

Abstract
The advances of genomics, sequencing, and high throughput technologies have led to the creation of large volumes of diverse datasets for drug discovery. Analyzing these datasets to better understand disease and discover new drugs is becoming more common. Recent open data initiatives in basic and clinical research have dramatically increased the types of data available to the public. The past few years have witnessed successful use of big data in many sectors across the whole drug discovery pipeline. In this review, we will highlight the state of the art in leveraging big data to identify new targets, drug indications, and drug response biomarkers in this era of precision medicine.
In 2013, the European Bioinformatics Institute hosted 15 petabytes data in their shared file systems.1 This increased to 25 petabytes in 2014, which is equal to the hard drive space of over 12,000 current‐day typical personal laptops (each with a 2 terabyte drive). These data were distributed in over 120,000 datasets available for searching and analysis in 2014. As voluminous as this data sounds, these numbers simply reflect the complexity and growth of the data from one single institute.
This growth in the digitalization of biomedical research is due to the advances and decreasing costs of genomics, sequencing, and the increasing use of high throughput technologies in the research enterprise. Large volumes of biomedical data are being produced every day, and much of these data are actually now becoming publicly available, owing to the initiatives of open data. Although the field of biomedical informatics is facing challenges in the storage and management of these datasets, this field is also embracing more exciting opportunities in the discovery of new knowledge from these data.2 Big datasets are now not only routinely analyzed to inform discovery and validate hypothesis, but also frequently repurposed to ask new biomedical questions. However, researchers are facing so many datasets that sometimes it is difficult to choose the appropriate one for their studies. In this review, we will first describe the data types commonly used in drug discovery and then list datasets publicly available. We will highlight some remarkable datasets that led to the discovery of new targets, drugs, or drug response biomarkers.
WHAT BIG DATA ARE AVAILABLE FOR DRUG DISCOVERY?
Drug discovery often starts with the classification and understanding of disease processes, followed by target identification and lead compound discovery. One trend of disease classification in drug discovery is moving from a symptom‐based disease classification system to a system of precision medicine based on molecular states.3, 4 Building a new classification of diseases requires molecular characterization of all diseases. In addition, an ideal level of disease understanding would characterize all levels of molecular changes, from DNA to RNA to protein, as well as the effects of environmental factors.
Each level of molecular change can be characterized by the analysis of relevant data points. Table 1 lists the data types frequently used in drug discovery and their current relevant technologies. At the DNA level, single‐nucleotide polymorphisms (SNPs) that occur specifically in the disease population is one type of DNA sequence variation widely used to characterize disease. Copy number variations (CNVs) reflect relatively large regions of genome alterations, which may be also associated with disease. Both SNPs and CNVs can be identified from the genome‐wide association studies (GWASs) and whole genome sequencing approaches. Mutations, particularly somatic mutations, are widely examined using next generation sequencing to find driver genes in cancer that confer a selective growth advantage of cells.
Table 1.
Common data types for drug discovery
| Data type | Description | Common techniques | Public availabilitya |
|---|---|---|---|
| SNP | A single nucleotide variation in a genetic sequence | SNP array: most widely used | **** |
| Whole genome sequencing | |||
| CNV | Variation of the number of copies of a particular gene in the genetic sequence | SNP array: most widely used; less sample DNA required; high probe density and coverage | **** |
| Comparative genome hybridization: high sensitivity and specificity; low spatial resolution | |||
| Whole genome sequencing: can detect smaller CNVs and novel types (e.g., inversions) | |||
| Mutation | A permanent change of the nucleotide sequence of the DNA; mostly somatic mutation that occurs in any of the cells except the germ cells | Whole exome sequencing: most widely used | **** |
| Whole genome sequencing: more expensive and more coverage | |||
| Gene expression | Mostly expression of mRNA but also includes expression of other transcripts | Microarray: most widely used | ***** |
| RNA‐Seq: can detect novel transcripts, low abundant transcripts and isoforms | |||
| Fluorescent in situ hybridization: can detect transcript abundance and spatial location in cells for a small number of genes | |||
| RT‐PCR: frequently used to confirm expression for a small number of genes | |||
| Protein expression | Can be expression of multiple isoforms or variations due to posttranslational modifications | Western blot: widely used to quantify protein expression for a small number of proteins | *** |
| ELISA: widely used to detect and quantitatively measure a protein in samples | |||
| Immunohistochemistry: can detect intracellular localization for a small number of proteins | |||
| Reverse phase protein array: can detect expression for a few hundred proteins | |||
| Mass spectrometry: can detect expression for a wide range of proteins | |||
| Protein‐protein interaction | Physical interactions between two or more proteins | Two‐hybrid screening: low‐tech; high false‐positive rate | **** |
| Mass spectrometry | |||
| Protein‐DNA interaction | Binding of a protein to a molecule of DNA | ChIP‐seq: combines chromatin immunoprecipitation with massively parallel DNA sequencing to identify the binding sites of DNA‐associated proteins | *** |
| Gene silencing | Effect of loss of gene function | RNAi: established method; knocks gene down at mRNA or non‐coding RNA level; can have transient effect (siRNA) or long‐term effect (shRNA) | ** |
| CRISPR‐Cas9: new method; modifies gene (via knockout/knockin) at the DNA level; causes permanent and heritable changes in the genome | |||
| Gene overexpression | Effect of gain of gene function | cDNAs/ORFs: provide clones of sequence | * |
| Drug efficacy | Effect of drug treatment; primarily represented as IC50/EC50/GI50 in vitro | HTS: rapidly assess the activity of a large number of compounds in biochemical assays or cell‐based assays | *** |
| MTT assay: often used to confirm activity for a small number of compounds | |||
| Drug‐target interaction | Physical interaction between a drug and a protein target | Affinity chromatography with mass spectrometry: most sensitive and unbiased method | *** |
| SPR | |||
| EMR/EHR | Patient response upon interventions | Digitalization | * |
CNV, copy number variation; CRISPR, clustered regularly interspaced short palindromic repeats; ELISA, enzyme‐linked immunosorbent assay; EMR/HER, electronic medical/health records; HTS, high throughput screening; MTT, methylthiazol tetrazolium; RT‐PCR, real‐time polymerase chain reaction; SNP, single‐nucleotide polymorphism; SPR, surface plasmon resonance.
Indicates the degree of public availability. For example, ***** shows researchers could easily access this type of data via public portals.
At the RNA level, gene expression (primarily mRNA) is arguably the most widely used feature for disease characterization. It has been used extensively to understand disease mechanism owing to the development of the microarray technology. The recent development of RNA‐Seq presents merits in the expanded coverage of transcripts and in the detection of low abundant transcripts.5 Protein expression is another critical feature used to characterize disease. Large‐scale quantification of protein expression is becoming possible recently because of the emerging new high throughput technologies, such as reverse‐phase protein arrays and mass spectrometry, although their coverage and quality remain limited. The interactions among DNA, RNA, and protein can be captured by ChIP‐Seq, mass spectrometry, and other techniques; however, most of those interactions have been captured only in cell lines or other in vitro models. More recently, next generation sequencing approaches have allowed sequencing environmental factors, such as microbial cells in the human body.
