

Abstract
Structures of RNA molecules are essential for their architectural, regulatory, and catalytic functions. Recent advances in high throughput sequencing enabled the development of methods for probing RNA structures on a transcriptome-wide scale – termed the RNA structurome. Here we review the state-of-the-art technologies for probing the RNA structurome, and highlight insights gained from these studies. We also point out the limits of current methods and discuss potential directions for future improvements.
Introduction
In addition to carrying genetic information like DNA, RNA molecules are folded into exquisite structures. The flexible and dynamic structures of RNA molecules underlie their versatile functions. Similar to the studies of proteins, structural analysis of RNA molecules has generated numerous insights into mechanisms of gene regulation on many different levels. Structural elements in mRNAs controls their subcellular localization, and internal ribosome entry site (IRES) form 3D structures to drive cap-independent translation (reviewed by [1]). Motif accessibility in mRNAs modulates the interaction with RNA binding proteins (RBPs) and microRNAs. RNA structure is also critical for noncoding RNAs. Structural motifs in long noncoding RNAs mediate their nuclear localization, stability, and interaction with chromatin modification machinery (reviewed by [2,3]). Unique structures of riboswitches allow them to bind specific metabolites and in turn transduce ligand binding to gene regulatory activities. Tertiary structures built upon the basic structural elements are required for ribozymes to catalyze a wide variety of biochemical reactions, including splicing, ligation and translation.
Traditional structure determination methods such as X-ray crystallography, nuclear magnetic resonance (NMR) and cryo-electron microscopy (cryo-EM) generate high-resolution structures in 3D, but they require purified, structurally stable and nearly static RNA molecules. Therefore they are not readily applied to study the vast majority of the RNAs, which are often in multiple conformations, dynamic, complex and constantly regulated by the cellular environment [4,5].
Another class of methods, chemical and enzymatic probing, tackles the RNA structure problem from a different angle. Nucleotides in RNA structures have different accessibilities depending on their base pairing and other interactions. Chemical probing employs a variety of compounds that react with nucleotides according to their environment and generate bulky adducts or break points that can be measured by Sanger or high throughput sequencing as stops or ends of reverse transcription [6]. Commonly used chemicals include Selective 2′ -hydroxyl acylation analyzed by primer extension (SHAPE) chemicals such as 1-methly-7-nitro-isatoic anhydride (1M7), N-methylisatoic anhydride (NMIA) that acylates 2′-OH in single-stranded (ss) regions (Table 1) [7], hydroxyl radicals that break unprotected phosphate backbones, dimethyl sulphate (DMS) that methylates ss adenosines and cytosines, 1-cyclohexyl-(2-morpholinoethyl) carbodiimide metho-p-toluene sulfonate (CMCT) that modifies ss uridines and guanines, kethoxal that modifies ss guanines, and in-line probing that takes advantage of the spontaneous degradation in ss regions. Similarly, enzymatic probing employs double-strand (ds) specific RNases to cut RNAs leaving ds/ss-specific break points that indicate the base pairing status (Table 1) [8–11]. The chemical and enzymatic probing data are then incorporated as constraints into a variety of RNA structure prediction algorithms to increase the accuracy of prediction [12]. Such experimental data add substantially to structural predictions based on free energy minimization or evolutionary co-variation.
Table 1.
