV-infected women who were not parasitemic. With the exception of one HIV-infected woman, all women reported taking at least one dose of SP during the antenatal period.
Peripheral blood samples were centrifuged, and the plasma and packed-cell fractions were stored at −80°C. Subsequently, frozen packed cells were thawed, and a few drops were transferred onto filter paper. These were shipped to the University of North Carolina at Chapel Hill, where DNA extraction was performed by using the QIAamp DNA minikit (Qiagen, Hilden, Germany).
Primers and probes.
All primers and probes in the present study were designed by using Primer Express software (Applied Biosystems, Courtaboeuf, France), with the defaults of the program, except that the minimum %G+C content was lowered to 20% due to the AT-rich nature of the P. falciparum genome. P. falciparum strain 3D7 sequences for DHFR (accession number AF248537) and DHPS (accession number AF250167) were used in probe and primer design. When we designed the primers and probes, we assumed that all codons within the amplicon were wild type except for the SNP of interest. All primers were synthesized by Qiagen. The probes were synthesized by Applied Biosystems with the fluorophores FAM (6-carboxyfluorescein) linked to the 5′ position of all wild-type probes and VIC (chemical name not released by Applied Biosystems) linked to the 5′ position of all mutant probes. A nonfluorescent quencher and a minor groove binder were linked to the 3′ ends of all probes (chemical names not released by Applied Biosystems).
Real-time PCR.
PCRs were carried out in duplicate in a 25-μl final volume containing 12.5 μl of Universal PCR Master Mix, 5 μl of DNA, forward and reverse primers at various concentrations, and both mutant and wild-type probes at a final concentration of 200 nM. Optimal primer concentrations were determined by running the reactions with all combinations of the forward and reverse probes at 300, 500, and 900 nM (Table 1). All reactions were run on an ABI Prism 7000 sequence detection system (Applied Biosystems) with the default settings; each sample was initially denatured at 95°C for 10 min and cycled 40 times, with each cycle consisting of 95°C for 15 s and 60°C for 60 s.
TABLE 1.
Real-time primer and probe sequences and optimal concentrations
| Target | Oligonucleotide (5′ → 3′)a | Final concn (nM) | Detection limit (copies/reaction) |
|---|---|---|---|
| DHFR-51 | F: TGAGGTTTTTAATAACTACACATTTAGAGGTCT | 300 | |
| R: TATCATTTACATTATCCACAGTTTCTTTGTT | 300 | ||
| WTP: AATGTAATTCCCTAGATATG | 10 | ||
| MP: AAATGTATTTCCCTAGATATG | 10 | ||
| DHFR-59 | F: Same as DHFR-51 | 300 | |
| R: Same as DHFR-51 | 500 | ||
| WTP: AATATTTTTGTGCAGTTACA | 100 | ||
| MP: TGAAATATTTTCGTGCAGTTA | 10 | ||
| DHFR-108 | F: TGGATAATGTAAATGATATGCCTAATTCTAA | 300 | |
| R: AATCTTCTTTTTTTAAGGTTCTAGACAATATAACA | 300 | ||
| WTP: AGAACAAGCTGGGAAA | 1,000 | ||
| MP: AGAACAAACTGGGAAAG | 10 | ||
| DHPS-437 | F: TGAAATGATAAATGAAGGTGCTAGTGT | 900 | |
| R: AATACAGGTACTACTAAATCTCTTTCACTAATTTTT | 900 | ||
| WTP: AGAATCCTCTGCTCCT | 10 | ||
| MP: AATCCTCTGGTCCTTT | 10 | ||
| DHPS-436 | Primers and wild-type probe same as DHPS-437 | ||
| MP: AGAATCCGCTGTC | NSb | ||
| DHPS-540 | F: AATGCATAAAAGAGGAAATCCACAT | 300 | |
| R: TCGCAAATCCTAATCCAATATCAA | 300 | ||
| WTP: CAATGGATAAACTAACAAA | 10 | ||
| MP: AATGGATGAACTAACAAA | 10 | ||
| DHPS-581 | F: CCTCGTTATAGGATACTATTTGATATTGGAT | 500 | |
| R: TGGGCAATAAATCTTTTTCTTGAATA | 300 | ||
| WTP: ATTTGCGAAGAAAC | 10 | ||
| MP: ATTTGGGAAGAAACAT | 100 | ||
| DHPS-613 | F: TGGATTAGGATTTGCGAAGAAAC | 500 | |
| R: GTTGTGTATTTATTACAACATTTTGATCATTC | 300 | ||
| WTP: ATTTATTGCCCATTGCAT | 10 | ||
| MP: AGATTTATTTCCCATTGCA (serine) | 10 | ||
| MP: AGATTTATTACCCATTGCA (threonine) | NS |
The SNP is underlined in the probes. F, forward primer; R, reverse primer; WTP, wild-type probe; MP, mutant probe.
