

Abstract
Patients with bilateral vestibular schwannomas associated with neurofibromatosis type 2 (NF2) experience significant morbidity such as complete hearing loss. We have recently shown that treatment with bevacizumab provided tumor stabilization and hearing recovery in a subset of NF2 patients with progressive disease. In the current study, we used two animal models to identify the mechanism of action of anti–vascular endothelial growth factor (VEGF) therapy in schwannomas. The human HEI193 and murine Nf2−/− cell lines were implanted between the pia and arachnoid meninges as well as in the sciatic nerve to mimic central and peripheral schwannomas. Mice were treated with bevacizumab (10 mg/kg/wk i.v.) or vandetanib (50 mg/kg/d orally) to block the VEGF pathway. Using intravital and confocal microscopy, together with whole-body imaging, we measured tumor growth delay, survival rate, as well as blood vessel structure and function at regular intervals. In both models, tumor vessel diameter, length/surface area density, and permeability were significantly reduced after treatment. After 2 weeks of treatment, necrosis in HEI193 tumors and apoptosis in Nf2−/− tumors were significantly increased, and the tumor growth rate decreased by an average of 50%. The survival of mice bearing intracranial schwannomas was extended by at least 50%. This study shows that anti-VEGF therapy normalizes the vasculature of schwannoma xenografts in nude mice and successfully controls the tumor growth, probably by reestablishing a natural balance between VEGF and semaphorin 3 signaling.
Introduction
Neurofibromatosis type 2 (NF2) is an autosomal dominant inherited disorder characterized by the development of multiple schwannomas, meningiomas, and ependymomas (1). Although most patients with sporadic schwannomas benefit from surgery and radiation therapy (2), NF2 patients have limited options due to the multiplicity and bilateral location of their tumors (3). Vestibular schwannomas are particularly challenging surgically due to the potential for brainstem or cranial nerve damage. Currently, there are no curative treatments and bilateral hearing loss is unfortunately common in patients affected by NF2.
Schwannomas, like other tumors, recruit blood vessels to sustain their growth. Indeed, a number of small studies have previously reported expression of the main angiogenic factor, vascular endothelial growth factor (VEGF), in schwannomas (4–7), a finding confirmed by our own analysis of a series of NF2-related and sporadic schwannomas at the Massachusetts General Hospital (8). The activity of VEGF is mostly mediated via two classes of receptors: the tyrosine kinases VEGFR1 and VEGFR2 and the neuropilins NRP1 and NRP2 (9, 10). The activity of VEGF in the peripheral nervous system is modulated by semaphorins, which are negative regulators that act through the neuropilin receptors (9–11).
Targeting the ligand, VEGF itself (12, 13), or blocking VEGF receptor activity (14–17) has led to some success in treating various cancers, including malignant gliomas. Antiangiogenic therapy has only recently been used to treat benign tumors that are generally thought to be resistant to standard cytotoxic chemotherapy. But in a recent study of 10 consecutive patients with bilateral vestibular schwannoma, we showed that anti-VEGF therapy results in improvement of hearing and stabilization of tumor growth (8). However, the retrospective nature of this study did not allow determination of the mechanism of action. We therefore developed two animal models of schwannoma to perform longitudinal studies of tumor growth. By using two therapeutic approaches, we show that targeting the VEGF pathway by blocking either VEGF or its main receptor (VEGFR2) can lead to tumor control and improved overall survival by decreasing permeability and inducing tumor cell death. Furthermore, we present evidence that loss of semaphorin by the Nf2−/− Schwann cell is responsible for shifting the balance between semaphorin and VEGF, leading ultimately to a proangiogenic state.
Materials and Methods
Chemical reagents and antibodies
Vandetanib [a dual tyrosine kinase inhibitor (TKI) of VEGFR2 and epidermal growth factor receptor (EGFR), a generous gift from AstraZeneca; ref. 18] was dissolved in DMSO for in vitro studies and in 1% Tween 80 (5 mg/mL) for in vivo studies. Bevacizumab (a humanized antibody to VEGF, obtained from Massachusetts General Hospital Pharmacy) was diluted with PBS (10 mg/mL) before use in animals. The following antibodies were used: phosphorylated EGFR (P-EGFR; 1:1,000; Cell Signaling Technology), phosphorylated AKR mouse T-cell lymphoma oncogene (P-AKT; 1:1,000; Cell Signaling Technology), phosphorylated extracellular signal-regulated kinase (P-ERK; 1:1,000; Cell Signaling Technology), EGFR (1:1,000; Cell Signaling Technology), AKT (1:1,000; Cell Signaling Technology), ERK1 (clone MK12, 1:1,000; BD Transduction Laboratories), and merlin (1:1,000; Santa Cruz Biotechnology). Recombinant mouse VEGF was obtained from the National Cancer Institute (NCI) via a materials transfer agreement and recombinant human EGF was obtained from R&D Systems.
Cell lines
Human HEI193 (a gift from Dr. Xandra Breakefield [Massachusetts General Hospital, Boston, MA]; refs. 19, 20) schwannoma cells were maintained in DMEM (Cellgro Mediatech) supplemented with 10% heat-inactivated fetal bovine serum (Atlanta Biologicals), 1× N2 (Invitrogen), 14 ng/mL glial cell line–derived neurotrophic factor, and 2 μmol/L forskolin (20). Murine Nf2−/− tumor Schwann cells were generated along the following guide lines: pure populations of embryonic Schwann cells were isolated from Nf2loxP/loxP− mice at embryonic day 13.5. The cells were then infected in vitro with an adenovirus expressing Cre recombinase to allow excision of exon 2 and thereby disruption of the Nf2 locus. The cells were injected in nude mice in the subcutaneous space in the flank, and the resulting tumor was harvested and dissociated. The resulting single-cell suspension was placed in culture to generate the first “line” used for all experiments in this study. Nf2−/− cells were maintained in DMEM supplemented with 100 ng/mL recombinant human neuregulin-β1 (PeproTech) and 5 μmol/L forskolin (Invitrogen).
Animal models
To recapitulate as closely as possible the microenvironment of vestibular schwannomas (intracranial but arising from a cranial nerve), we modified the previously published model for gliomas (21). In brief, Nf2−/− or HEI193 cells were implanted between the pia and arachnoid meninges in nude mice (8 weeks old) bearing transparent cranial windows and allowed to reach 2-mm diameter (approximately 6 and 11 days for Nf2−/− and HEI193 cells, respectively) before initiation of treatment. In the second model, we reproduced the nerve microenvironment using the mouse sciatic nerve. This environment had previously been shown to be suitable for schwannoma growth (22). To mimic the clinical situation where treatment would be initiated when tumor volume is already significant (between 2 and 20 cm3), we started treating the sciatic nerve tumors after they were macroscopically visible (7 days for Nf2−/− and 4–5 weeks for HEI193 following implantation). In both models, approximately a million cells were injected with a 28.5-gauge needle to generate schwannomas.