Today, a snapshot of the molecular changes in disease can be quickly modeled by the variety of datasets collected using multiple techniques. The recent development of single cell sequencing adds another layer of molecular changes. The number of layers dramatically increases as we consider the dynamic process of disease progression. Moreover, other than disease samples from patients, diverse preclinical models (e.g., cell lines, animal models) could be molecularly characterized in order to understand disease and validate hypothesis.
On the drug side, molecular changes in disease models perturbed by chemical or genetic agents can be captured to understand disease and drug mechanism. Gene function and gene regulatory networks can be studied via genome‐wide functional screens, such as RNAi and clustered regularly interspaced short palindromic repeats (CRISPR)‐Cas9.6 In addition, cellular responses of thousands of chemical compounds in a large of number of disease models can be quickly detected by high throughput screening. Patient response upon drug intervention can be tracked and analyzed recently owing to the availability of electronic medical records (EMRs) and clinical trials. In addition to the molecular and clinical data, free‐text data presented in literature are also useful in drug discovery.
WHAT BIG DATA SOURCES ARE PUBLICLY AVAILABLE?
No single laboratory, institute, or consortium is able to produce the data fully capturing all the layers of the complex disease systems. In addition, understanding of these systems relies on a large number of samples, such that statistical power could be reached. Integrative analysis of multiple layers of data points from different sources is thus essential to understand disease and discover new drugs. Hence, it is of utmost importance that the data should be open to the public, such that every piece of information can be easily connected.
Many important reference datasets have recently been created and released, and can be used for drug discovery.7 Notable examples are listed in Table 2. Arguably, public datasets can be used to inform every step of preclinical drug discovery. Clinical datasets are becoming increasingly open as well.8 Figure 1 shows a list of public datasets that can be leveraged to identify new targets, drug indications, and drug response biomarkers. Not only have public datasets been widely used as a source of reference, but also they have been intensively analyzed to ask new questions, discover new findings, or even validate hypothesis. In this study, we selectively review some outstanding cases in the past few years in which discoveries were made primarily through the analysis of big data and validated rigorously through experimental approaches.
Table 2.
Common public databases for drug discovery
| Database | Description (as of October 2015) | URL |
|---|---|---|
| dbSNP | SNPs for a wide range of organisms, including >150M human reference SNPs. | http://www.ncbi.nlm.nih.gov/snp |
| dbVar | Genomic structural variations (primarily CNVs) generated mostly by published studies of various organisms, including >2.1M human CNVs. | http://www.ncbi.nlm.nih.gov/dbvar |
| COSMIC | Primarily somatic mutations from expert curation and genome‐wide screening, including >3.5M coding mutations. | http://cancer.sanger.ac.uk/cosmic |
| 1000 Genomes Project | Genomes of a large number of people to provide a comprehensive resource on human genetic variation, including >2.5K samples. | http://www.1000genomes.org |
| TCGA | Genomics and functional genomics data repository for >30 cancers across >10K samples. Primary data types include mutation, copy number, mRNA, and protein expression. | https://tcga‐data.nci.nih.gov/tcga |
| GEO | Functional genomics data repository hosted by NCBI, including >1.6M samples. | http://www.ncbi.nlm.nih.gov/geo |
| ArrayExpress | Functional genomics data repository hosted by EBI, including >1.8M samples. | https://www.ebi.ac.uk/arrayexpress |
| GTEx | Transcriptomic profiles of normal tissues, including >7K samples across >45 tissue types. | http://www.gtexportal.org |
| CCLE | Genetic and pharmacologic characterization of >1,000 cancer cell lines. | http://www.broadinstitute.org/ccle |
| Human Protein Atlas | Expression of >17K unique proteins in cell lines, normal, and cancer tissues. | http://www.proteinatlas.org |
| Human Proteome Map | Expression of >30K proteins in normal tissues. | http://humanproteomemap.org |
| StringDB | Protein‐protein interactions for >9M proteins from >2K organisms. | http://string‐db.org |
| ENCODE | Protein‐DNA interactions, including >1.4K ChIP‐Seq experiments across ∼200 cell lines. | http://genome.ucsc.edu/ENCODE |
| Project Achilles | Genetic vulnerabilities across >100 genomically characterized cancer cell lines by genome‐wide genetic perturbation reagents (shRNAs or Cas9/sgRNAs), including >11.2K genes. | http://www.broadinstitute.org/achilles |
| LINCS | Cellular responses upon the treatment of chemical/genetic perturbagen, including >1M gene expression profiles representing >5,000 compounds and >3,500 genes (shRNA and overexpression) in >15 cell lines. | http://lincscloud.org |
| Genomics of Drug Sensitivity in Cancer project | Drug sensitivity data of 140 drugs in >700 cancer cell lines. | http://www.cancerrxgene.org |
| ChEMBL | Bioactivities for drug‐like small molecules, including >10K targets, >1.7M distinct compounds, and >13.5M activities. | https://www.ebi.ac.uk/chembl |
| PubChem | Chemical compounds and bioassay experiments, including >60M unique chemical compounds and >1.1M assays. | http://pubchem.ncbi.nlm.nih.gov |
| CMap | >6,000 drug gene expression profiles representing 1,309 compounds tested in 3 main cell lines. | http://www.broadinstitute.org/cmap |
| CTRP | Links genetic, lineage, and other cellular features of cancer cell lines to small‐molecule sensitivity, including 860 cell lines and 461 compounds. | http://www.broadinstitute.org/ctrp.v2.2 |
| ImmPort | Clinical assessments in immunology along with molecular profiles, including 143 clinical studies/trials and 799 experiments on >22.4K subjects. | https://immport.niaid.nih.gov |
| ClinicalTrials.gov | Registry and results database of publicly and privately supported clinical studies, including >201.7K studies. | https://clinicaltrials.gov |
| PharmGKB | Genetic variations on drug response, including >3K diseases, >27K genes, and >3K drugs. | https://www.pharmgkb.org |
CCLE, Cancer Cell Line Encyclopedia; CMap, Connectivity Map; CNVs, copy number variants; COSMIC, catalog of somatic mutations in cancer; CTRP, Cancer Therapeutics Response Portal; dbSNP, Single Nucleotide Polymorphism Database; dbVar, database of genomic structural variation; EBI, European Bioinformatics Institute; ENCODE, Encyclopedia of DNA Elements; GEO, Gene Expression Omnibus; GTEx, Genotype‐Tissue Expression; IMMPORT, Immunology Database and Analysis Portal; LINCS, Library of Integrated Network‐based Cellular Signatures; NCBI, National Center for Biotechnology Information; SNPs, single‐nucleotide polymorphisms; TCGA, The Cancer Genome Atlas.
Figure 1.
Public datasets can be leveraged to identify new targets, drug indications, and drug response biomarkers.
LEVERAGE BIG DATA TO IDENTIFY NEW TARGETS FOR PRECLINICAL STUDIES
Using big data to select targets for preclinical studies often starts with the identification of molecular changes between disease samples and healthy samples. The molecular changes are implicated in gene expression change, genetic variation, or other features, and are furthermore used to inform target discovery. Figure 2 illustrates three common big data approaches that use different molecular features to discover targets, and basic experimental approaches to validate targets. We will first discuss these three approaches and then suggest that public datasets can be used to validate targets before time‐consuming experiments.