Methods for global determination of RNA structures. Methods are listed not by the labs that developed them, but by the strategy and chemical used. Regular SHAPE is very inefficient for in vivo analysis but is one of the earliest method for structure probing. Enzymatic probing is not useful for in vivo analysis but provided the first global analysis of the RNA structurome. RING-MaP and SHAPE-MaP were developed for single-RNA analysis but the strategy is useful for in vivo structurome analysis. HiCLIP and RPL has yet to achieve global analysis of RNA structurome, but represent interesting future directions
| Methods | Probe | Data | Inferred | Features and limitations | References |
|---|---|---|---|---|---|
| Secondary structure in vitro | |||||
| Chemical probing | 1M7, DMS, CMCT, hydroxyl, etc. | 1D | 2D | All nucleotides, mostly in vitro, no direct base pairing information | [6,7] |
| Enzymatic | RNase I, V1, P1, S1 | 1D | 2D | All nucleotides, in vitro only, no direct base pairing information | [8–11] |
| SHAPE-MaP, RING-MaP | 1M7 and DMS | 1D | 2D/3D | Additional correlation for bases but only for short distance. Adaptable to in vivo and global | [26•,27•] |
| Secondary structure in vivo | |||||
| DMS-seq | DMS | 1D | 2D | In vivo, only interrogates A/C, no direct base pairing information | [16••,17••,20••] |
| icSHAPE | NAI, NAI-N3 | 1D | 2D | In vivo, all nucleotides, minimal background, no direct base pairing information | [18,19••] |
| Secondary and higher order structure in vivo | |||||
| CLASH, hiCLIP | UV crosslinking | 2D | In vivo, direct duplex determination, only dsRBP-bound structures | [28,29,30••] | |
| RPL | No crosslinking | 2D | Lysed cells, direct duplex determination, low efficiency and accuracy | [31] | |
Several excellent reviews have been devoted to the chemistry and computational analysis of the chemical and enzymatic probing experiments [12–15]. Here we focus on the in vivo transcriptome-wide applications of chemical probing. These methods have provided important insights into the physiological states of RNA molecules. Nevertheless, these methods only generate one-dimensional averaged structure information, which is far from enough for a complete description of the RNA structurome. In addition, we also discuss recent efforts in developing methods that examine long range, alternative and complex RNA structures.
Methods for in vivo RNA structurome probing
Recent advances in high throughput sequencing have brought chemical and enzymatic probing experiments to a global scale. Although enzymatic and most chemical probes can only be applied to in vitro RNA samples, some of the reagents such as DMS and the recently developed NAI (2-methylnicotinic acid imidazolide) and NAI–N3 have appropriate half-lives (∼34 min for NAI and NAI–N3) and readily enter the cell, thus enabling in vivo probing of RNA structures (Table 1) [16••,17••,18,19••]. Together, the high throughput sequencing technologies and in vivo probing chemicals made it possible to determine the RNA structurome in vivo.
The first reports of in vivo structurome probing used DMS, an alkylation reagent, to modify adenines and cytosines that are detected as reverse transcription stops (Table 1) [16••,17••,20••]. Although these studies differ in some of the experimental details, they are similar in concept and major steps. The alkylation of ss bases blocks reverse transcription, and thus reports the base pairing status of the RNA sequence. One of the disadvantages is that the DMS-based methods only interrogate two out of the four bases. Since modification of bases affects their base pairing abilities, the modification of RNA molecules were titrated to achieve ‘single-hit’ kinetics, where approximately one in 200 nucleotides are modified. The single-hit kinetics results in another disadvantage: most RNA fragments are unmodified and thus sequencing these RNA produces high background.
Conventional SHAPE reagents modify all four nucleotides uniformly but are only applicable to in vitro samples. To overcome this limitation, Spitale and colleagues synthesized new types of SHAPE reagents that readily enters the cell and efficiently react with single-stranded nucleotides [18]. These chemicals were shown to work in bacteria, yeast, fly, mouse and human cells. To further reduce the high background due to the single-hit modification conditions, Spitale and colleagues introduced a clickable azide handle to the acylation agent NAI, which allows easy purification of modified RNA fragments using the biotin–steptavidin system (icSHAPE: in vivo click SHAPE) [19••]. This additional selection step greatly reduces background and enables more sensitive and specific analysis of modified RNA fragments.
Since each RNA molecule and conformation is modified at very low frequencies in chemical probing experiments, a large number of sequencing reads are needed to generate a structure profile, even with biotin selection. Therefore the structure information obtained represents an average of all conformations. Despite their limitations, DMS and NAI–N3 are the best reagents so far for chemical probing of in vivo RNA structurome.