To capitalize on the advantages of each model, we used the cranial window models to study kinetic changes in vessel (diameter and permeability) and overall survival, whereas the peripheral implantation was used to study growth kinetics and vessel morphology (number and maturation state of vessels, and apoptosis and proliferation of endothelial cells).
Due to the species specificity of bevacizumab, the Nf2 murine model was treated with the TKI vandetanib, whereas the human HEI193 model was treated with bevacizumab. In the peripheral model, HEI193 was treated with both regimens.
Multiphoton laser scanning microscopy angiography and vessel morphometry quantification
Intracranially implanted tumors were allowed to grow for 6 days (Nf2−/− cells) or 11 days (HEI193 cells) before treatment with vandetanib at 50 mg/kg/d orally or bevacizumab at 10 mg/kg/wk i.v., respectively. At time points of interest, mice were anesthetized with 0.3 mL ketamine/xylazine, and multiphoton laser scanning microscopy (MPLSM) angiography of blood vessels was performed following i.v. injection of 0.1 mL FITC-dextran (2 million molecular weight; Sigma) at 10 mg/mL. Three-dimensional profiles (200-μm stacks) of the tumor vasculature were collected before as well as 1 and 6 days after treatment. A vessel-tracing algorithm was used to quantify vessel diameter, length density, and surface area density of each angiograph (23).
Blood vessel permeability measurement
Changes in vessel permeability during anti-VEGF therapy were measured in tumors grown in the cranium. Nf2−/− or HEI193 cells were infected with green fluorescent protein (GFP) retroviruses for visual tracking of tumor volume within the cranial window. Measurement of the effective vascular permeability was performed as described previously (24). In brief, GFP-expressing tumor cells were first located, and the fluorescence intensity of rhodamine within the tumor tissue was measured every 2 minutes for 24 minutes after injection of 0.1 mL tetramethylrhodamine-labeled bovine serum albumin (rho-BSA; 10 mg/mL). Permeability of the vessels to albumin was calculated as P = (1 − HT) V/S [1/(I0 − Ib)*dI/dt + 1/K], where I is the average fluorescence intensity of the whole image, I0 is the value of I immediately after the filling of all vessels by rho-BSA, and Ib is the background fluorescence intensity. The average hematocrit (HT) of tumor vessels was estimated to be 19%. V and S are the total volume and surface area of vessels within the tissue volume covered by the surface image, respectively. The time constant of BSA plasma clearance (K) is 9.1 × 103 seconds (24).
Whole-body imaging
Longitudinal studies to investigate the effect of anti-VEGF therapies on modulating size of Nf2−/− and HEI193 schwannomas within the sciatic nerve were performed using the whole-body imaging system from Xenogen (IVIS Lumina II). Tumor cells were first infected with lentiviruses expressing firefly luciferase (mCherry; provided by Dr. Xandra Breakefield). Tumor cells (~1 million cells) stably expressing luciferase were implanted into the sciatic nerve of 8-week-old nude mice. Bioluminescence intensity was first measured 2 and 30 days after implantation of Nf2−/− and HEI193, and bioluminescence was then measured every 2 days for the next 2 weeks to follow tumor growth. To image tumors, D-luciferin at 33 mg/mL was injected i.p. Five minutes after injection, bioluminescence images were collected for a length of 20 minutes. Data were analyzed with Living Image software version 3.0.4, retaining the same region of interest for all tumors within a given experiment. The total photon flux per second was compared for each tumor accordingly.
Semiquantitative polymerase chain reaction
Total RNA was extracted from Nf2−/− or HEI193 cells using the RNeasy Mini kit (Qiagen) according to the manufacturer’s instructions. Genomic DNA was digested using the RNase-free DNase I (Qiagen) to ensure the complete removal of DNA contamination. The amount of total RNA was quantified by absorbance at 260 nm (NanoDrop). Five hundred nanograms of RNA from each sample were transcribed to cDNA using SuperScript III RNase H− Reverse Transcriptase (Invitrogen). Polymerase chain reaction (PCR) was performed in a thermal cycler (Dyad). PCR products were then resolved in a 2% agarose gel by electrophoresis, and DNA images were captured with the U.V. transilluminator (Bio-Rad). The glyceraldehyde-3-phosphate dehydrogenase (Gapdh) gene was amplified in each PCR experiment as an internal loading control.
Western blot analysis
Cells were rinsed with cold PBS and lysed in an appropriate volume of lysis buffer. Protein concentration was measured with bicinchoninic acid protein assay kit (Pierce) according to the manufacturer’s instructions. Appropriate volumes of protein lysates were then mixed with 4× SDS gel-loading buffer, boiled at 100°C for 5 minutes, and resolved in 4% to 12% gradient polyacrylamide gels (Invitrogen). Proteins were subsequently transferred onto polyvinylidene difluoride membrane (0.45 μm; Thermo Scientific), and the membrane was blocked with 5% nonfat milk in TBS with Tween 20 for 1 hour. Blots were then incubated with appropriately diluted primary and horseradish peroxidase–conjugated secondary antibodies, each for 1 hour at room temperature. Finally, blots were developed with enhanced chemiluminescence solutions and exposed onto HyBlot CL (Denville Scientific).
Immunohistochemistry
Mice were perfusion fixed with 4% formaldehyde in PBS (25). Perfused tumors were extracted, postfixed in formaldehyde at 4°C for 4 to 5 hours, and incubated with 30% sucrose overnight at 4°C with constant rocking. Brain or sciatic nerve tumors were then embedded in OCT freezing medium and frozen for sectioning. Sections (10 μm thick) were first air dried for 30 minutes at room temperature and postfixed with acetone for 5 minutes at −20°C. Individual stains were performed using the following antibodies: CD31 (1:150; Chemicon), platelet-derived growth factor receptor β (PDGFRβ; 1:100; eBioscience), desmin (1:300; DAKO), and collagen IV (1:2,000; Chemicon). Detection was carried out using secondary antibodies (or streptavidin) labeled with fluorophores with different wavelengths (FITC, Cy3, and Cy5). For apoptosis, the ApopTag kit (Invitrogen) was used according to the manufacturer’s recommendations. Confocal images were acquired using a FluoView 500 confocal microscope (Olympus). A constant surface area (0.4 mm2) was imaged in all cases.
Quantification of perivascular cells and basement membrane coverage
Quantification of the stained area was performed using an in-house segmentation algorithm (26). Analysis of basement membrane thickness was performed by fitting the fraction of collagen IV–stained area at a range of distances (1–15 μm) from the vessel wall (determined by CD31 staining segmentation). The profile away from the vessel wall was fit to an exponential decay function (F = Ae−x/L + C), where F = fraction of collagen IV–positive area at distance x from the vessels, x = distance from vessels (1–10 μm), A = constant related to the extent of pericyte/basement membrane coverage, and L = characteristic length related to the distance between pericyte and the vessel wall when applied to desmin-stained tissue and to basement membrane thickness when applied to collagen IV–stained tissue.