Figure 2.
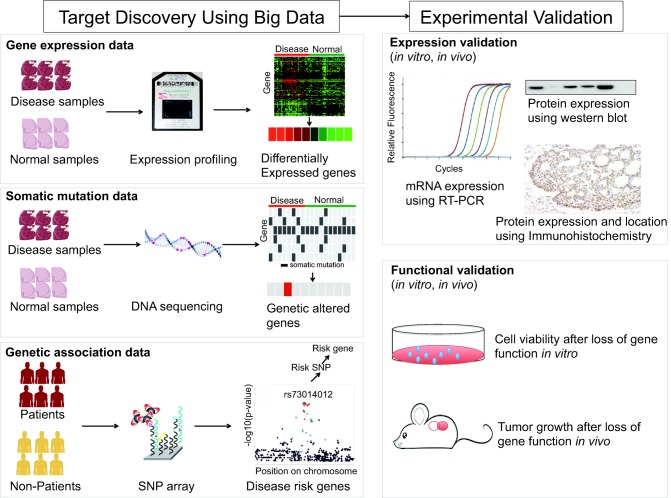
An illustration of big data approaches to identifying new targets.
Target discovery using gene expression data
Among molecular features, gene expression is the most widely used feature and has been extensively explored to inform target selection. As an example, Grieb et al.9 found that MTBP was significantly elevated in breast cancer samples compared with normal breast tissues by examining mRNA expression of 844 breast cancer samples from The Cancer Genome Atlas (TCGA). Analysis of survival data revealed that increased MTBP levels are significantly linked with poor patient survival. They further stratified patients into clinically relevant subgroups: estrogen‐receptor positive, HER2 positive, and triple negative breast cancer (TNBC) tumors and observed that MTBP is expressed higher in the triple negative tumor subgroup than in the other two subgroups. Further knockdown of MTBP significantly impaired TNBC tumor growth in vivo. In another example, analysis of clear cell renal cell carcinoma samples from TCGA indicated that TPL2 overexpression was significantly related to the presence of metastases and poor outcome in clear cell renal cell carcinoma.10 Silencing of TPL2 inhibited cell proliferation, clonogenicity, anoikis resistance, migration, and invasion capabilities and inhibited orthotopic xenograft growth and lung metastasis, demonstrating the significant role of TPL2 in disease progression. In addition, public gene expression databases, such as TCGA, were regularly used as a source of reference to confirm gene expression. One example includes the confirmation of HMMR in the study of glioblastoma.11
The targets in these previous examples were first proposed by authors and were then confirmed by the analysis of public gene expression data. By contrast, targets can also be directly discovered through the primary analysis of gene expression data. Without any specific targets in mind, Hsu et al.12 sought for druggable kinases, which are oncogenic in TNBC. By analyzing gene expression data from CCLE and National Cancer Institute‐60 panel of cancer cell lines, and gene expression profiles of breast tumor initiating cells, they found 13 kinases with higher mRNA expression in TNBC cell lines than in non‐TNBC cell lines. Subsequent protein expression validation reduced the candidate list to eight kinases, which were further correlated to TNBC clinical subtype samples in TCGA. Among these eight kinases, three kinases (PKC‐α, CDK6, and MET) with high expression were associated with shorter overall survival in patients with TNBC, suggesting their potential as prognostic markers and therapeutic targets. In the subsequent functional validation, two‐drug combinations targeting these three kinases inhibited TNBC cell proliferation and tumorigenic potential and a combination of PKC‐α−MET inhibitors attenuated tumor growth in vivo.
Analyzing the samples from a single data source may limit the broader application of the findings because of biological and technical bias. Meta‐analysis that is aimed at detecting consistent changes across multiple data sources may increase statistical power and further mitigate the bias. The availability of public datasets enables researchers to perform meta‐analysis of microarray datasets for many diseases. Our colleagues Kodama et al.13 proposed a meta‐analysis approach: a gene expression‐based GWAS that searches for genes repeatedly implicated in multiple experiments. They carried out an expression‐based GWAS for type 2 diabetes by using 1,175 samples collected from 130 independent microarray experiments and identified the immune‐cell receptor CD44 as the top candidate. They further validated that CD44 deficiency ameliorated adipose tissue inflammation and insulin resistance and anti‐CD44 treatment decreased blood glucose levels and adipose macrophage infiltration. In another example, our colleagues Chen et al.14 analyzed 13 independent non‐small cell lung cancer (SCLC) gene expression datasets consisting of 2,026 lung samples collected from Gene Expression Omnibus (GEO). They identified 11 genes that were consistently overexpressed across all the samples, among which protein kinase PTK7 was found. Immunostaining revealed that PTK7 was highly expressed in primary adenocarcinoma patient samples. They verified that RNA interference‐mediated attenuation of PTK7 decreased cell viability and increased apoptosis in a subset of adenocarcinoma cell lines and loss of PTK7 impaired tumor growth in xenotransplantation assays, suggesting its potential as a novel therapeutic target in non‐SCLC.
Target discovery using somatic mutation data
Many complex diseases are caused by alterations of DNA sequences. Targeting genetic alterations is thus an ideal approach to find therapeutic solutions. Recent advances in DNA sequencing technologies enabled large‐scale characterization of disease samples. Analyzing molecular data of these samples plays an essential role in identifying alterations responsible for disease. TCGA is one notable example that molecularly characterized >10,000 tumor samples across over 30 cancers across multiple technologies.15 The large‐scale analysis of tumor samples suggested that an average of 33 to 66 genes harbor somatic mutations that could alter the function of their protein targets and ∼140 genes can promote tumorigenesis.16 Most human cancers are caused by two to eight sequential alterations that lead to a selective growth advantage of the cell where it resides.16 These alterations have been widely explored as therapeutic targets. Representative examples include EGFR amplification in lung cancer,17 BRAF mutation in melanoma,18 and ALK translocations in lung cancer.19
A cancer that possesses a genomic alteration may be treated by a drug that targets this alteration, even though this drug was not originally discovered for this tumor type. For instance, KIT was discovered as a target for chronic myelogenous leukemia and later it was discovered as a target in gastrointestinal stromal tumors, leading to the repositioning of the KIT inhibitor, Imatinib, for treating patients with KIT‐positive gastrointestinal stromal tumors.20 Rubio‐Perez et al.21 recently collected and analyzed somatic mutations, copy‐number alterations, fusion genes, and RNA‐Seq expression data of 4,068 tumors in 16 cancer types in TCGA and collected somatic mutations for 2,724 additional tumors. They identified 459 mutational driver genes and 38 drivers acting via copy‐number alterations or fusions. After mapping these driver genes to drug databases, including ChEMBL and ClinicalTrials.gov, they found that up to 73.3% of patients could benefit from agents in clinical stages. This in silico analysis showed the potential of targeting genomic alterations for individual tumors, yet experimental validation is expected for wide clinical applications. The recent launch of the National Cancer Institute‐Molecular Analysis for Therapy Choice program that aims to identify targets and therapeutics for individual patients solely based on mutations demonstrates a broad interest of this approach in target selection.