NS, not successful.
Data were transferred into Microsoft Excel version 2002, where the baseline (minimum) fluorescence was subtracted and replicates were averaged. To determine the detection limit for the wild-type and mutant probes, 10-fold dilutions of the relevant plasmids were run in triplicate. The detection limit was taken as the lowest copy number at which the standard deviation of the fluorescence of the wild-type and mutant genotype did not overlap. To assess the compatibility of this assay with mixed samples, two primer-probe sets (DHFR-51 and DHFR-59) were run with DNA from different proportions (0:1, 1:9, 2:8, 5:5, 8:2, 9:1, and 1:0) of wild-type and mutant plasmids. A total of 10,000 copies of plasmid DNA was used in each reaction.
To determine the specificity of the primer-probe sets and to ensure their compatibility with genomic DNA, seven strains of P. falciparum (3D7, Dd2, FCR3, HB3, K1, VI/S, and W2) were genotyped by using real-time PCR. The DHFR and DHPS genotypes of these strains, presented in Table 2, have been reported elsewhere (3, 6, 7, 18, 22). Real-time PCR was performed as described above except that only 1 μl of DNA was used per reaction. The genotype was determined by visually comparing the change in fluorescence of the wild-type and mutant probes.
TABLE 2.
DHFR and DHPS genotypes for P. falciparum strains and clinical samples as determined by sequencinga
| Strain or sample | Amino acid at:
|
|||||||
|---|---|---|---|---|---|---|---|---|
| DHFR codon:
|
DHPS codon:
|
|||||||
| 51 | 59 | 108 | 436 | 437 | 540 | 581 | 613 | |
| Strains | ||||||||
| 3D7 | Asn | Cys | Ser | Ser | Gly | Lys | Ala | Ala |
| Dd2 | Ile | Arg | Asn | Phe | Gly | Lys | Ala | Ser |
| HB3 | Asn | Cys | Asn | Ser | Ala | Lys | Ala | Ala |
| W2 | Ile | Arg | Asn | Phe | Gly | Lys | Ala | Ser |
| FCR3 | Asn | Cys | Thr | Ser | Ala | Lys | Ala | Ala |
| K1 | Asn | Arg | Asn | Ser | Gly | Lys | Gly | Ala |
| VI/S | Ile | Arg | Asn | Phe | Gly | Lys | Ala | Thr |
| Clinical samples | ||||||||
| CS1318 | Ile | Arg | Asn | Ser | Ala | Lys* | Ala | Ala |
| CS1338 | Ile* | Arg | Asn | Ser | Gly | Glu | Ala* | Ala |
| CS1452 | Ile | Arg | Asn | Ser | Ala* | Glu | Gly* | Ala |
| CS1648 | Ile | Cys | Asn | Ser | Gly | Glu | Ala | Ala |
| CS1915 | Ile* | Arg | Asn | Ser | Gly | Glu | Ala | Ala |
| Reference | ||||||||
| WT | Asn | Cys | Ser | Ser | Ala | Lys | Ala | Ala |
| M | Ile | Arg | Asn/Thr | Ala/Phe | Gly | Glu | Gly | Ser/Thr |
The last two rows portray the wild-type (WT) and mutant (M) genotypes for each polymorphism. See the text for citations. Clinical samples that were identified as mixed with real-time PCR are indicated by an asterisk.