Statistical analysis
We used the unpaired, two-tailed Student’s t test for comparison between two samples. One-way ANOVA Fisher’s test followed by Tukey’s honestly significant difference test was used for multiple comparisons with a 95% confidence level. For survival rate, we plotted the survival distribution curve with the Kaplan-Meier method followed by log-rank testing (XLSTAT software). We considered the difference between comparisons to be significant when P < 0.05 for all the statistical analysis.
Results
Establishment and characterization of schwannoma models suitable for kinetic analysis
The prerequisite for these studies was to develop new models to study the effect of antiangiogenic therapy on schwannoma tumor vessels. We used a newly established murine cell line (Nf2−/− Schwann cells) to recapitulate the genetic abnormality found in NF2-related vestibular schwannomas and the human cell line (HEI193 cells), which was cultured from a schwannoma resected from a NF2 patient. Although of different origin, both cell lines showed similarities in that they lacked wild-type (wt) merlin expression (Fig. 1A), did not express VEGFR1 or VEGFR2, but did express EGFR that could be phosphorylated on stimulation with recombinant EGF (Fig. 1B). In addition, both cell lines secreted VEGF in vitro (data not shown). Furthermore, analysis of expression levels of semaphorin 3 (SEMA3A to SEMA3G) and neuropilins (NRP1 and NRP2) in murine Schwann cells revealed a downregulation of SEMA3D, SEMA3F, SEMA3G, and NRP1 on loss of the Nf2 gene (Fig. 1C). Downregulation of SEMA3A, SEMA3F, and NRP1 was also observed in HEI193 cells compared with normal human Schwann cells (Fig. 1C). Furthermore, reintroduction of Nf2 gene in the Nf2−/− Schwann cells induced reexpression not only of merlin but also of SEMA3 and NRP1 (Fig. 1D). This combination of receptors and ligands provided a system, particularly relevant to the peripheral nervous system, with an overall proangiogenic balance by loss of the naturally secreted inhibitor induced by loss of Nf2 gene.
Figure 1.
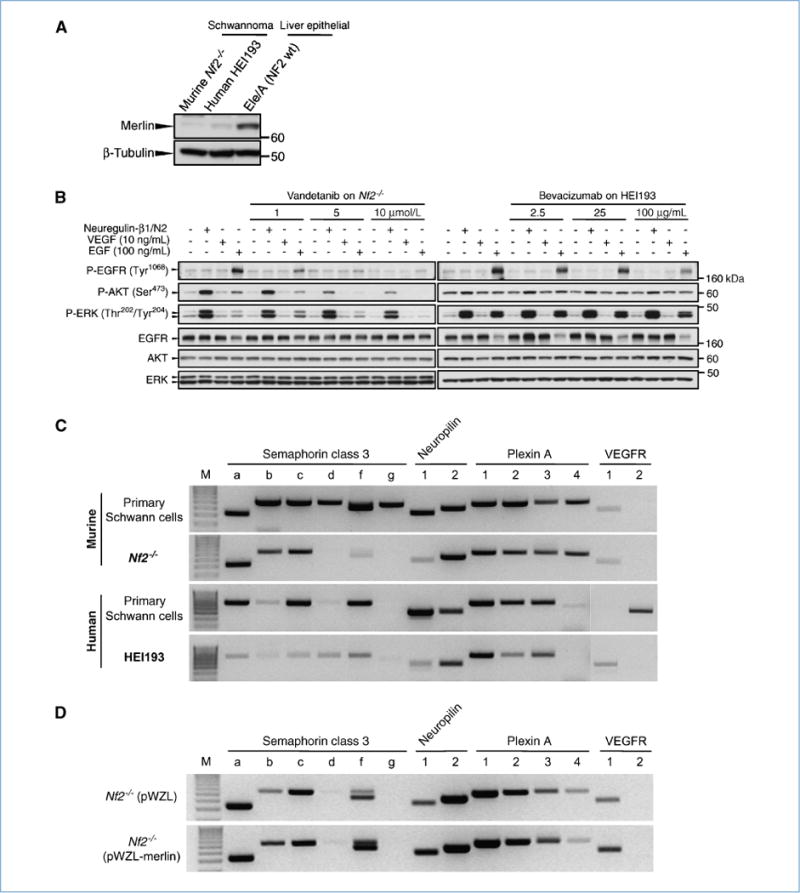
Characterization of a novel schwannoma line and the effect of VEGF inhibitors on these cell lines in vitro. A, wt merlin is below the detection limit both in the newly established murine schwannoma line (Nf2−/−) and in the human HEI193. Liver epithelial cells derived from the Nf2 wt mice served as a merlin-positive control. B, effect of vandetanib and bevacizumab on Nf2−/− and HEI193 signaling. Nf2-deficient or HEI193 schwannoma at density of 2.5 × 106 cells were starved overnight for 12 hours with DMEM and incubated with full medium, recombinant mouse VEGF (10 ng/mL), or human EGF (100 ng/mL) with various concentration of vandetanib or bevacizumab for 15 minutes at 37°C as indicated in the diagram. Vandetanib decreased P-EGFR, P-AKT, and P-ERK in a concentration-dependent manner, whereas bevacizumab did not affect HEI193 signaling unless at very high concentration. C, expression pattern for SEMA3, its receptors, and the VEGF receptors in murine wt Schwann cells and their Nf2−/− counterpart (top) and human Schwann cells compared with HEI193 (bottom). Loss of the Nf2 gene is concurrent with a loss of SEMA3 (b, d, f, and g in murine, all in human), NRP1, and VEGFR1 expression, whereas NRP2 and plexins retain their initial expression. VEGF receptors are not typically expressed by Schwann cells, and oncogenic transformation did not seem to change this feature in vitro. D, reintroduction of the Nf2 gene is accompanied by a reexpression of SEMA3, NRP1, and VEGFR1, supporting the hypothesis of a link between loss of merlin and a change in balance within the angiogenic pathway.
Due to the species specificity of bevacizumab to human VEGF, the murine NF2−/− model could not be treated with the VEGF inhibitor. However, we believe that the NF2−/− Schwann cells are more representative of the molecular evolution of vestibular schwannoma than HEI193 cells. For example, the NF2−/− cells lose expression of the semaphorin and NRP1, precisely recapitulating the pattern seen in patients. On the other hand, bevacizumab is the agent currently used in the clinic (8, 27). We therefore took two parallel approaches to extract an overall picture of “VEGF withdrawal” mechanisms based on the respective strengths of the two distinct but biologically related models: administration of bevacizumab (a monoclonal antibody against human VEGF, as a single agent) or administration of vandetanib (a TKI specific to EGFR and VEGF), which is safe in patients in phase I/II trials (28) and efficacious in a murine model. Furthermore, EGFR has been reported to play a critical role in Schwann cell proliferation (29–31), and treatment with an inhibitor has shown efficacy in patients (27).
We then characterized the antitumor and antivascular effects of vandetanib and bevacizumab in vitro by stimulating Nf2−/− cells with recombinant EGF before adding vandetanib or bevacizumab. There was a concentration-dependent inhibition of P-EGFR, P-AKT, and P-ERK by vandetanib but no notable pharmacologic effect by bevacizumab (Fig. 1B). These results suggest that vandetanib likely targets both tumor cells and host endothelium, whereas bevacizumab targets only the endothelium in our in vivo experimental settings.