Analysis of genomic features from a wide range of cancers revealed that a large fraction of driver genes are either undruggable or are tumor suppressors, which usually cannot be interfered by drugs.16 Targeting their downstream or upstream‐dependent components may bypass this problem. For example, inactivating mutations of the tumor suppressors BRCA1 or BRCA2 lead to activation of a downstream pathway required to repair DNA damage. Poly ADP‐ribose polymerase, a family of protein involved in the DNA repair, was subsequently developed as a therapeutic target for those with absence of BRCA function.22 In addition to BRCA, defects in the DNA‐damage response, a complex network of proteins required for cell‐cycle checkpoint and DNA repair, have been associated with tumorigenesis, yet are undruggable. Squatrito et al.23 assessed genes encoding key components of the DNA‐damage response from the glioma samples in TCGA and found that 3.2% of these samples showed somatic mutations in ATR, ATM, or CHEK1 and 36% of these samples presented genomic loss of at least one copy of ATR, ATM, CHEK1, or CHEK2, suggesting tumor suppressor activity of the ATM/Chk2/p53 pathway. Further experiments confirmed that the loss of ATM/Chk2/p53 pathway components accelerate tumor development. Hence, it would be interesting to target the components involved in this pathway.
Target discovery using genetic association data
Recent GWASs have identified common DNA sequence variants that contribute to many human diseases. An increasing number of studies demonstrate that genes with disease‐associated alleles may be promising drug targets as shown by the list of targets validated by genetics.24 In one example, the analysis of patients with familial hypercholesterolemia reveals mutations in the low‐density lipoprotein receptor gene causes high levels of low‐density lipoprotein cholesterol and an increased risk of heart disease, leading to the subsequent discovery of the statin class of HMG‐CoA reductase inhibitors. In another example, rare gain‐of‐function mutations in the PCSK9 gene were found in the families with high low‐density lipoprotein levels and an increased incidence of coronary heart disease, and subsequent functional studies and clinical trials revealed that the loss of function of PCSK9 significantly reduced low‐density lipoprotein cholesterol levels.
By evaluating ∼10 million SNPs, Okada et al.25 recently performed a GWAS meta‐analysis in a total of >100,000 subjects of European and Asian ancestries comprising 29,880 rheumatoid arthritis cases and 73,758 controls. They discovered 42 novel rheumatoid arthritis risk loci, adding up to a total of 101 total rheumatoid arthritis risk loci. These loci were connected to 98 genes. They demonstrated the gene list expanded from those 98 genes via protein‐protein interaction networks significantly overlap with the targets of the drugs approved for rheumatoid arthritis. This suggested that other targets among those 98 genes might be therapeutic targets. Nelson et al.26 performed a large‐scale evaluation of genetic support in target selection. They collected 16,459 gene‐medical subject heading pairs consisting of 2,531 traits and 7,253 genes associated with traits from public genetic databases, and collected 19,085 target‐medical subject heading pairs from drug databases. The significant enrichment of known targets in the list of variant genes suggested that selecting genetically supported targets could increase the success rate in clinical development.
Because GWASs are often not able to identify the causal relation between variant and disease, combining genetic analysis with other types of evidence may increase the likelihood of selecting a good target. We recently integrated gene expression with disease‐associated SNPs and therapeutic target datasets across a diverse set of 56 diseases in 12 disease categories.27 We systematically evaluated how successful differentially expressed genes, disease‐associated SNPs, or the combination of both could recover known disease targets. We observed the combination of differentially expressed genes and SNPs has more predictive power than each feature alone. This suggested that linking differentially expressed genes with SNPs improves the accuracy of prioritizing candidate targets.
Leveraging public datasets for target validation
The de novo analysis of data discussed above can be used to produce a list of candidate targets. In order to prioritize targets for time‐consuming experimental validation, one needs to first assess their novelty and commercialization potential.28 In addition, a good target in a preclinical study should satisfy the following criteria: (1) it should be druggable; (2) it should be expressed only in the abnormal cells of clinical samples and not, or barely, expressed in normal cells; and (3) the modulation of the target has the potential to reverse disease phenotype.
Public datasets can be leveraged to assess these criteria. The druggability can be assessed through an integrative analysis of protein functional class, homology to targets of approved drugs, three‐dimensional structure, and the existence of published active small molecules.29 We may search its mRNA expression in cell‐lines (data from CCLE and ref. 30), patients (data from TCGA and GEO), and normal tissues (data from GTEx). We may also search its protein expression in cell lines (data from The Human Protein Atlas), patient tissues (data from The Human Protein Atlas and TCGA), and normal tissues (data from The Human Protein Atlas and the Human Proteome Map31). We may further infer its function through the recent high throughput experiments. For example, Cowley et al.32 used a genome‐scale, lentivirally delivered shRNA library to perform massively parallel pooled shRNA screens in 216 cancer cell lines and identified genes that are essential for cell proliferation and/or viability. Essential genes in 72 breast, pancreatic, and ovarian cancer cell lines were inferred using a lentiviral shRNA library targeting ∼16,000 genes.33 Essential genes in a few human cancer cell lines were also characterized recently using the bacterial CRISPR system.34 Target function can be even inferred through the measurement of gene expression changes upon genetic perturbation (data available in Library of Integrated Network‐based Cellular Signatures).
Outstanding challenges
First, measurements made from disease samples may have poor quality. Recent studies indicated that a large number of tumor samples are impure because of the mixed immune cells and stromal cells.35 Second, large technical and biological variation of samples exists. Third, the quality of reagents, especially antibodies, varies widely.36 The misuse of antibodies may directly lead to the failure of experiments. Last, although the dataset from high throughput experiments is useful either as a reference tool to detect expression or as a tool to infer biological function, they occasionally give false signals, resulting in the misclassification of potentially good targets.
LEVERAGE BIG DATA TO IDENTIFY NEW DRUG INDICATIONS FOR PRECLINICAL STUDIES
Since discovering a new chemical entity is a very long and complicated process, we will mainly discuss the reuse of existing drugs (referred as drug repositioning), which offers a relatively short approval process and straightforward path to clinical translation. Computational approaches for drug repositioning have been reviewed previously.37, 38 Figure 3 illustrates three common big data approaches that use different features to discover new drug indications, and basic experimental approaches to validate them. We will first discuss these three approaches and then discuss the discovery of new drug combinations. Finally, we will argue that public datasets can be used to validate drug indications before time‐consuming experiments.
Figure 3.
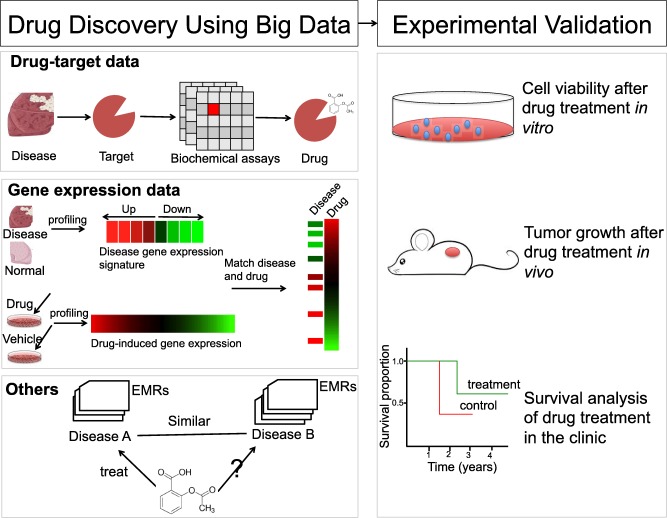
An illustration of big data approaches to identifying new drug indications.