Validation of method by genotyping clinical samples.
The first step in genotyping the clinical samples involved the use of real-time PCR to detect the lactate dehydrogenase (ldh) gene under the conditions described previously (17). Serial dilutions of P. falciparum 3D7 DNA at a known concentration were run on the same plate as the clinical samples. A standard curve of the 3D7 DNA was constructed and used to extrapolate the DNA concentration of the clinical samples. P. falciparum DNA was not detected in the two control samples from nonparasitemic women and therefore were not processed further. The remaining 22 samples were genotyped by real-time PCR with the optimal primer and probe concentrations (Table 1). The amount of DNA added per reaction varied (1 to 3 μl) depending on the detection limit of the primer-probe set and the concentration of the sample. Samples of low concentration were run for 45 cycles instead of 40 cycles. Each plate contained DNA from P. falciparum strains at relevant concentrations to act as wild-type and mutant controls.
The genotype was determined by comparing the cycle threshold (CT) values of the wild-type and mutant probe for each sample. The CT is the number of PCR cycles needed before the fluorescent signal surpasses a certain threshold (usually 20 to 40% of maximum). The more DNA of a specific genotype, the smaller the CT for that genotype-specific probe. For example, a sample was deemed to be wild type when the wild-type probe CT was less than the mutant probe CT and the difference was similar to that seen with the wild-type plasmid DNA. A sample was considered to be mixed when the CT's of both wild-type and mutant probes were closer and intermediate between the patterns seen for pure and wild-type plasmid DNA.
Five clinical samples were chosen to be sequenced based on the following criteria. (i) Real-time results were available for all seven SNPs. (ii) Both wild-type and mutant genotypes were included, when possible. (iii) If the sample was mixed, a dominant genotype could be distinguished from real-time PCR (which eliminated three samples from consideration).
Sequencing.
The five clinical samples were amplified with the same primers as in the MGB real-time PCR assay. Reactions were carried out in 25-μl final volume containing 12.5 μl of SYBR Green Master Mix, 1 μl of DNA, and forward and reverse primers at 300 nM. All reactions were run on an ABI Prism 7000 sequence detection system (Applied Biosystems) with the default settings with the dissociation step added at 60°C. The dissociation curves were examined to ensure that each reaction contained only one product. The resulting amplicon was purified by using CentriSpin-10 columns (Princeton Separations, Adelphia, N.J.). Sequencing was performed at the University of North Carolina Sequencing Core by using the ABI Prism BigDye terminator cycle sequencing ready reaction kit (Applied Biosystems).
RESULTS
Real-time PCR was both sensitive and specific for P. falciparum DHFR and DHPS genetic polymorphisms. The reaction conditions were effective for a wide range of P. falciparum DNA concentrations and had detection limits ranging from 10 to 1,000 copies (Table 1).
For the seven polymorphisms shown in Fig. 1, the wild-type probe generated fluorescence with the wild-type sequence but not the mutant sequence, while the mutant probe generated fluorescence with the mutant sequence but not the wild-type sequence. No amplification was ever seen in the absence of template (data not shown).
FIG. 1.

Real-time PCR detection of P. falciparum genotypes. Plasmid DNA containing wild-type and mutant template DNA (as labeled) were amplified with mixtures of wild-type probe (FAM labeled [solid line]) and mutant probe (VIC labeled [broken line]). Relative fluorescence was calculated by subtracting the minimal fluorescence from each value.
Background fluorescence (wild-type probe with mutant sequence and vice versa) was minimal, except in the case of the DHFR wild-type (Cys-59) probe. However, this background reaction did not prevent genotype determination, since the wild-type sequence still clearly amplified more rapidly than the mutant sequence. The original batch of the Cys-59 probe had a weak signal but provided sufficient discriminating ability. Subsequent batches of Cys-59 wild-type probe from Applied Biosystems had a much stronger signal and more background binding. However, the discriminating ability was similar to the original batch (data not shown).