Schwannomas are vascularized; anti-VEGF therapies decrease vessel size and number
By 10 days to 2 weeks following subdural injection of 105 tumor cells, 2-mm-diameter tumors with established vasculature were visible (Fig. 2A and B, D0 panels). Treatment of intracranial schwannomas with vandetanib (50 mg/kg/d) or bevacizumab (10 mg/kg/wk) induced vascular changes as early as 24 hours after the first dose (Fig. 2A and B, D1 panels). By 6 days of treatment, both vandetanib and bevacizumab decreased the vessel surface area (S/V) compared with controls (34% and 33%, respectively, graphs SA density). However, the biological pathways responsible for the decrease were different between the two treatments. Vandetanib resulted in a 32% decrease of the number of vessels (length density) without affecting the vessel diameter, whereas bevacizumab decreased vessel diameter by 22% as early as 24 hours following the first injection but did not change the number of vessels. In both cases, total vascular volume in the tumor was significantly decreased by the 6th day of treatment.
Figure 2.
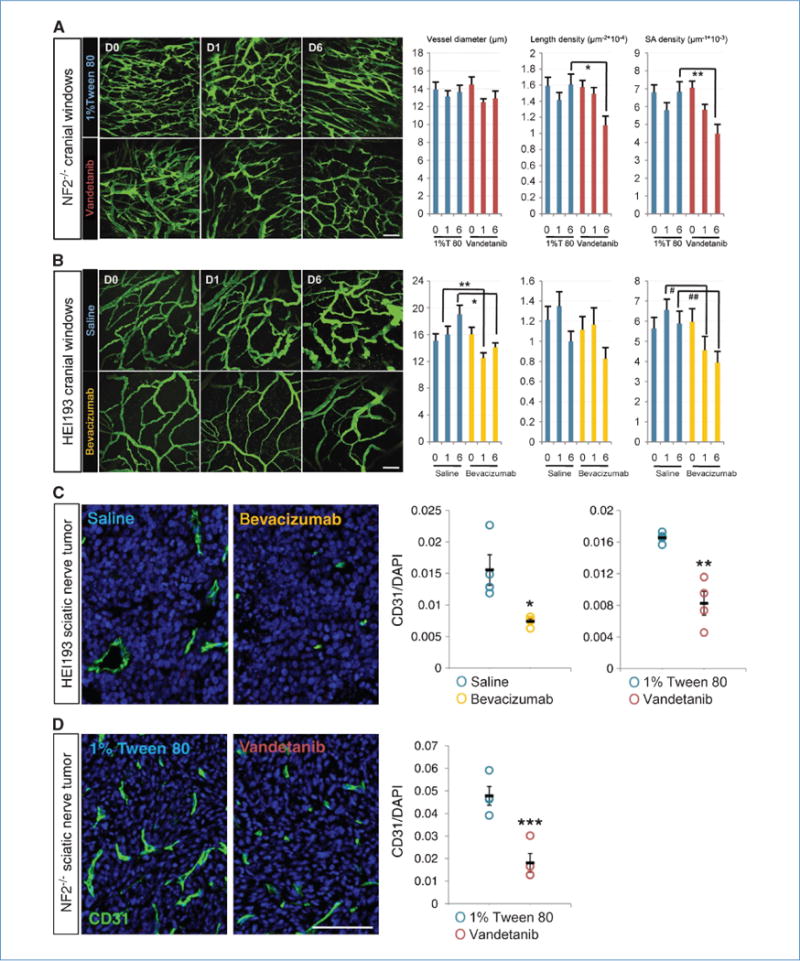
Inhibition of VEGF pathways reduces number and density of schwannoma vessels in brain (A and B) and nerve microenvironment (C and D). Vandetanib reduced vessel length and surface area density of brain Nf2−/− schwannoma without affecting vessel diameter, as shown by two-photon MPLSM in vivo 6 days after treatment. 1% Tween 80, n = 11; vandetanib, n = 12. *, P = 9.11 × 10−5; **, P = 8 × 10−4. Bevacizumab, on the other hand, reduced vessel diameter and vessel surface area density of HEI193 as short as 1 day after treatment, maintained up to 6 days without affecting the length density. Saline, n = 7; bevacizumab, n = 8. *, P = 6.6 × 10−5; **, P = 1.32 × 10−5; #, P = 9.07 × 10−5; ##, P = 5 × 10−4. Scale bars, 100 μm. In sciatic nerve tumor, both vandetanib and bevacizumab reduced CD31-positive fraction of HEI193 schwannoma 6 days after treatment (C), so did vandetanib on Nf2−/− (D). All n = 4. *, P = 0.0428; **, P = 0.0097; ***, P = 0.0023. Scale bar, 100 μm.
Tumors implanted in the sciatic nerve showed similar changes in overall microvascular density (with an initial average of 53 and 250 vessels per mm2 for HEI193 and Nf2−/−, respectively; see Supplementary Fig. S1) with a decrease of 50% in HEI193 (36 vessels per mm2; Fig. 2C; Supplementary Fig. S1B) and 62% in Nf2−/− (average of 98 vessels per mm2; Fig. 2D; Supplementary Fig. S1A) after treatment with bevacizumab and vandetanib, respectively. To distinguish the response of tumor cells from that of the host endothelium, we also treated HEI193 with vandetanib. After 6 days of treatment with vandetanib, a 50% decrease in total number of vessels was also seen (Fig. 2C; Supplementary Fig. S1B). Blocking tumor-derived VEGF or VEGFR2 in host endothelium produced virtually the same antiangiogenic outcome.
Anti-VEGF therapy delays tumor growth and improves overall survival in mice with intracranial schwannomas
Tumor growth delay
Figure 3A shows the physical appearance of tumors 2 weeks after treatment. Vandetanib and bevacizumab decreased Nf2−/− and HEI193 tumor size 3- and 7-fold, respectively, after 2 weeks of treatment (Fig. 3B). To determine how anti-VEGF therapy affects growth rate, we transfected tumor cells with luciferase and followed tumor growth with a whole-body imager. Although tumors had to be measured at a much earlier stage (due to saturation of the signal), by day 6, vandetanib and bevacizumab resulted in 2.5-and 4-fold decreases in tumor volume, respectively (Fig. 3C and D). To further investigate the mechanism by which anti-VEGF therapies delay schwannoma growth, we assessed tumor tissue viability. We found that vandetanib increased apoptosis in Nf2−/− by 6-fold (Fig. 4A), whereas necrosis was not noticeable at the measured time points. Interestingly, instead of inducing apoptosis, both bevacizumab and vandetanib treatment increased the necrotic fraction in HEI193 by 3- to 4-fold (Fig. 4B). From these data, we conclude that anti-VEGF treatment delays tumor progression by causing tumor cell death primarily through the antivascular effect.
Figure 3.