New insights into the RNA structurome
Chemical probing data for RNA structures are typically analyzed and used in two ways: probe-and-predict (PnP) and probe-and-average (PnA). For the PnP approach, reactivity profiles are incorporated as constraints into secondary structure prediction methods to produce structure models that best explain the reactivity profile (for review see [12]). While this approach increases the accuracy of structure prediction, reliable models can only be obtained for very short RNA molecules or regions with stable single structure conformations [21]. Therefore application of this approach to the entire RNA structurome is not feasible. On the other hand, the PnA approach typically aligns large numbers of pre-defined RNA sequence motifs to derive a meta-gene reactivity profile, which is an average of each already averaged RNA structure. Using the PnA approach, recent studies have revealed interesting features of the RNA structurome in vivo and provided many novel insights into the regulatory functions of RNA structures (Figure 1). Several studies also compared in vivo structures to in vitro structures and discovered features that are programmed by sequence alone and those influenced by the cellular environment.
Figure 1.
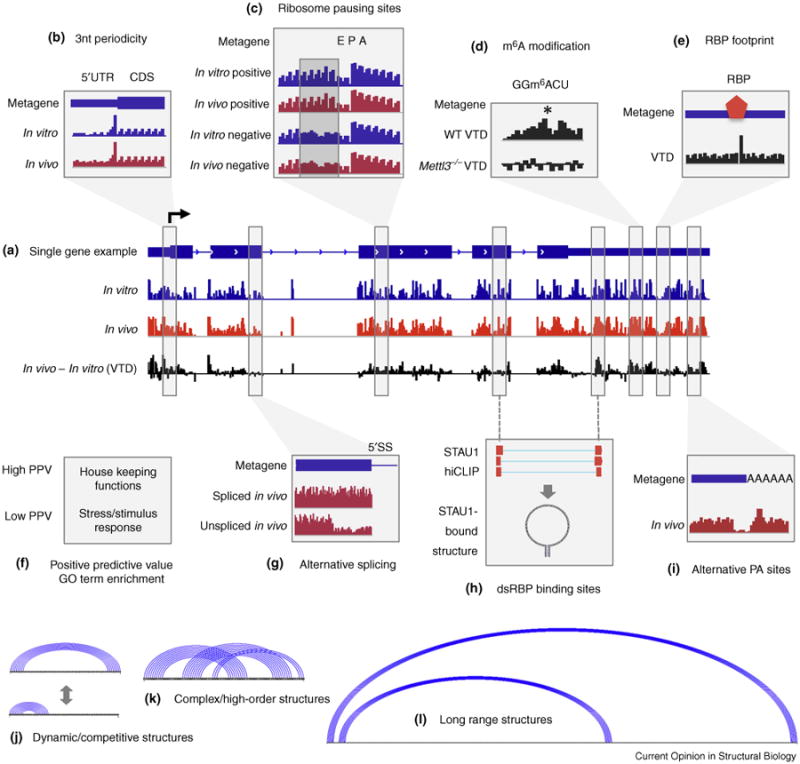
The RNA strucutrome: novel insights, yet incomplete picture. (a–i) New findings about the RNA structurome from recent global structure probing studies. Most of these structure features are obvious on a metagene level but not on a single gene level. (a) An example set of chemical probe reactivity tracks for a typical mRNA gene, including in vivo and in vitro probing and the difference between them (VTD track). Several studies have observed less structure in vivo than in vitro and the active unfolding of structures in vivo [17••,19••]. (b) Structures in the CDS, but not UTRs, exhibit obvious 3 nt periodicity both in vitro and in vivo [8,17••,19••,22]. The region immediately upstream of the start codon and near the stop codon is less structured than average [16••,17••,19••]. However the relative structure content between the UTR and CDS differs in various organisms, more in the CDS for yeast and plant, but more in the UTR for animals [8]. (c) Ribosome pausing sites, rather than negative controls, exhibit obvious 3 nt periodicity before the position where the ribosome resides (indicated by E (exit), P (peptidyl-tRNA) and A (aminoacyl-tRNA)) [19••]. (d) m6A modification makes the surrounding sequence less structured [19••] than negative controls (Mettl3−/− loses most m6A). (e) RBP binding alters the chemical environment of target RNA, therefore leaving a footprint [19••]. (f) Gene ontology analysis of A. thaliana reveals that PPV (positive predictive value), a measure for how close the in silico predicted structure is to chemically probed structure, is high for RNAs encoding house keeping proteins, but low for those encoding stress/stimulus response proteins [16••]. (g) Metagene analysis of alternative splicing reveals higher structure (lower reactivity) before the 5′ splice site (5′SS) before the skipped exon [16••]. (h) hiCLIP analysis of STAU1 in human cells reveals large numbers of secondary structures, some of which span long distances [30••]. (i) Metagene analysis of