Attempts to distinguish polymorphisms at DHPS-436 were unsuccessful. Real-time PCR was successful in determining the genotype of the P. falciparum strains for DHFR-51, DHFR-59, DHPS-540, and DHPS-581. For DHFR-108, the probe was designed to detect the asparagine mutant. However, the mutant probe also picked up the Thr-108 mutant, though the CT value was greater than that for Asn-108. Similarly, the DHPS-613 mutant probe, designed to identify the Ser-613 mutation, also bound to the Thr-613 mutation. However, in this case the CT values for the mutant probe were similar for both Ser-613 and Thr-613, and therefore they could not be distinguished. In both cases, wild type could be distinguished from mutant sequences. The genotyping of DHPS-437 was successful in the presence of Ser-436. There was no binding of the wild-type or mutant probe for DHPS-437 in the presence of the Ala-436 and Phe-436 mutations.
For two of the loci (DHFR-51 and DHFR-59), various mixtures of wild-type and mutant plasmids were prepared and then PCR amplified. In all cases, a mixture could be differentiated from a pure sample if the minor component was present at ≥10% (Fig. 2).
FIG. 2.

Real-time PCR genotyping of a mixed genotype sample. Various mixtures of wild-type and mutant plasmid DNA were amplified. The results for the 51 VIC probe are presented as the averages of three replicates from which the minimal fluorescence has been subtracted. The solid line represents a 100% mutant sample. The gray line represents a 10% mutant and a 90% wild-type sample. The broken line represents a 100% wild-type sample.
This method was validated with clinical samples. Complete DHFR genotypes were determined by using real-time PCR for all 22 samples taken from parasitemic women. An example of the real-time amplification of a clinical sample is shown in Fig. 3. In regards to DHPS, 21 samples were fully genotyped; the DHPS-540 genotype could not be ascertained for one sample. The five selected clinical samples were successfully sequenced for all seven loci. For the three DHFR loci and four DHPS loci, there was a 100% concordance of the dominant genotype between sequencing and real-time PCR (Table 2). Sequencing was unable to detect mixed infections that were identified by using real-time PCR.
FIG. 3.
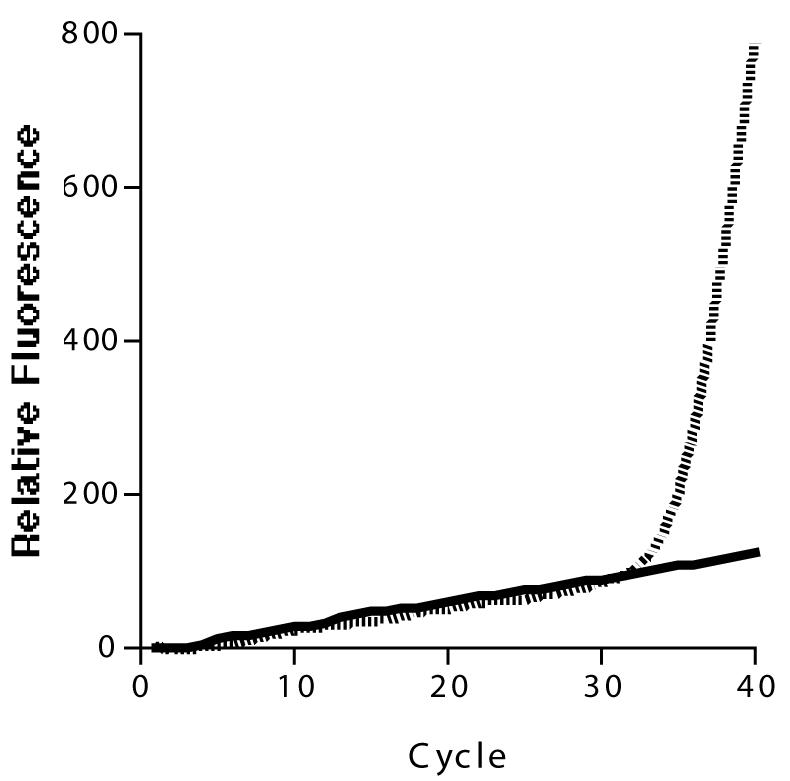
Real-time PCR genotyping of polymorphism 108 in clinical sample CS1338. The FAM-labeled probe (solid line) is designed to detect the wild-type sequence. The VIC-labeled probe (broken line) is designed to detect the mutant sequence. The change of fluorescence of the two probes was compared to controls run at similar concentrations. The genotype was determined to be mutant (Asn/Thr) with no wild-type component. The relative fluorescence was calculated by subtracting the minimal fluorescence from each value.