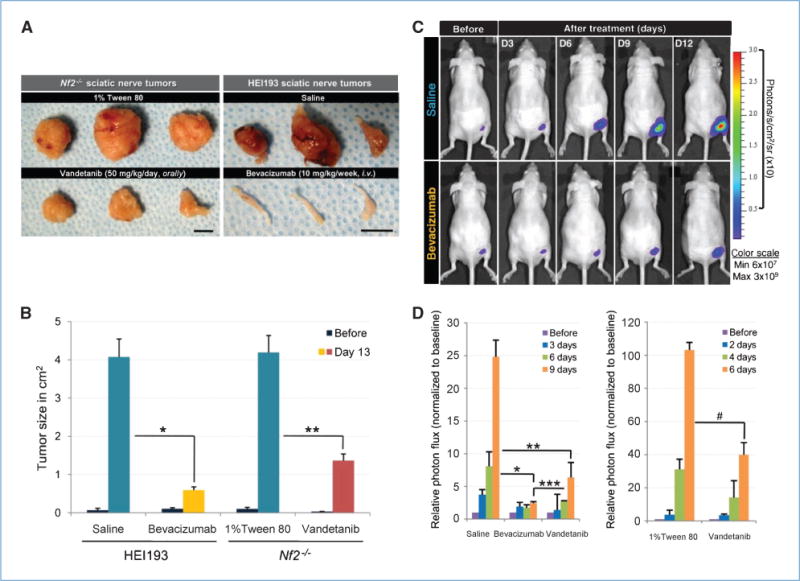
Inhibition of VEGF pathways delays schwannoma growth. Both vandetanib and bevacizumab delayed growth of schwannoma in sciatic nerve using either caliper (A and B) or whole-body imaging (C and D) as a tool for size measurement. A, photographs showing images of sciatic nerve tumors 12 days after treatment. Scale bars, 1 cm. B, keeping track of tumor size using caliper. Tumor sizes were measured every 2 days in both controls and the treated, and there was a marked decrease of tumor size 13 days after treatment. Controls, n = 9; vandetanib, n = 8; bevacizumab, n = 3. *, P = 0.0019; **, P = 0.0082. C, representative bioluminescence images showing time-dependent growth of schwannoma in sciatic nerve and growth delay of tumors that have been treated with bevacizumab. D, tumor sizes were expressed as the total number of photon flux. Both vandetanib and bevacizumab significantly reduced HEI193 tumor size, and it seemed that bevacizumab produced a more long-term suppression (left graph). Saline, n = 9; bevacizumab, n = 8; vandetanib, n = 7. *, P = 1.175 × 10−6; **, P = 5.988 × 10−5; ***, P = 0.008. Vandetanib had a similar effect on Nf2−/− (right graph). 1% Tween 80, n = 3; vandetanib, n = 4. #, P = 0.0076.
Figure 4.
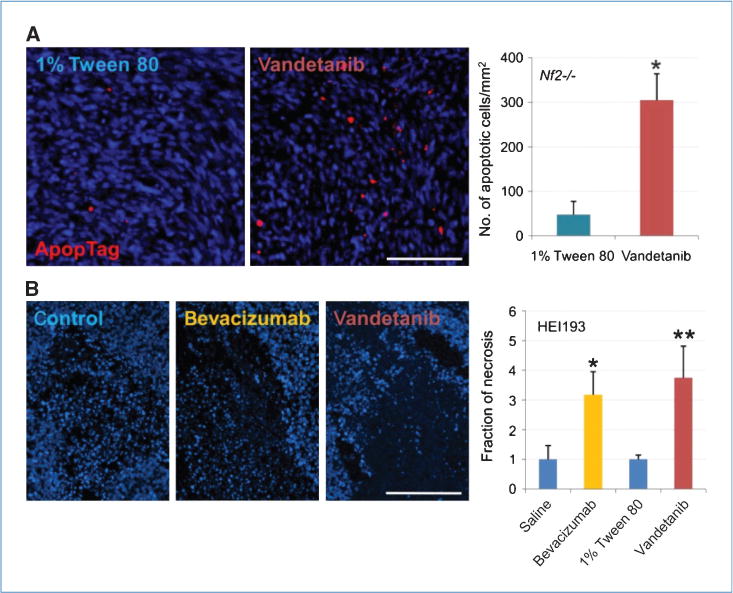
Inhibition of VEGF pathway increased tumor cell death. A, vandetanib increased Nf2−/− cell apoptosis by 6-fold 6 days after treatment, revealed by ApopTag. n = 4 in both control and treated. *, P = 6.946 × 10−5. B, bevacizumab increased necrotic fraction of tumor instead, there being a 3-fold increase 6 days after treatment. n = 4 in control and the treated. *, P = 0.0119; **, P = 0.0174. Scale bar, 100 μm.
Overall survival
Human vestibular schwannomas can be life threatening if they compress vital structures such as the brain stem. Indeed, the ability of the brain to absorb increases in volume is much more limited than other organs. In a similar fashion, tumors implanted within the cranium of nude mice have a strong effect on their survival. Kaplan-Meier curves showed that inhibiting the VEGF pathway in both schwannoma cell lines had a significant effect on overall survival. Bevacizumab extended survival by 95% in nude mice bearing HEI193 tumors, whereas vandetanib increased survival by 48% in mice bearing Nf2−/− tumors (Fig. 5).
Figure 5.
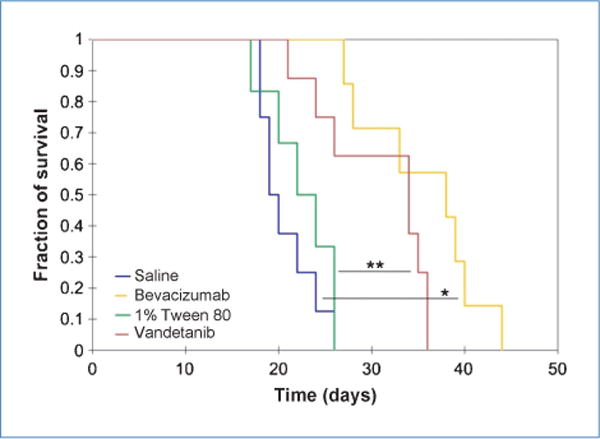
Both vandetanib and bevacizumab markedly extended survival of mice bearing brain schwannoma. Median survival for saline, 19.5 days (n = 8); for bevacizumab, 38 days (n = 7); for 1% Tween 80, 23 days (n = 6); and for vandetanib, 34 days (n = 8). *, P < 0.001; **, P = 0.016, log-rank test.