Indication discovery using drug‐target data
Targeting an individual alteration using either a small or a large molecule remains the main paradigm in drug discovery. This approach has led to the discovery of many successful drugs, such as trastuzumab (HER2 in breast cancer), crizotinib (ALK in non‐SCLC), and dabrafenib (BRAF in melanoma). When a new target is proposed for a disease, existing drugs that interfere with this target can be searched from the literature or drug‐target databases (e.g., DrugBank39 and ChEMBL) and their potential new usage is further validated by experiments. This approach is commonly practiced. If there is no drug available for this target, structure‐based design, such as homology modeling, can be used to infer new drug hits.
Indication discovery using gene expression data
Another common approach is to look for inverse drug‐disease relationships by comparing disease molecular features and drug molecular features, such as gene expression. This approach starts with the creation of a disease gene expression signature by comparing disease samples and normal tissue samples, followed by querying drug‐gene expression databases, such as Connectivity Map (CMap) and Library of Integrated Network‐based Cellular Signatures. For example, our colleagues Dudley et al.40 and Sirota et al.41 performed large‐scale analysis of gene expression profiles across over 100 diseases using microarray data from GEO and mapped disease signatures to over 100 drugs signatures in CMap. Using this system's approach, they repurposed the anticonvulsant topiramate for the treatment of inflammatory bowel disease and the antiulcer drug cimetidine for the treatment of lung adenocarcinoma. Our colleagues Jahchan et al.42 used a similar systematic drug‐repositioning bioinformatics approach to query a large compendium of gene expression profiles using a SCLC expression signature derived from GEO. They predicted antidepressant drugs for the treatment of SCLC and validated that this group of drugs potently induce apoptosis in both chemotherapy naive and chemotherapy resistant SCLC cells in culture, in mouse and human SCLC tumors transplanted into immunocompromised mice, and in endogenous tumors from a mouse model for human SCLC. This finding even led to the launch of a clinical trial (NCT01719861).
Van Noort et al.43 systematically assessed how well the known disease‐drug indications were recapitulated by the expression‐based inverse correlation of disease‐drug relations for 40 individual diseases. They found that colorectal cancer is one of the diseases in which known disease‐drug indications could be well recapitulated. This finding, together with the unmet clinical need in the treatment of metastasized colorectal cancer, led them to look for drugs that inhibit metastasis in colorectal cancer. Instead of a signature built by comparing disease samples and normal samples, they built a gene signature of metastatic potential by comparing nonmetastatic tumors vs. metastatic primary tumors. By querying the CMap V2 using this signature, they predicted three novel compounds against colorectal cancer: citalopram, troglitazone, and enilconazole, and verified these compounds by in vitro assays of clonogenic survival, proliferation, and migration and in a subcutaneous mouse model.
Although drugs in these previous examples were validated in preclinical models, the question of whether the disease gene expression was really reversed in disease models remains unknown. A recent study in a mouse model of dyslipidemia found that treatments that restore gene expression patterns to their norm are associated with the successful restoration of physiological markers to their baselines, providing a sound basis to this computational approach.44
Other studies have used slightly different approaches. For example, instead of building a universal signature for one disease, Zerbini et al.45 considered the variation of individual patients. They built a disease signature for individual patients with clear cell renal cell carcinoma and predicted drugs for individual patients. Pentamidine, one of the common drugs shared by all the patients, showed its efficacy in vitro and in the 786‐O human clear cell renal cell carcinoma xenograft mouse model. Brum et al.46 profiled gene expression in human mesenchymal stromal cells toward osteoblasts and created significantly regulated genes. They found that the signature of parbendazole matches the expression changes observed for osteogenic human mesenchymal stromal cells, suggesting that parbendazole could stimulate osteoblast differentiation. They further validated that parbendazole induced osteogenic differentiation through a combination of cytoskeletal changes.
Indication discovery using other sources
Many other molecular and clinical features, including side effect, genetic variation, and chemical structure, have been leveraged for drug repositioning. We highlight some exciting findings here and refer other findings to our recent review on the trend of computational drug repositioning.38 Our colleagues Paik et al.47 extracted clinical features from over 13 years of EMRs, including >9.4 M laboratory tests of >530,000 patients, in addition to diverse genomics features. With these features, they computed drug‐drug similarity and disease‐disease similarity. Based on the assumption that similar diseases can be treated with similar drugs, they inferred 3,891 new indications that were previously not known to be associated. Among those new indications, terbutaline sulfate was indicated as a potential drug for amyotrophic lateral sclerosis treatment and was further validated in an in vivo zebrafish model of amyotrophic lateral sclerosis.
Iorio et al.48 built a drug‐drug similarity matrix using the gene expression data from CMap and verified an unexpected similarity between CDK2 inhibitors and topoisomerase inhibitors. They also found that a Rho‐kinase inhibitor might be reused as an enhancer of cellular autophagy, potentially applicable to several neurodegenerative disorders. This work was further extended in a recent study in which glipizide and splitomicin were found to perturb microtubule function through a semisupervised approach.49
Discovery of new drug combinations
As many diseases are driven by complex molecular and environmental interactions, targeting a single component may not be sufficient to disrupt these complex interactions; thus, there is increasing interest in targeting multiple molecules using combined drugs or multitarget inhibitors. Using big data to predict drug synergy is appealing, yet challenging. In a recent community‐based open challenge for drug synergy predictions, among the 31 submitted methods, only three methods performed significantly better than random chance.50 Nevertheless, a few interesting combinations have been found through a big data mining approach. Mitrofanova et al.51 assumed that if a drug could downregulate the activated target genes and upregulate the repressed targets of a master regulator (e.g., a key transcription factor), then the drug could reverse the activity of the master regulator. Using the drug signatures derived from genetically engineered mouse models, they identified drugs to reverse the master regulator pair, FOXM1/CENPF, which is essential for prostate tumor malignancy. They further extended the concept that effective drug combinations should induce a more significant reversal of master regulator‐specific regulon expression, compared to the individual drugs. The combination of rapamycin + PD0325901 was predicted to have the strongest reversion of the FOXM1/CENPF activity, both with respect to the total number of targets affected by both drugs and the number of unique targets affected by each drug. Their synergistic effect was validated in mouse and human prostate cancer models. Sun et al.52 demonstrated that using genomic and network characteristics could lead to a good performance of predicting synergistic drugs for cancer. They confirmed 63.6% of their predictions for breast cancer through experimental validation and literature search, and identified that the combination of erlotinib and sorafenib has strong synergy and low toxicity in a zebrafish MCF7 xenograft model.