A. thaliana alternative polyadenylation sites reveals a pattern of low reactivity followed by high reactivity [16••]. (j–l) Important aspects of the structurome that are still poorly understood. Diagrams were drawn using the linear structure format. (j) The same sequence may adopt different structure states in vivo that compete with each other or with RBP and miRNAs. (k) Complex and high order structures, that is, an interlocked pseudoknot. (l) Long range base pairing interactions that may span many kilobases.
Long before RNA structurome analysis was possible, Shabalina et al. performed in silico folding of human and mouse mRNAs and found a surprising trinucleotide periodicity of base pairing probability in the coding regions, which is absent from the UTRs [22]. This prediction was first validated in vitro for yeast mRNAs [8], and later shown to be a ubiquitous phenomenon in vivo in several organisms (Figure 1b) [16••,17••,19••]. These studies also reported a tendency for the start codon region, either canonical or non-canonical, to have less structure, a property that likely facilitates translation initiation.
Applying the PnA approach to A. thaliana seedlings, Ding and colleagues discovered structural features in mRNAs around alternative splicing and polyadenylation sites (Figure 1g and i) [16••]. The data suggest that, in addition the sequence motifs, structures around mRNA processing sites play important roles in guiding the processing. Gene ontology analysis showed that A. thaliana mRNAs that encode stress/stimulus response proteins tend to be less structured in vivo than those encoding housekeeping proteins. However, these data should be interpreted with caution, since it is known that GC content and many other factors could affect the relative structure content of RNA molecules.
Based on the idea that cellular components affect RNA structures and may lead to local structural rearrangements, Spitale et al. analyzed the difference between in vitro and in vivo icSHAPE data and observed specific VTD (in vivo in vitro difference) signatures for several sequence motifs [19••]. Notably, the VTD signal enabled the detection of focal rearrangements of structures induced by RBP binding such that the presence or absence of the VTD at each instance of a RBP binding motif can be used to predict RBP binding in vivo (Figure 1d). This concept of the RNA structural imprint is analogous to transcription factor ‘footprint’ on DNA Similar to the VTD approach, Smola et al. used 1M7 to detect protein-binding sites on several abundant RNAs by comparing modification status between in cellulo and ex vivo conditions [23]. Together, these studies highlight the power of SHAPE-based methods in RNA–protein interactions. Spitale et al. further discovered that the N6-methyladenosine (m6A) RNA modification is a structural switch in vivo that leads to unpairing of RNA duplexes (Figure 1e) [19••], a result independently verified by in vitro assays and detailed biophysical characterization [24,25]. Surprisingly, although in silico and in vitro studies of RNA structure detected more significant structure around ribosome pause sites [8], this pattern was not seen in the in vivo studies [17••,19••]. This could reflect the influences of the cellular environment on the RNA structure.