Improved function and morphology contribute to slower tumor growth
We recently showed that vascular permeability can play a role in overall survival in mice bearing tumors growing within the cranium (32). Treatment of Nf2−/− tumors with vandetanib decreased vessel permeability to BSA by 69.3% at 24 hours and by 74.9% after 6 days (Fig. 6A). Similarly, bevacizumab reduced HEI193 vessel permeability by 43% 24 hours after the first injection. Again, the effect was sustained, with permeability reduced by 51.7% 6 days after treatment (Fig. 6B). The stronger effect seen in the murine Nf2−/− tumors treated with vandetanib is most likely due to a more complete inhibition because bevacizumab is unable to block the host-derived VEGF. In addition, EGFR can be expressed by tumor endothelium (33) and the dual blockade induced by vandetanib might be beneficial. These functional changes were accompanied by morphologic changes of the vessels. Analysis of the tumor vessels in the sciatic nerve (the slower-growing model) showed a 40% increase in perivascular coverage (defined as PDGFRβ-positive perivascular cells) in HEI193 tumors treated with either bevacizumab or vandetanib (Fig. 6C) and a 60% increase in pericyte coverage in Nf2−/− tumors that were treated with vandetanib (Fig. 6D).
Figure 6.
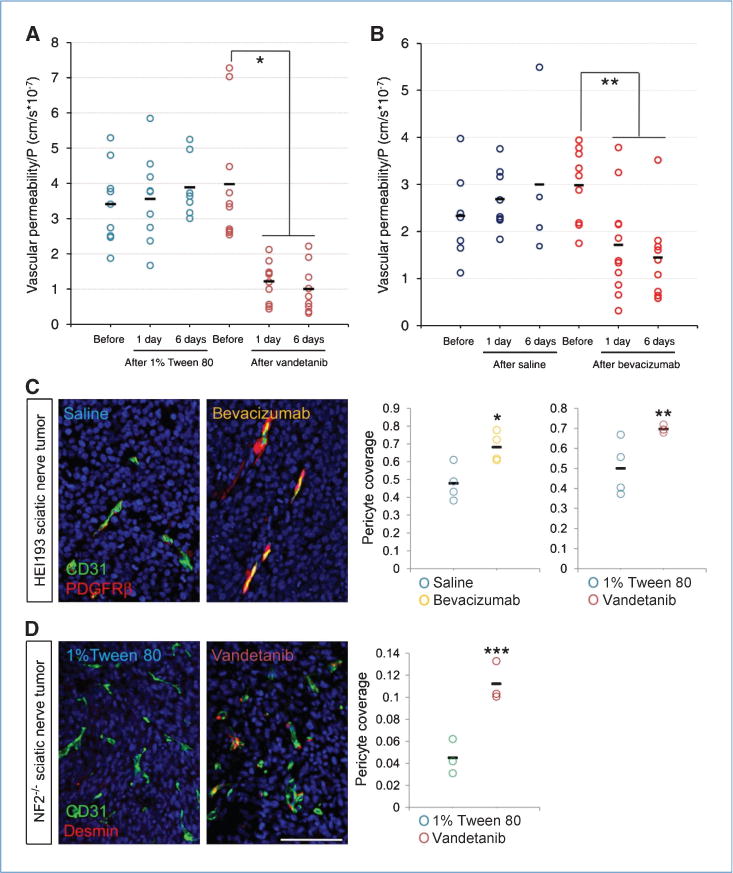
Inhibition of VEGF pathway improved function and recovered molecular composition of schwannoma vessels. A and B, both vandetanib and bevacizumab reduced vessel leakiness to rho-BSA 24 hours after treatment and the suppression was maintained for up to 6 days of treatment. For 1% Tween 80: before, n = 9; 1 day, n = 9; and 6 days, n = 7; for vandetanib: before, n = 10; 1 day, n = 11; and 6 days, n = 9; for saline: before, n = 8; 1 day, n = 8; and 6 days, n = 4; for bevacizumab: before, n = 9; 1 day, n = 11; and 6 days, n = 9. *, P < 0.001; **, P = 0.01, analyzed by ANOVA Fisher’s F test. C and D, anti-VEGF treatment increased pericyte coverage of schwannoma blood vessels, defined by PDGFRβ in HEI193 (C) and desmin in Nf2−/− (D) at day 6. All n = 4, except G = 3. *, P = 0.0194; **, P = 0.0303; ***, P = 0.0082. Scale bar, 100 μm.
Interestingly, we did not observe changes in basement membrane thickness (defined as collagen IV thickness) in either cranial or sciatic nerve schwannomas (data not shown). This is very different from the “classic” normalization seen in brain tumors treated with anti-VEGF therapies (34). Whether the normalization mechanism (35) is different in benign tumors or whether this observation is specific to Schwann cells warrants further study.
Discussion
Although most cancer patients treated with anti-VEGF monotherapy have derived some benefit in progression-free survival, it generally has had little effect on the overall survival in malignant diseases (36). In a retrospective series of 10 patients with benign vestibular schwannomas treated with bevacizumab, 6 of 10 patients had a radiographic response and 4 of 10 had a sustained response (at least a 20% decrease in tumor volume and/or significant improvement in word recognition; ref. 8). Six of the patients have been on treatment for >12 months (longest treatment period: 18 months) and no grade 3 or 4 adverse events have been reported. These promising results suggest that anti-VEGF therapy might be more effective in treating benign NF2-related tumors than for malignant disease. Thus, it is essential to understand the biological mechanisms of response to guide treatment properly. To this end, we established two animal models with relevance to the microenvironment of human tumors.
Here, we showed that inhibition of VEGF or its receptor modified the vascular network in a sustained fashion. Independent of the tumor type or the site of tumor implantation, the number of vessels was significantly decreased by 6 days of treatment and the remaining vessels normalized morphologically (decreased in diameter) and functionally (increased pericyte coverage and reduced permeability), ultimately leading to decreased tumor growth rate and increased survival. One potential explanation for the profound response of benign tumors to anti-VEGF therapy lies in their inability to invade the surrounding tissue. Cancer cells that cannot migrate to co-opt host vessels are completely dependent on vessel recruitment and, thus, more sensitive to antiangiogenic therapy.
Schwannomas typically progress slowly and are not considered to be angiogenic tumors. However, a recent review of surgical archival specimen revealed a proangiogenic profile defined by number and size of vessels combined with quantification of VEGF expression and SEMA3F loss (8). In this study, we found that loss of the Nf2 gene was accompanied by the loss of SEMA3 and that treatment with anti-VEGF therapies resulted in a sustained decreased growth rate and vessel permeability. This suggests that VEGF might have a different effect or mechanism of action in the context of peripheral nerve tumors.
Although permeability in schwannomas is supposed to be of minor importance, because the eighth cranial nerve is not within the blood-brain barrier, a blood-nerve barrier has been shown (37). Indeed, the permeability coefficient of various electrolytes, nonelectrolytes, perm selectivity, and transporters between cerebral and endoneurial capillaries is very similar (38), implying that anti-VEGF therapies are very likely modulating the permeability in the nerve as well. Therefore, controlling the extent of permeability and subsequent edema might still contribute to the overall clinical benefits. Indeed, nerve edema is an important component of Wallerian degeneration (39). One of the most important forms of nerve edema is induced by external compression, similar to the one that can be induced by tumor growth (40). Control of permeability has been beneficial in improving the condition of patients with diabetic polyneuropathy (41). It is therefore logical to hypothesize that anti-VEGF might have a dual effect by (a) decreasing the induction of angiogenesis, thereby slowing rate of growth, and (b) decreasing the nerve edema, ultimately slowing down nerve degeneration and restoring normal physiology. These factors may explain the benefit seen in hearing ability in patients (8).