Leveraging public datasets to validate new drug indications
Public datasets can be leveraged to validate drug hits and understand drug mechanisms. For example, drug efficacy and toxicity in vitro or in vivo may be searched from the drug‐sensitivity databases (e.g., CCLE, ChEMBL, canSAR53) and toxicity databases (e.g., CTD54), respectively. Drug efficacy can be inferred from EMRs as well. Xu et al.55 recently demonstrated the usage of EMRs in the validation of drug‐disease pairs through a case study of metformin associated with reduced cancer mortality. Our colleagues Khatri et al.56 validated the beneficial effect of atorvastatin on graft survival by retrospective analysis of EMRs of a single‐center cohort of 2,515 renal transplant patients followed for up to 22 years.
To understand drug mechanisms, the models,57, 58 which were built by leveraging public datasets, can be used. Woo et al.58 recently built a computational model called DEMAND to infer drug targets in a disease model (e.g., cell line) by using drug‐gene expression profiles and a regulatory network of the disease model. Their model recovered the established proteins involved in the mechanism of action for 70% of the tested compounds and revealed altretamine, an anticancer drug, as an inhibitor of GPX4 lipid repair activity.
Outstanding challenges
First, selecting appropriate preclinical models from a large number of available models is often challenging during the validation stage, as some validation models may not be reliable per se, or the molecular features of some models may be quite different with those used for the prediction.59 We recently identified that half of the hepatocellular carcinoma cell lines are not significantly correlated to the hepatocellular carcinoma tumors from TCGA using gene expression features.60 Domcke et al.61 identified a few rarely used ovarian cancer cell lines that more closely resembled ovarian tumors than commonly used cell lines by analyzing a variety of genomic features. In addition to choosing the appropriate preclinical models, moving preclinical findings into the clinic is challenging. One drug or one drug combination validated successfully in preclinical models may fail to translate into the clinic because of the concerns of high toxicity, high cost, low bioavailability, or many other factors. Our recent following analysis of the previous work on drug combinations in clinical trials62 revealed that a drug is more likely to be combined with existing therapies and a brand name drug is rarely combined with another brand name drug (unpublished), suggesting the necessity of considering the characteristics of clinical trials during preclinical studies.
LEVERAGE BIG DATA TO IDENTIFY DRUG RESPONSE BIOMARKERS IN THE ERA OF PRECISION MEDICINE
Because drugs are mostly discovered based on disease molecular features, it is natural that they should be applied to those patients possessing these molecular features. A number of existing drugs have been proven to be effective only for a group of patients with specific molecular features: for example, trastuzumab for patients with HER2‐positive breast cancer. Identifying molecular features (or biomarkers) for predicting drug response is critical to identify the right patient populations for any drug under investigation.63 Figure 4 shows two big data approaches to identify biomarkers for predicting drug response, and experimental approaches to validate biomarkers.
Figure 4.
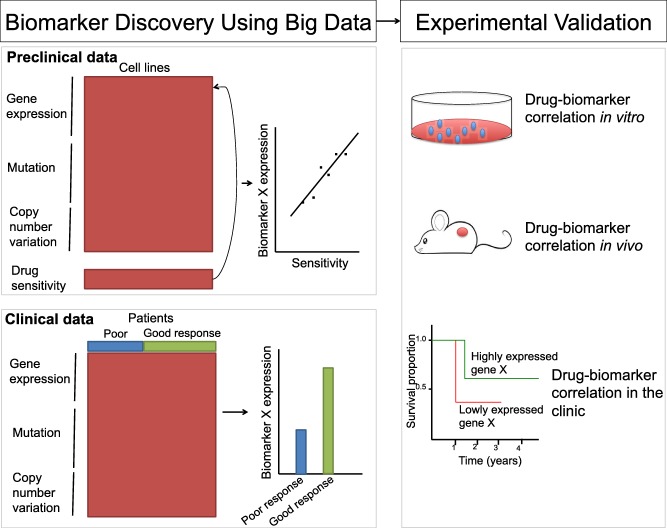
An illustration of big data approaches to identifying new drug response biomarkers.
Biomarker discovery using genomic and pharmacogenomics data from preclinical samples
The recent large‐scale generation of pharmacogenomics data in preclinical disease models (especially cell lines) and molecular characterization of these models enable researchers to identify biomarkers for predicting drug response. By integrating pharmacological profiles for 24 anticancer drugs across 479 cell lines with the gene expression, copy number, and mutation data of these cell lines, Barretina et al.64 identified genetic, lineage, and gene‐expression‐based biomarkers of drug sensitivity. They highlighted a few cases: plasma cell lineage for IGF1 receptor inhibitors, AHR expression for MEK inhibitors, and SLFN11 expression for topoisomerase inhibitors. Kim et al.65 identified three distinct target/response‐indicator pairings including NLRP3 mutation/inflammasome activation for FLIP addiction, co‐occurring KRAS and LKB1 mutation for COPI addiction, and a seven‐gene expression signature for a synthetic indolotriazine. Basu et al.66 quantitatively measured the sensitivity of 242 molecularly characterized cancer cell lines to 354 small molecules and created the Cancer Therapeutics Response Portal that enables researchers to correlate genetic features to sensitivity. Using their portal, they identified that activating mutations in the oncogene β‐catenin could predict sensitivity of the BCL‐2 family antagonist navitoclax. Their subsequent work expanded the portal to 860 cell lines and 481 compounds including 70 US Food and Drug Administration‐approved agents, 100 clinical candidates, and 311 small‐molecule probes,67 allowing researchers to identify biomarkers for a larger number of drugs. Several other similar sources include 77 therapeutic compounds in ∼50 breast cancer cell lines68 and 90 drugs in 51 stable cancer cell lines.69
Biomarker discovery using genomic data from clinical samples
Biomarkers can be also detected by comparing genomic profiles of clinical samples. Outstanding examples include the finding of EGFR mutations as a predictor of sensitivity to gefitinib,70 and a 12‐gene colon cancer recurrence score as a predictor of recurrence in patients with stage II and III colon cancer treated with fluorouracil and leucovorin.71 O'Connell et al.71 performed quantitative reverse transcription polymerase chain reaction of 375 genes in four independent cohorts consisting of 1,851 patients with stage II or III colon cancer. These patients were either treated with surgery alone or surgery plus fluorouracil/leucovorin and their recurrence‐free interval at three years were observed. Of 375 genes, 48 genes were significantly associated with risk of recurrence and 66 genes were significantly associated with fluorouracil/leucovorin benefit. From these genes, seven genes were selected based on their biology and the strength of association with outcomes. Expression of these seven genes was normalized against five reference genes, leading to the development of a recurrence score used to predict the risk of recurrence. The recurrence score was subsequently validated in independent clinical studies.72
Outstanding challenges
Lack of effective biomarkers may lead to the failure of clinical trials, whereas biomarkers are only detected or confirmed through clinical trials. The complexity and large variation of clinical trials may cause some important biomarkers to be missed in the original study. This issue can be mitigated through an integrative analysis of clinical trials across multiple studies. Unfortunately, a large number of trials are still not available to the public. Open clinical trial data becomes necessary in order to identify more effective biomarkers for current therapies or even rescue failed drugs via identifying the right patient populations.