DISCUSSION
We describe a real-time PCR assay to detect polymorphisms associated with drug resistance in P. falciparum. We could successfully discriminate between wild-type and resistant alleles in seven of the eight loci important in Africa.
As a surveillance tool, real-time PCR has certain advantages over classical PCR. First, the assay is rapid and can reliably distinguish between two alleles after a single 3-h experiment. There is no need to run gels after the reaction. Second, the risk of contamination is far lower than with nested PCR because it is a closed-tube reaction. Third, the assay can be automated and processed in a high-throughput fashion. The initial cost of real-time PCR (consisting of buying the actual machine and optimizing the assay) is higher than normal PCR and may not be available in all countries where malaria is endemic. However, part of this cost could be made up if used for high-throughput processing since reagents for a single reaction cost between $0.40 and $2 (U.S. dollars).
Recently, a fluorogenic PCR assay using FRET probes was developed to genotype codons 50 to 60 of DHFR in P. falciparum (5). This technique has an advantage of low risk of contamination similar to that of the assay we describe in the present study. However, it has yet to be expanded to detect the other important mutations associated with drug resistance.
There are several limitations to the real-time PCR system described here. The most serious deficiency is its inability to distinguish alleles at position 436, and alleles at 437 when there is a mutation at the 436 position. However, the 436 mutations are rare in Africa and appear not to be involved in the “quintuple mutants” (Asn-108, Ile-51, and Arg-59 in DHFR and Gly-437 and Glu-540 in DHPS) that are especially important in Africa (18). Also, although real-time PCR can theoretically distinguish three or more alleles, we were not able to accomplish this at DHFR positions 108 and 613. Although we can distinguish the wild type from mutants at DHFR position 108, we could not distinguish between Asn-108, associated with SP resistance, and Thr-108, associated with cycloguanil resistance. Thus, further study is needed to enable the detection of these other polymorphisms. In addition, it should be noted that the detection limit differs among the primer-probe sets. This could potentially lead to an under-representation of certain genotypes. Nevertheless, given the advantages of this method, real-time PCR measurement of seven DHFR and DHPS polymorphisms could serve as a useful tool for the surveillance of SP-resistant malaria.
Acknowledgments
We thank W. Ngrenngarmlert, A. Ponguta, A. Purfield, and P. Wilson for assistance in the laboratory.
This study was supported by NIH grants AI45426 and AI49084, NIH-FIC grant 5 D43 TW00908, ASPH/CDC cooperative agreement S1935-21/22, and the Center for AIDS research at the University of North Carolina. A. Alker was supported by the M.D./Ph.D. program at UNC Chapel Hill for part of this work.