In conclusion, Nf2 loss increases the proliferation rate of Schwann cells but with slow, more gradual progression compared with malignant tumors. Schwannomas are not typically necrotic and are therefore less likely to trigger hypoxia-driven overexpression of VEGF. Instead, we propose that the loss of merlin may result in loss of semaphorin, tipping the angiogenic balance toward angiogenesis. The loss of neurofibromin in NF1-derived tumors has also been linked to increased angiogenesis, but there is still relatively little information on the role of angiogenesis in NF2-derived tumors and the putative relationship between merlin and angiogenic pathways.
Our study supports the hypothesis that decreasing vascular supply and normalizing the remaining vessels can induce a growth delay and extend survival. The preclinical models validated in this study could now be used to screen potential new class of inhibitors, chosen based on our understanding of the molecular evolution of these tumors. We found in these benign tumors that anti-VEGF therapy seems to follow the original hypothesis proposed by Folkman that stopping vessel recruitment significantly slows tumor growth. However, there is still a lack of understanding of the angiogenesis process in these NF2 tumors. Clinically, they can be stable for long intervals before entering periods of growth; identification and characterization of the hypothetical “angiogenic switch” responsible for their progression may help improve patient monitoring and lead to new treatments.
Supplementary Material
Acknowledgments
We thank Sylvie Roberge for her technical expertise, Yannis Perentes for his critical input in developing the sciatic nerve model, Dr. Xandra Breakefield for her generous gift of HEI193, the molecular neuropathology core for the luciferase mCherry transfection, and Lance Munn and Ned Kirkpatrick for their critical review of the manuscript.
Grant Support
Claflin Award (E. di Tomaso), Drug Discovery Initiative Award from Children’s Tumor Foundation (E. di Tomaso), Damon Runyon Cancer Research Foundation (J. Lahdenranta), The Flight Attendant Medical Research Institute (A.W. Chan), The Federal Share/NCI Proton Beam Program income grants (E. di Tomaso and R.K. Jain), and PO1CA80124 (R.K. Jain).
Footnotes
Disclosure of Potential Conflicts of Interest
S.R. Plotkin: commercial research grant, PTC Therapeutic and Pfizer. R.K. Jain: commercial research grant, AstraZeneca and Dyax; consultant/advisory board, AstraZeneca, Dyax, Enlight, and Millenium; lecture fee from Roche Pharmaceutical. The other authors disclosed no potential conflicts of interest.
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
References
- 1.Giovannini M, Robanus-Maandag E, van der Valk M, et al. Conditional biallelic Nf2 mutation in the mouse promotes manifestations of human neurofibromatosis type 2. Genes Dev. 2000;14:1617–30. [PMC free article] [PubMed] [Google Scholar]
- 2.Harner SG, Laws ER., Jr Diagnosis of acoustic neurinoma. Neurosurgery. 1981;9:373–9. doi: 10.1227/00006123-198110000-00004. [DOI] [PubMed] [Google Scholar]
- 3.Mrugala MM, Batchelor TT, Plotkin SR. Peripheral and cranial nerve sheath tumors. Curr Opin Neurol. 2005;18:604–10. doi: 10.1097/01.wco.0000179507.51647.02. [DOI] [PubMed] [Google Scholar]
- 4.Caye-Thomasen P, Baandrup L, Jacobsen GK, Thomsen J, Stangerup SE. Immunohistochemical demonstration of vascular endothelial growth factor in vestibular schwannomas correlates to tumor growth rate. Laryngoscope. 2003;113:2129–34. doi: 10.1097/00005537-200312000-00014. [DOI] [PubMed] [Google Scholar]
- 5.Caye-Thomasen P, Werther K, Nalla A, et al. VEGF and VEGF receptor-1 concentration in vestibular schwannoma homogenates correlates to tumor growth rate. Otol Neurotol. 2005;26:98–101. doi: 10.1097/00129492-200501000-00017. [DOI] [PubMed] [Google Scholar]
- 6.Uesaka T, Shono T, Suzuki SO, et al. Expression of VEGF and its receptor genes in intracranial schwannomas. J Neurooncol. 2007;83:259–66. doi: 10.1007/s11060-007-9336-0. [DOI] [PubMed] [Google Scholar]
- 7.Koutsimpelas D, Stripf T, Heinrich UR, Mann WJ, Brieger J. Expression of vascular endothelial growth factor and basic fibroblast growth factor in sporadic vestibular schwannomas correlates to growth characteristics. Otol Neurotol. 2007;28:1094–9. doi: 10.1097/MAO.0b013e31814b2787. [DOI] [PubMed] [Google Scholar]
- 8.Plotkin SR, Stemmer-Rachamimov AO, Barker FG, II, et al. Hearing improvement after bevacizumab in patients with neurofibromatosis type 2. N Engl J Med. 2009;361:358–67. doi: 10.1056/NEJMoa0902579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klagsbrun M, Eichmann A. A role for axon guidance receptors and ligands in blood vessel development and tumor angiogenesis. Cytokine Growth Factor Rev. 2005;16:535–48. doi: 10.1016/j.cytogfr.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 10.Klagsbrun M, Takashima S, Mamluk R. The role of neuropilin in vascular and tumor biology. Adv Exp Med Biol. 2002;515:33–48. doi: 10.1007/978-1-4615-0119-0_3. [DOI] [PubMed] [Google Scholar]
- 11.Bielenberg DR, Hida Y, Shimizu A, et al. Semaphorin 3F, a chemorepulsant for endothelial cells, induces a poorly vascularized, encapsulated, nonmetastatic tumor phenotype. J Clin Invest. 2004;114:1260–71. doi: 10.1172/JCI21378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Willett CG, Boucher Y, di Tomaso E, et al. Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat Med. 2004;10:145–7. doi: 10.1038/nm988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Willett CG, Boucher Y, Duda DG, et al. Surrogate markers for antiangiogenic therapy and dose-limiting toxicities for bevacizumab with radiation and chemotherapy: continued experience of a phase I trial in rectal cancer patients. J Clin Oncol. 2005;23:8136–9. doi: 10.1200/JCO.2005.02.5635. [DOI] [PubMed] [Google Scholar]
- 14.Batchelor TT, Sorensen GA, di Tomaso E, et al. AZD2171, a pan-VEGF and PDGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates vasogenic edema in glioblastoma patients. Cancer Cell. 2007;11:83–95. doi: 10.1016/j.ccr.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rich JN, Sathornsumetee S, Keir ST, et al. ZD6474, a novel tyrosine kinase inhibitor of vascular endothelial growth factor receptor and epidermal growth factor receptor, inhibits tumor growth of multiple nervous system tumors. Clin Cancer Res. 2005;11:8145–57. doi: 10.1158/1078-0432.CCR-05-0319. [DOI] [PubMed] [Google Scholar]