PERSPECTIVES
One belief of the current drug discovery paradigm is that thoroughly understanding molecular changes of diseases will ultimately lead to the discovery of new therapeutics. In order to capture molecular changes of disease and changes upon drug interventions, the molecular profiles have to be presented in an accessible format, which we now consider big data. There is no doubt that the profiles we have created will quickly become small sets because of rapid advances in technologies. In the near future, much larger volumes and complex datasets will be created to characterize disease systems: from single cells to organs, from cancer cells to microorganisms, from cell lines to genetically modified mice to individual patients, and from one time point to the longitudinal course of treatment. The incredible number of targets, drugs, and biomarkers discovered by leveraging big datasets in the past years suggests an unprecedented opportunity to leverage them to transform discovery now.
Given the volumes and complexity of datasets for drug discovery, no single person or team could comprehend or use all of them; therefore, it is necessary to reengineer the entire pipeline of drug discovery, where every step is driven by data and rigorous data models. Example steps include the selection of appropriate tissue samples to profile, the selection of appropriate models to validate hypothesis, etc. In addition, high performance computing allows us to generate hypotheses very quickly, but the current experimental settings limit the validation efforts. It is often true that the validation of a drug in preclinical models takes over 10 times longer than the prediction. New sharing economy inspired sources for biomedical research, such as Science Exchange (http://scienceexchange.com) and Assay Depot (http://assaydepot.com) could facilitate running experiments using external sources. More efficient ways are expected to quickly transform big data discoveries into clinical applications.
ACKNOWLEDGMENTS
We thank Drs. Marina Sirota, Mei‐Sze Chua, Hyojung Paik, Dvir Aran, and Shann‐Ching Chen for critical comments on the manuscript.
SOURCE OF FUNDING
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R01GM079719. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
CONFLICT OF INTEREST/DISCLOSURE
Atul Butte is a scientific advisor to Assay Depot, and a founder and scientific advisor to NuMedii, Inc. Bin Chen is a consultant to NuMedii, Inc.
References
- 1. EMBL–European Bioinformatics Institute EMBL‐EBI Annual Scientific Report 2014. <https://www.embl.de/aboutus/communication_outreach/publications/ebi_ar/ebi_ar_2014.pdf>.
- 2. Marx, V. Biology: the big challenges of big data. Nature 498, 255–260 (2013). [DOI] [PubMed] [Google Scholar]
- 3. Barabási, A.L. , Gulbahce, N. & Loscalzo, J. Network medicine: a network‐based approach to human disease. Nat. Rev. Genet. 12, 56–68 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen, B. & Butte, A.J. Network medicine in disease analysis and therapeutics. Clin. Pharmacol. Ther. 94, 627–629 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mantione, K.J. et al Comparing bioinformatic gene expression profiling methods: microarray and RNA‐Seq. Med. Sci. Monit. Basic Res. 20, 138–142 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barrangou, R. , Birmingham, A. , Wiemann, S. , Beijersbergen, R.L. , Hornung, V. & Smith, A. Advances in CRISPR‐Cas9 genome engineering: lessons learned from RNA interference. Nucleic Acids Res. 43, 3407–3419 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kannan, L. et al Public data and open source tools for multi‐assay genomic investigation of disease. Brief. Bioinform. (2015); e-pub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Doshi, P. , Goodman, S.N. & Ioannidis, J.P. Raw data from clinical trials: within reach? Trends Pharmacol. Sci. 34, 645–647 (2013). [DOI] [PubMed] [Google Scholar]
- 9. Grieb, B.C. , Chen, X. & Eischen, C.M. MTBP is overexpressed in triple‐negative breast cancer and contributes to its growth and survival. Mol. Cancer Res. 12, 1216–1224 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee, H.W. et al Tpl2 kinase impacts tumor growth and metastasis of clear cell renal cell carcinoma. Mol. Cancer Res. 11, 1375–1386 (2013). [DOI] [PubMed] [Google Scholar]
- 11. Tilghman, J. et al HMMR maintains the stemness and tumorigenicity of glioblastoma stem‐like cells. Cancer Res. 74, 3168–3179 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hsu, Y.H. et al Definition of PKC‐α, CDK6, and MET as therapeutic targets in triple‐negative breast cancer. Cancer Res. 74, 4822–4835 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kodama, K. et al Expression‐based genome‐wide association study links the receptor CD44 in adipose tissue with type 2 diabetes. Proc. Natl. Acad. Sci. USA 109, 7049–7054 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen, R. et al A meta‐analysis of lung cancer gene expression identifies PTK7 as a survival gene in lung adenocarcinoma. Cancer Res. 74, 2892–2902 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. International Cancer Genome Consortium et al International network of cancer genome projects. Nature 464, 993–998 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vogelstein, B. , Papadopoulos, N. , Velculescu, V.E. , Zhou, S. , Diaz, L.A. Jr. & Kinzler, K.W. Cancer genome landscapes. Science 339, 1546–1558 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sharma, S.V. , Bell, D.W. , Settleman, J. & Haber, D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 7, 169–181 (2007). [DOI] [PubMed] [Google Scholar]
- 18. Chapman, P.B. et al Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 364, 2507–2516 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kwak, E.L. et al Anaplastic lymphoma kinase inhibition in non‐small‐cell lung cancer. N. Engl. J. Med. 363, 1693–1703 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. de Silva, C.M. & Reid, R. Gastrointestinal stromal tumors (GIST): C‐kit mutations, CD117 expression, differential diagnosis and targeted cancer therapy with imatinib. Pathol. Oncol. Res. 9, 13–19 (2003). [DOI] [PubMed] [Google Scholar]
- 21. Rubio‐Perez, C. et al In silico prescription of anticancer drugs to cohorts of 28 tumor types reveals targeting opportunities. Cancer Cell 27, 382–396 (2015). [DOI] [PubMed] [Google Scholar]
- 22. Farmer, H. et al Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921 (2005). [DOI] [PubMed] [Google Scholar]
- 23. Squatrito, M. , Brennan, C.W. , Helmy, K. , Huse, J.T. , Petrini, J.H. & Holland, E.C. Loss of ATM/Chk2/p53 pathway components accelerates tumor development and contributes to radiation resistance in gliomas. Cancer Cell 18, 619–629 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Plenge, R.M. , Scolnick, E.M. & Altshuler, D. Validating therapeutic targets through human genetics. Nat. Rev. Drug Discov. 12, 581–594 (2013). [DOI] [PubMed] [Google Scholar]
- 25. Okada, Y. et al Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506, 376–381 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nelson, M.R. et al The support of human genetic evidence for approved drug indications. Nat. Genet. 47, 856–860 (2015). [DOI] [PubMed] [Google Scholar]
- 27. Fan‐Minogue, H. , Chen, B. , Sikora‐Wohlfeld, W. , Sirota, M. & Butte, A.J. A systematic assessment of linking gene expression with genetic variants for prioritizing candidate targets. Pac. Symp. Biocomput. 383–394 (2015). [PMC free article] [PubMed] [Google Scholar]
- 28. Knowles, J. & Gromo, G. A guide to drug discovery: target selection in drug discovery. Nat. Rev. Drug Discov. 2, 63–69 (2003). [DOI] [PubMed] [Google Scholar]
- 29. Patel, M.N. , Halling‐Brown, M.D. , Tym, J.E. , Workman, P. & Al‐Lazikani, B. Objective assessment of cancer genes for drug discovery. Nat. Rev. Drug Discov. 12, 35–50 (2013). [DOI] [PubMed] [Google Scholar]
- 30. Klijn, C. et al A comprehensive transcriptional portrait of human cancer cell lines. Nat. Biotechnol. 33, 306–312 (2015). [DOI] [PubMed] [Google Scholar]