REFERENCES
- 1.Afonina, I. A., M. Reed, E. Lusby, I. G. Shishkina, and Y. S. Belousov. 2002. Minor groove binder-conjugated DNA probes for quantitative DNA detection by hybridization-triggered fluorescence. BioTechniques 32:940-949. [DOI] [PubMed] [Google Scholar]
- 2.Berglez, J., P. Iliades, W. Sirawaraporn, P. Coloe, and I. Macreadie. 2004. Analysis in Escherichia coli of Plasmodium falciparum dihydropteroate synthase (DHPS) alleles implicated in resistance to sulfadoxine. Int. J. Parasitol. 34:95-100. [DOI] [PubMed] [Google Scholar]
- 3.Brooks, D. R., P. Wang, M. Read, W. M. Watkins, P. F. Sims, and J. E. Hyde. 1994. Sequence variation of the hydroxymethyldihydropterin pyrophosphokinase: dihydropteroate synthase gene in lines of the human malaria parasite, Plasmodium falciparum, with differing resistance to sulfadoxine. Eur. J. Biochem. 224:397-405. [DOI] [PubMed] [Google Scholar]
- 4.Chaparro, J., M. O. Rojas, and M. Wasserman. 2001. Plasmodium falciparum: underestimation of dihydrofolate reductase and dihydropteroate synthase polymorphism in field samples: a technical shortcoming of nested PCR assays with mutation-specific primers. Exp. Parasitol. 99:115-122. [DOI] [PubMed] [Google Scholar]
- 5.Decuypere, S., E. Elinck, C. Van Overmeir, A. O. Talisuna, U. D'Alessandro, and J. C. Dujardin. 2003. Pathogen genotyping in polyclonal infections: application of a fluorogenic polymerase-chain-reaction assay in malaria. J. Infect. Dis. 188:1245-1249. [DOI] [PubMed] [Google Scholar]
- 6.Duraisingh, M. T., J. Curtis, and D. C. Warhurst. 1998. Plasmodium falciparum: detection of polymorphisms in the dihydrofolate reductase and dihydropteroate synthetase genes by PCR and restriction digestion. Exp. Parasitol. 89:1-8. [DOI] [PubMed] [Google Scholar]
- 7.Foote, S. J., D. Galatis, and A. F. Cowman. 1990. Amino acids in the dihydrofolate reductase-thymidylate synthase gene of Plasmodium falciparum involved in cycloguanil resistance differ from those involved in pyrimethamine resistance. Proc. Natl. Acad. Sci. USA 87:3014-3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.García de Viedma, D., M. del Sol Díaz Infantes, F. Lasala, F. Chaves, L. Alcalá, and E. Bouza. 2002. New real-time PCR able to detect in a single tube multiple rifampin resistance mutations and high-level isoniazid resistance mutations in Mycobacterium tuberculosis. J. Clin. Microbiol. 40:988-995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jelinek, T., A. H. D. Kilian, J. Curtis, M. T. Duraisingh, G. Kabagambe, F. von Sonnenburg, and D. C. Warhurst. 1999. Plasmodium falciparum: selection of serine 108 of dihydrofolate with co-trimoxazole in Ugandan children. Am. J. Trop. Med. Hyg. 61:125-130. [DOI] [PubMed] [Google Scholar]
- 10.Kearns, A. M., C. Graham, D. Burdess, J. Heatherington, and R. Freeman. 2002. Rapid real-time PCR for determination of penicillin susceptibility in pneumococcal meningitis, including culture-negative cases. J. Clin. Microbiol. 40:682-684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kublin, J. G., R. S. Witzig, A. H. Shankar, J. Q. Zurita, R. H. Gilman, J. A. Guarda, J. F. Cortese, and C. V. Plowe. 1998. Molecular assays for surveillance of antifolate-resistant malaria. Lancet 351:1629-1630. [DOI] [PubMed] [Google Scholar]
- 12.Kutyavin, I., I. Afonina, A. Mills, V. Gorn, E. Lukhtanov, E. Belousov, M. Singer, D. Walburger, S. Lokhov, A. Gall, R. Dempcy, M. Reed, R. Meyer, and J. Hedgpeth. 2000. 3′-Minor groove binder-DNA probes increase sequence specificity at PCR extension temperatures. Nucleic Acids Res. 28:655-661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Livak, K. J. 1999. Allelic discrimination using fluorogenic probes and the 5′ nuclease assay. Genet. Anal. 14:143-149. [DOI] [PubMed] [Google Scholar]