- 16.Ryan AJ, Wedge SR. ZD6474—a novel inhibitor of VEGFR and EGFR tyrosine kinase activity. Br J Cancer. 2005;92(Suppl 1):S6–13. doi: 10.1038/sj.bjc.6602603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pan Q, Chanthery Y, Liang WC, et al. Blocking neuropilin-1 function has an additive effect with anti-VEGF to inhibit tumor growth. Cancer Cell. 2007;11:53–67. doi: 10.1016/j.ccr.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 18.Wedge SR, Ogilvie DJ, Dukes M, et al. ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Res. 2002;62:4645–55. [PubMed] [Google Scholar]
- 19.Hung G, Li X, Faudoa R, et al. Establishment and characterization of a schwannoma cell line from a patient with neurofibromatosis 2. Int J Oncol. 2002;20:475–82. [PubMed] [Google Scholar]
- 20.Prabhakar S, Messerli SM, Stemmer-Rachamimov AO, et al. Treatment of implantable NF2 schwannoma tumor models with oncolytic herpes simplex virus G47ΔHSV vector treatment of schwannomas. Cancer Gene Ther. 2007;14:460–7. doi: 10.1038/sj.cgt.7701037. [DOI] [PubMed] [Google Scholar]
- 21.Winkler F, Kozin SV, Tong R, et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: role of oxygenation, angiopoietin-1 and matrix metalloproteinases. Cancer Cell. 2004;6:553–63. doi: 10.1016/j.ccr.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 22.Lee JK, Sobel RA, Chiocca EA, Kim TS, Martuza RL. Growth of human acoustic neuromas, neurofibromas and schwannomas in the subrenal capsule and sciatic nerve of the nude mouse. J Neurooncol. 1992;14:101–12. doi: 10.1007/BF00177613. [DOI] [PubMed] [Google Scholar]
- 23.Tyrrell JA, di Tomaso E, Fuja D, et al. Robust 3-D modeling of vasculature imagery using superellipsoids. IEEE Trans Med Imaging. 2007;26:223–37. doi: 10.1109/TMI.2006.889722. [DOI] [PubMed] [Google Scholar]
- 24.Yuan F, Chen Y, Dellian M, Safabakhsh N, Ferrara N, Jain RK. Time-dependent vascular regression and permeability changes in established human tumor xenografts induced by an anti-vascular endothelial growth factor/vascular permeability factor antibody. Proc Natl Acad Sci U S A. 1996;93:14765–70. doi: 10.1073/pnas.93.25.14765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang YS, di Tomaso E, McDonald DM, Jones R, Jain RK, Munn LL. Mosaic blood vessels in tumors: frequency of cancer cells in contact with flowing blood. Proc Natl Acad Sci U S A. 2000;97:14608–13. doi: 10.1073/pnas.97.26.14608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.di Tomaso E, London N, Fuja D, et al. PDGF-C induces maturation of blood vessels in a model of glioblastoma and attenuates the response to anti-VEGF treatment. PLoS One. 2009;4:e5123. doi: 10.1371/journal.pone.0005123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Plotkin SR, Singh MA, O’Donnell CC, Harris GJ, McClatchey AI, Halpin C. Audiologic and radiographic response of NF2-related vestibular schwannoma to erlotinib therapy. Nat Clin Pract Oncol. 2008;5:487–91. doi: 10.1038/ncponc1157. [DOI] [PubMed] [Google Scholar]
- 28.Sathornsumetee S, Rich JN. Vandetanib, a novel multitargeted kinase inhibitor, in cancer therapy. Drugs Today (Barc) 2006;42:657–70. doi: 10.1358/dot.2006.42.10.1025318. [DOI] [PubMed] [Google Scholar]
- 29.Curto M, Cole BK, Lallemand D, Liu C-H, McClatchey AI. Contact-dependent inhibition of EGFR signaling by Nf2/Merlin. J Cell Biol. 2007;177:893–903. doi: 10.1083/jcb.200703010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maher PA, Schubert D. Schwannoma-derived growth factor interacts with the epidermal growth factor receptor. J Neurochem. 1995;65:1895–8. doi: 10.1046/j.1471-4159.1995.65041895.x. [DOI] [PubMed] [Google Scholar]
- 31.Scoles DR, Qin Y, Nguyen V, Gutmann DH, Pulst SM. HRS inhibits EGF receptor signaling in the RT4 rat schwannoma cell line. Biochem Biophys Res Commun. 2005;335:385–92. doi: 10.1016/j.bbrc.2005.07.083. [DOI] [PubMed] [Google Scholar]
- 32.Kamoun WS, Ley CD, Farrar CT, et al. Edema control by cediranib, a vascular endothelial growth factor receptor-targeted kinase inhibitor, prolongs survival despite persistent brain tumor growth in mice. J Clin Oncol. 2009;27:2542–52. doi: 10.1200/JCO.2008.19.9356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Amin DN, Bielenberg DR, Lifshits E, Heymach JV, Klagsbrun M. Targeting EGFR activity in blood vessels is sufficient to inhibit tumor growth and is accompanied by an increase in VEGFR-2 dependence in tumor endothelial cells. Microvasc Res. 2008;76:15–22. doi: 10.1016/j.mvr.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 34.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 35.Tong RT, Boucher Y, Kozin SV, Winkler F, Hicklin DJ, Jain RK. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res. 2004;64:3731–6. doi: 10.1158/0008-5472.CAN-04-0074. [DOI] [PubMed] [Google Scholar]
- 36.Jain RK, Duda DG, Willett CG, et al. Biomarkers of response and resistance to antiangiogenic therapy. Nat Rev Clin Oncol. 2009;6:327–38. doi: 10.1038/nrclinonc.2009.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rechthand E, Rapoport SI. Regulation of the microenvironment of peripheral nerve: role of the blood-nerve barrier. Prog Neurobiol. 1987;28:303–43. doi: 10.1016/0301-0082(87)90006-2. [DOI] [PubMed] [Google Scholar]
- 38.Wadhwani KC, Rapoport SI. Transport properties of vertebrate blood-nerve barrier: comparison with blood-brain barrier. Prog Neurobiol. 1994;43:235–79. doi: 10.1016/0301-0082(94)90002-7. [DOI] [PubMed] [Google Scholar]
- 39.Mizisin AP, Kalichman MW, Myers RR, Powell HC. Role of the blood-nerve barrier in experimental nerve edema. Toxicol Pathol. 1990;18:170–85. doi: 10.1177/019262339001800123. [DOI] [PubMed] [Google Scholar]
- 40.Padera TP, Stoll BR, Tooredman JB, Capen D, di Tomaso E, Jain RK. Pathology: cancer cells compress intratumour vessels. Nature. 2004;427:695. doi: 10.1038/427695a. [DOI] [PubMed] [Google Scholar]
- 41.Poduslo JF, Curran GL. Increased permeability across the blood-nerve barrier of albumin glycated in vitro and in vivo from patients with diabetic polyneuropathy. Proc Natl Acad Sci U S A. 1992;89:2218–22. doi: 10.1073/pnas.89.6.2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.