Recent advances in vitro RNA structure mapping methods can complement in vivo RNA structurome data. Systematic ‘mutate-and-map’ can identify specific base-pairing interactions, and can be combined with multiplexed hydroxyl radical cleavage analysis of tertiary proximity to model three dimensional structures of RNA at near atomic resolution [32]. A powerful strategy going forward may be to nominate RNA structural motifs by in vivo structurome data, followed by detailed in vitro dissection and 3D modelling of the top candidates.
Direct determination of helices and complex and dynamic structures
The one-dimensional data from enzymatic and chemical probing of RNA structures have provided interesting insights into the collective features of RNA structures, yet their direct use in structure modelling has been quite difficult. First, base pairing interactions predicted with the reactivity data are far from accurate, especially for long range interactions and complex pseudoknots. Secondly, structures of most RNA molecules are heterogeneous, flexible and dynamic in living cells, and thus the structures are better described as an ensemble. However, current methods for in vivo chemical probing of the RNA structurome detect the average structure in the dynamic cellular environment. Deconvolution of the structure ensemble into their individual states from the one-dimensional data is impractical. Several recent studies started addressing these problems using new computational and experimental approaches.
Nucleotides that interact with each other display correlated chemical reactivity and this correlation can be used to detect interactions and classify distinct structure states. Weeks and colleagues recently implemented this principle in two methods, SHAPE-Mutational Profiling (SHAPE-MaP) and RNA INteracting Group-MaP (RING-MaP) [26•,27•]. Both methods used special conditions to facilitate read through of reverse transcription at modified nucleotides to introduce mutations, usually more than one for each RNA template. Correlation and cluster analysis of the mutations on sequenced reads generate restraints for building 2D and 3D models of the RNA interrogated. These methods have been applied to a few in vitro folded RNAs and yielded improved structure models. Although this strategy has only been tested on a few molecules, they can be potentially adapted to the entire transcriptome. However, using these methods for direct deconvolution of large and complex RNAs might be challenging.
Direct base pairing information can also be obtained by measuring physical proximity between nucleotides. Tollervey and colleagues discovered ligated RNA fragments in crosslinking and sequencing dsRNAs that are bound by proteins. Based on this discovery, they developed a method called Crosslinking, Ligation and Sequencing Hybrids (CLASH), for the analysis of RNA structures and RNA-RNA interactions [28,29]. Sugimoto and colleagues applied this principle to iCLIP, developing the hybrid iCLIP (hiCLIP) method, to identify the structures bound by a dsRBP STAU1 [30••]. In this approach, the STAU1-bound dsRNA fragments are captured by the proximity ligation, and each sequenced read represents a single molecule measurement of the structure. The hiCLIP method revealed many STAU1-associated long-range interactions — separated by thousands of bases or more on the linear transcript — that are especially hard to detect using conventional methods. Since each sequencing read comes from a single structure conformation, the proximity ligation also enables the discovery of individual structural states in a small subset of the structurome, rather than an average of the structural ensemble [30••]. One of the major limitations in the CLASH/hiCLIP methods is the low percentage of reads that provide direct contact information (less than 2%). Since most eukaryotes encode a large number of RBPs (in fact a greater number than DNA binding transcription factors), applying hiCLIP to them one by one is laborious. Furthermore, many RNA structures are not bound by RBPs at all and therefore impossible to identify using hiCLIP.
Similar to the use of proximity ligation to capture RNA structures, Ramani and Shendure reported a more general method, RNA Proximity Ligation (RPL) [31]. This approach employed in situ RNase digestion of RNA and proximity ligation to join duplexes in physical proximity. Proof of concept results were presented for a few highly abundant noncoding RNAs, such as the ribosomal RNAs and snoRNAs. However, since no crosslinking is used and cells were either digested by endogenous nucleases or lysed and digested with exogenous nucleases, promiscuous ligation events lead to low accuracy and high noise. Since no selection is performed, the percentage of chimeric fragments is very low (less than 0.5%), making the method difficult to scale for transcriptome-wide applications. Although the correlation and proximity-ligation based methods described here have yet to achieve analysis of whole RNA structuromes, they represent interesting attempts from different angles other than the chemical probing methods.