- 14.Mendez, F., A. Munoz, G. Carrasquilla, D. Jurado, M. Arevalo-Herrera, J. Cortese, and C. Plowe. 2002. Determinants of treatment response to sulfadoxine-pyrimethamine and subsequent transmission potential in falciparum malaria. Am. J. Epidemiol. 156:230-238. [DOI] [PubMed] [Google Scholar]
- 14a.Mwapasa, V., S. J. Rogerson, M. E. Molyneux, E. T. Abrams, D. D. Kamwendo, V. M. Lema, E. Tadesse, E. Chaluluka, P. E. Wilson, and S. R. Meshnick. 2004. The effect of Plasmodium falciparum malaria on peripheral and placental HIV-1 RNA concentrations in pregnant Malawian women. AIDS 18:1051-1059. [DOI] [PubMed]
- 15.Nzila, A. M., E. K. Mberu, J. Sulo, H. Dayo, P. A. Winstanley, C. H. Sibley, and W. M. Watkins. 2000. Towards an understanding of the mechanism of pyrimethamine-sulfadoxine resistance in Plasmodium falciparum: genotyping of dihydrofolate reductase and dihydropteroate synthase of Kenyan parasites. Antimicrob. Agents Chemother. 44:991-996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peterson, D. S., W. K. Milhous, and T. E. Wellems. 1990. Molecular basis of differential resistance to cycloguanil and pyrimethamine in Plasmodium falciparum malaria. Proc. Natl. Acad. Sci. USA 87:3018-3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pickard, A. L., C. Wongsrichanalai, A. Purfield, D. Kamwendo, K. Emery, C. Zalewski, F. Kawamoto, R. S. Miller, and S. R. Meshnick. 2003. Resistance to antimalarials in Southeast Asia and genetic polymorphisms in pfmdr1. Antimicrob. Agents Chemother. 47:2418-2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Plowe, C. 2001. Folate antagonists and mechanisms of resistance, p. 173-190. In P. J. Rosenthal (ed.), Antimalarial chemotherapy: mechanisms of action, resistance, and new directions in drug discovery. Humana Press, Totowa, N.Y.
- 19.Plowe, C., J. F. Cortese, A. Djimde, O. C. Nwanyanwu, W. M. Watkins, P. A. Winstanley, J. G. Estrada-Franco, R. E. Mollinedo, J. C. Avila, J. L. Cespedes, D. Carter, and O. K. Doumbo. 1997. Mutations in Plasmodium falciparum dihydrofolate reductase and dihydropteroate synthase and epidemiologic patterns of pyrimethamine-sulfadoxine use and resistance. J. Infect. Dis. 176:1590-1596. [DOI] [PubMed] [Google Scholar]
- 20.Vet, J., A. Majithia, S. Marras, S. Tyagi, S. Dube, B. Poiesz, and F. R. Kramer. 1999. Multiplex detection of four pathogenic retroviruses using molecular beacons. Proc. Natl. Acad. Sci. USA 96:6394-6399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang, P., C. S. Lee, R. Bayoumi, A. Djimde, O. Doumbo, G. Swedberg, L. D. Dao, H. Mshinda, M. Tanner, W. M. Watkins, P. F. Sims, and J. E. Hyde. 1997. Resistance to antifolates in Plasmodium falciparum monitored by sequence analysis of dihydropteroate synthetase and dihydrofolate reductase alleles in a large number of field samples of diverse origins. Mol. Biochem. Parasitol. 89:161-177. [DOI] [PubMed] [Google Scholar]
- 22.Wang, P., M. Read, P. F. Sims, and J. E. Hyde. 1997. Sulfadoxine resistance in the human malaria parasite Plasmodium falciparum is determined by mutations in dihydropteroate synthetase and an additional factor associated with folate utilization. Mol. Microbiol. 23:979-986. [DOI] [PubMed] [Google Scholar]
- 23.Wongsrichanalai, C., A. L. Pickard, W. H. Wernsdorfer, and S. R. Meshnick. 2002. Epidemiology of drug-resistant malaria. Lancet Infect. Dis. 2:209-218. [DOI] [PubMed] [Google Scholar]