- 31. Kim, M.S. et al A draft map of the human proteome. Nature 509, 575–581 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cowley, G.S. et al Parallel genome‐scale loss of function screens in 216 cancer cell lines for the identification of context‐specific genetic dependencies. Sci. Data 1, 140035 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Marcotte, R. et al Essential gene profiles in breast, pancreatic, and ovarian cancer cells. Cancer Discov. 2, 172–189 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang, T. et al Identification and characterization of essential genes in the human genome. Science 350, 1096–1101 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yoshihara, K. et al Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 4, 2612 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Baker, M. Reproducibility crisis: blame it on the antibodies. Nature 521, 274–276 (2015). [DOI] [PubMed] [Google Scholar]
- 37. Dudley, J.T. , Deshpande, T. & Butte, A.J. Exploiting drug‐disease relationships for computational drug repositioning. Brief. Bioinform. 12, 303–311 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li, J. , Zheng, S. , Chen, B. , Butte, A.J. , Swamidass, S.J. & Lu, Z. A survey of current trends in computational drug repositioning. Brief. Bioinform. (2015); e-pub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Knox, C. et al DrugBank 3.0: a comprehensive resource for ‘omics’ research on drugs. Nucleic Acids Res. 39(Database issue), D1035–D1041 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dudley, J.T. et al Computational repositioning of the anticonvulsant topiramate for inflammatory bowel disease. Sci. Transl. Med. 3, 96ra76 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sirota, M. et al Discovery and preclinical validation of drug indications using compendia of public gene expression data. Sci. Transl. Med. 3, 96ra77 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jahchan, N.S. et al A drug repositioning approach identifies tricyclic antidepressants as inhibitors of small cell lung cancer and other neuroendocrine tumors. Cancer Discov. 3, 1364–1377 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. van Noort, V. et al Novel drug candidates for the treatment of metastatic colorectal cancer through global inverse gene‐expression profiling. Cancer Res. 74, 5690–5699 (2014). [DOI] [PubMed] [Google Scholar]
- 44. Wagner, A. et al Drugs that reverse disease transcriptomic signatures are more effective in a mouse model of dyslipidemia. Mol. Syst. Biol. 11, 791 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zerbini, L.F. et al Computational repositioning and preclinical validation of pentamidine for renal cell cancer. Mol. Cancer Ther. 13, 1929–1941 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Brum, A.M. et al Connectivity Map‐based discovery of parbendazole reveals targetable human osteogenic pathway. Proc. Natl. Acad. Sci. USA 112, 12711–12716 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Paik, H. et al Repurpose terbutaline sulfate for amyotrophic lateral sclerosis using electronic medical records. Sci. Rep. 5, 8580 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Iorio, F. et al Discovery of drug mode of action and drug repositioning from transcriptional responses. Proc. Natl. Acad. Sci. USA 107, 14621–14626 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Iorio, F. et al A semi‐supervised approach for refining transcriptional signatures of drug response and repositioning predictions. PLoS One 10, e0139446 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bansal, M. et al A community computational challenge to predict the activity of pairs of compounds. Nat. Biotechnol. 32, 1213–1222 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mitrofanova, A. , Aytes, A. , Zou, M. , Shen, M.M. , Abate‐Shen, C. & Califano, A. Predicting drug response in human prostate cancer from preclinical analysis of in vivo mouse models. Cell Rep. 12, 2060–2071 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sun, Y. et al Combining genomic and network characteristics for extended capability in predicting synergistic drugs for cancer. Nat. Commun. 6, 8481 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Halling‐Brown, M.D. , Bulusu, K.C. , Patel, M. , Tym, J.E. & Al‐Lazikani, B. canSAR: an integrated cancer public translational research and drug discovery resource. Nucleic Acids Res. 40(Database issue), D947–D956 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Davis, A.P. et al The Comparative Toxicogenomics Database's 10th year anniversary: update 2015. Nucleic Acids Res. 43(Database issue), D914–D920 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Xu, H. et al Validating drug repurposing signals using electronic health records: a case study of metformin associated with reduced cancer mortality. J. Am. Med. Inform. Assoc. 22, 179–191 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Khatri, P. et al A common rejection module (CRM) for acute rejection across multiple organs identifies novel therapeutics for organ transplantation. J. Exp. Med. 210, 2205–2221 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Keiser, M.J. et al Predicting new molecular targets for known drugs. Nature 462, 175–181 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Woo, J.H. et al Elucidating compound mechanism of action by network perturbation analysis. Cell 162, 441–451 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Day, C.P. , Merlino, G. & Van Dyke, T. Preclinical mouse cancer models: a maze of opportunities and challenges. Cell 163, 39–53 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chen, B. , Sirota, M. , Fan‐Minogue, H. , Hadley, D. & Butte, A.J. Relating hepatocellular carcinoma tumor samples and cell lines using gene expression data in translational research. BMC Med. Genomics 8 (suppl. 2), S5 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Domcke, S. , Sinha, R. , Levine, D.A. , Sander, C. & Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 4, 2126 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wu, M. , Sirota, M. , Butte, A.J. & Chen, B. Characteristics of drug combination therapy in oncology by analyzing clinical trial data on ClinicalTrials.gov. Pac. Symp. Biocomput. 68–79 (2015). [PMC free article] [PubMed] [Google Scholar]
- 63. Collins, F.S. & Varmus, H. A new initiative on precision medicine. The N. Engl. J. Med. 372, 793–795 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Barretina, J. et al The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kim, H.S. et al Systematic identification of molecular subtype‐selective vulnerabilities in non‐small‐cell lung cancer. Cell 155, 552–566 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Basu, A. et al An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 154, 1151–1161 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Seashore‐Ludlow, B. et al Harnessing connectivity in a large‐scale small‐molecule sensitivity dataset. Cancer Discov. 5, 1210–1223 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Heiser, L.M. et al Subtype and pathway specific responses to anticancer compounds in breast cancer. Proc. Natl. Acad. Sci. USA 109, 2724–2729 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Martins, M.M. et al Linking tumor mutations to drug responses via a quantitative chemical‐genetic interaction map. Cancer Discov. 5, 154–167 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Paez, J.G. et al EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304, 1497–1500 (2004). [DOI] [PubMed] [Google Scholar]
- 71. O'Connell, M.J. et al Relationship between tumor gene expression and recurrence in four independent studies of patients with stage II/III colon cancer treated with surgery alone or surgery plus adjuvant fluorouracil plus leucovorin. J. Clin. Oncol. 28, 3937–3944 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yothers, G. et al Validation of the 12‐gene colon cancer recurrence score in NSABP C‐07 as a predictor of recurrence in patients with stage II and III colon cancer treated with fluorouracil and leucovorin (FU/LV) and FU/LV plus oxaliplatin. J. Clin. Oncol. 31, 4512–4519 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]