Summary and future perspectives
A full understanding of the RNA structurome requires complete description of the structures and their dynamics. Recent development of sequencing-based chemical probing methods has afforded a global view of local RNA structure profiles. It is anticipated that further development of basepairing detection methods would provide the much-needed information on more complex and dynamic RNA architectures. Both nucleic acid specific or protein– RNA crosslinkers can be further explored to target different kinds of secondary or tertiary structures. The combination of these two distinct classes of experimental methods is necessary for obtaining the full picture of the RNA structurome, from nucleotides to high-level architectures. Although 3D modelling of RNA structures is currently limited to short RNAs [32], the integration of chemical probing and crosslinking methods should help deconvolve structural ensembles of large RNAs and facilitate the isolation of structural domains so that 3D modelling may be applied to them. Phylogenetic analysis of RNA structures often provides support for their functional importance, but suffer from the sparseness of covariation in most RNAs. Integration of phylogenetic analysis with experimental methods will combine the strengths of both approaches and help understand functions of newly identified RNA structures.
The long-term goal for the study of RNA structurome is to not only create a catalogue of the structures, but also infer their functions based on classification and correlation with known sequence motifs and protein binding, gene ontology, and then prioritize them for functional and mechanistic studies. In many biological processes, RNA structures can be viewed as a hub that integrates input from cellular components and extracellular environments. Perturbations of the underlying sequence or these input sources regulate important RNA-based functions or lead to diseases. For example, stress conditions promote selective translation of mRNAs, a process that depends on structures on mRNAs [33]. Furthermore, mutations in many RNA helicases lead to cancers [34]. Application of the RNA structure determination methods to these diverse problems will shed light on both basic mechanisms of gene expression and potential therapeutic opportunities for treating diseases.
Acknowledgments
We thank members of the Chang lab for discussion and apologize to colleagues whose works are not discussed due to space limitation. We acknowledge support from NIH (R01-HG004361 and P50-HG007735) and California Institute for Regenerative Medicine (to HYC). ZL is a Layton Family Fellow of the Damon Runyon-Sohn Foundation Pediatric Cancer Fellowship Award (DRSG-14-15).
Footnotes
Conflict of interest statement: HYC is an inventor on patent held by Stanford University on in vivo SHAPE technology.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Mortimer SA, Kidwell MA, Doudna JA. Insights into RNA structure and function from genome-wide studies. Nat Rev Genet. 2014;15:469–479. doi: 10.1038/nrg3681. [DOI] [PubMed] [Google Scholar]
- 2.Batista PJ, Chang HY. Cytotopic localization by long noncoding RNAs. Curr Opin Cell Biol. 2013;25:195–199. doi: 10.1016/j.ceb.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wan Y, Kertesz M, Spitale RC, Segal E, Chang HY. Understanding the transcriptome through RNA structure. Nat Rev Genet. 2011;12:641–655. doi: 10.1038/nrg3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wiebe NJ, Meyer IM. TRANSAT – method for detecting the conserved helices of functional RNA structures, including transient, pseudo-knotted and alternative structures. PLoS Comput Biol. 2010;6:e1000823. doi: 10.1371/journal.pcbi.1000823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jaeger JA, SantaLucia J, Jr, Tinoco I., Jr Determination of RNA structure and thermodynamics. Annu Rev Biochem. 1993;62:255–287. doi: 10.1146/annurev.bi.62.070193.001351. [DOI] [PubMed] [Google Scholar]
- 6.Ehresmann C, Baudin F, Mougel M, Romby P, Ebel JP, Ehresmann B. Probing the structure of RNAs in solution. Nucleic Acids Res. 1987;15:9109–9128. doi: 10.1093/nar/15.22.9109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Merino EJ, Wilkinson KA, Coughlan JL, Weeks KM. RNA structure analysis at single nucleotide resolution by selective 2′-hydroxyl acylation and primer extension (SHAPE) J Am Chem Soc. 2005;127:4223–4231. doi: 10.1021/ja043822v. [DOI] [PubMed] [Google Scholar]
- 8.Kertesz M, Wan Y, Mazor E, Rinn JL, Nutter RC, Chang HY, Segal E. Genome-wide measurement of RNA secondary structure in yeast. Nature. 2010;467:103–107. doi: 10.1038/nature09322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wan Y, Qu K, Zhang QC, Flynn RA, Manor O, Ouyang Z, Zhang J, Spitale RC, Snyder MP, Segal E, et al. Landscape and variation of RNA secondary structure across the human transcriptome. Nature. 2014;505:706–709. doi: 10.1038/nature12946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Underwood JG, Uzilov AV, Katzman S, Onodera CS, Mainzer JE, Mathews DH, Lowe TM, Salama SR, Haussler D. FragSeq: transcriptome-wide RNA structure probing using high-throughput sequencing. Nat Methods. 2010;7:995–1001. doi: 10.1038/nmeth.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng Q, Ryvkin P, Li F, Dragomir I, Valladares O, Yang J, Cao K, Wang LS, Gregory BD. Genome-wide double-stranded RNA sequencing reveals the functional significance of base-paired RNAs in Arabidopsis. PLoS Genet. 2010;6:e1001141. doi: 10.1371/journal.pgen.1001141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seetin MG, Mathews DH. RNA structure prediction: an overview of methods. Methods Mol Biol. 2012;905:99–122. doi: 10.1007/978-1-61779-949-5_8. [DOI] [PubMed] [Google Scholar]
- 13.Kwok CK, Tang Y, Assmann SM, Bevilacqua PC. The RNA structurome: transcriptome-wide structure probing with next-generation sequencing. Trends Biochem Sci. 2015;40:221–232. doi: 10.1016/j.tibs.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 14.Spitale RC, Flynn RA, Torre EA, Kool ET, Chang HY. RNA structural analysis by evolving SHAPE chemistry. Wiley Interdiscip Rev RNA. 2014;5:867–881. doi: 10.1002/wrna.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weeks KM. Advances in RNA structure analysis by chemical probing. Curr Opin Struct Biol. 2010;20:295–304. doi: 10.1016/j.sbi.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16••.Ding Y, Tang Y, Kwok CK, Zhang Y, Bevilacqua PC, Assmann SM. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature. 2014;505:696–700. doi: 10.1038/nature12756. This paper along with Refs. [17••,20••] combined DMS modification of RNA and high throughput sequencing to analyze in vivo RNA structurome for the first time. These analyses reveal many interesting structural features associated with distinct functional motifs and principles that govern structure formation in cells. [DOI] [PubMed] [Google Scholar]
- 17••.Rouskin S, Zubradt M, Washietl S, Kellis M, Weissman JS. Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature. 2014;505:701–705. doi: 10.1038/nature12894. See annotation to Ref. [16••] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spitale RC, Crisalli P, Flynn RA, Torre EA, Kool ET, Chang HY. RNA SHAPE analysis in living cells. Nat Chem Biol. 2013;9:18–20. doi: 10.1038/nchembio.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19••.Spitale RC, Flynn RA, Zhang QC, Crisalli P, Lee B, Jung JW, Kuchelmeister HY, Batista PJ, Torre EA, Kool ET, et al. Structural imprints in vivo decode RNA regulatory mechanisms. Nature. 2015;519:486–490. doi: 10.1038/nature14263. This paper reports a newly designed SHAPE reagent that enables purification and selective sequencing of modified RNA fragments, increasing specificity and sensitivity of in vivo SHAPE. The dramatic improvement of probing accuracy made it possible to examine genetically programmed RNA structural features and structural imprints of RBP binding and RNA modifications. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20••.Talkish J, May G, Lin Y, Woolford JL, Jr, McManus CJ. Mod-seq: high-throughput sequencing for chemical probing of RNA structure. RNA. 2014;20:713–720. doi: 10.1261/rna.042218.113. See annotation to Ref. [16••] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kladwang W, VanLang CC, Cordero P, Das R. Understanding the errors of SHAPE-directed RNA structure modeling. Biochemistry. 2011;50:8049–8056. doi: 10.1021/bi200524n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shabalina SA, Ogurtsov AY, Spiridonov NA. A periodic pattern of mRNA secondary structure created by the genetic code. Nucleic Acids Res. 2006;34:2428–2437. doi: 10.1093/nar/gkl287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smola MJ, Calabrese JM, Weeks KM. Detection of RNA–protein interactions in living cells with SHAPE. Biochemistry. 2015;54:6867–6875. doi: 10.1021/acs.biochem.5b00977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA–protein interactions. Nature. 2015;518:560–564. doi: 10.1038/nature14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roost C, Lynch SR, Batista PJ, Qu K, Chang HY, Kool ET. Structure and thermodynamics of N6-methyladenosine in RNA: a spring-loaded base modification. J Am Chem Soc. 2015;137:2107–2115. doi: 10.1021/ja513080v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26•.Homan PJ, Favorov OV, Lavender CA, Kursun O, Ge X, Busan S, Dokholyan NV, Weeks KM. Single-molecule correlated chemical probing of RNA. Proc Natl Acad Sci U S A. 2014;111:13858–13863. doi: 10.1073/pnas.1407306111. This paper along with Ref. [27•] reports new chemical probing conditions that introduce mutations to sites of modifications. They further use correlation analysis to reveal through-space contacts and cluster analysis to reveal distinct states in the structural ensemble. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27•.Siegfried NA, Busan S, Rice GM, Nelson JA, Weeks KM. RNA motif discovery by SHAPE and mutational profiling (SHAPE-MaP) Nat Methods. 2014;11:959–965. doi: 10.1038/nmeth.3029. See annotation to Ref. [26••] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Helwak A, Kudla G, Dudnakova T, Tollervey D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell. 2013;153:654–665. doi: 10.1016/j.cell.2013.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kudla G, Granneman S, Hahn D, Beggs JD, Tollervey D. Cross-linking, ligation, and sequencing of hybrids reveals RNA–RNA interactions in yeast. Proc Natl Acad Sci U S A. 2011;108:10010–10015. doi: 10.1073/pnas.1017386108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30••.Sugimoto Y, Vigilante A, Darbo E, Zirra A, Militti C, D'Ambrogio A, Luscombe NM, Ule J. hiCLIP reveals the in vivo atlas of mRNA secondary structures recognized by Staufen 1. Nature. 2015;519:491–494. doi: 10.1038/nature14280. This paper uses crosslinking and proximity ligation to determine RNA structures bound by the dsRBP STAU1, and revealed long range and alternative RNA structures, which are impossible to find using chemical probing methods. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramani V, Qiu R, Shendure J. High-throughput determination of RNA structure by proximity ligation. Nat Biotechnol. 2015;33:980–984. doi: 10.1038/nbt.3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng CY, Chou FC, Kladwang W, Tian S, Cordero P, Das R. Consistent global structures of complex RNA states through multidimensional chemical mapping. Elife. 2015;4:e07600. doi: 10.7554/eLife.07600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thakor N, Holcik M. IRES-mediated translation of cellular messenger RNA operates in eIF2alpha-independent manner during stress. Nucleic Acids Res. 2012;40:541–552. doi: 10.1093/nar/gkr701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fuller-Pace FV. DEAD box RNA helicase functions in cancer. RNA Biol. 2013;10:121–132. doi: 10.4161/rna.23312. [DOI] [PMC free article] [PubMed] [Google Scholar]